
Abstract
Background
Oligodendroglioma is a rare type of primary brain tumor which, like other malignant gliomas, metastasizes very rarely even when in high-grade form.
Case report
A 36-year-old white man diagnosed 29 months previously as having 1p/19q codeleted anaplastic oligodendroglioma presented bilateral cruralgia and lower limb motor deficits. A computed tomography scan showed multiple osteoblastic bone lesions. The presence of oligodendroglial cells was revealed by bone marrow biopsy and confirmed by immunohistochemical analyses. A positon emission tomography-computed tomography scan confirmed the exclusive involvement of bones.
Conclusion
This case joins less than 20 other reported cases of oligodendroglioma bone marrow metastasis, and is one of only a handful of cases of diffuse bone metastases beyond the axial skeleton. To the best of our knowledge, the early relapse of 1p/19q codeleted anaplastic oligodendroglioma with this distribution of metastases has never been described in the literature.
Similar content being viewed by others


Background
It has generally been thought that primary brain tumors rarely metastasize, and that these rare cases only occur inside the central nervous system (CNS). However, a rapid review of the literature demonstrates that this is not true. In a reported series of 116 extracranial metastasis cases, approximately 40% were glioblastomas, 27% medulloblastomas, 16% ependymomas, 10% astrocytomas, and 5% oligodendrogliomas [1]. The oligodendroglioma is a rare neoplasm in adults, and accounts for less than 5% of all primary brain tumors and 5–20% of all glial neoplasms [2]. It can be low grade (grade II) or high grade (grade III, or anaplastic). The prognosis is better than for glioblastoma, with a protracted natural history and a greater response to therapy [3]. The genetic loss of 1p/19q chromosomes corresponds to a distinct tumor entity in oligodendroglioma, which is characterized by a prolonged natural history irrespective of treatment and by a high sensitivity to both radiotherapy (RT) and chemotherapy [3, 4]. As shown in the aforementioned reported series, metastatic relapse is much rarer in oligodendrogliomas than for other malignant gliomas, with fewer than 50 cases reported in the literature [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42]. Bone seems to be one of the most frequent metastatic sites outside the CNS in this type of glioma, with approximately 20 instances reported since 1970, compared to sites in the lymph nodes, liver, lungs, spleen, or pancreas [12, 14, 29, 31,32,33,34,35,36,37,38,39,40,41,42]. However, to the best of our knowledge, few cases of patients with diffuse bony metastases extending beyond the axial skeleton have been described to date [34, 36,37,38,39,40,41].
We report a case of bone metastases from a 1p/19q codeleted and IDH1-mutant anaplastic oligodendroglioma 29 months after first diagnosis of a brain tumor.
Case presentation
A 36-year-old white man presented in April 2017 with a 2-week history of bilateral cruralgia. Following a diagnosis in December 2014 of a right temporoparietal grade III oligodendroglioma with IDH1 mutation and 1p/19q codeletion, he underwent emergency surgical cerebral decompression for a comatose state secondary to brain herniation, with incomplete resection due to massive cerebral edema. A second surgical resection 1 month later remained incomplete, with residual in-depth disease. He was treated with cranial RT with concomitant temozolomide chemotherapy. Identical chemotherapy treatment was continued from March to December 2015 (standard protocol for high-grade gliomas), receiving six series of treatment that ended 11 months after the second surgery [43]. In January 2016, a cranioplasty was carried out to treat infected craniotomy bone flaps. He was monitored for the following 9 months with regular MRI scans. In August 2016, that is 20 months after surgical resection, a local tumor recurred and was treated with a third subtotal resection. Second-line procarbazine, lomustine, and vincristine (PCV) chemotherapy was initiated following surgery, 4 months before the current presentation.
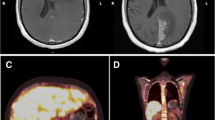
A physical examination revealed motor deficits of the lower limbs in addition to pre-existing left-sided hemiparesis and a swollen left supraclavicular lymph node. A computed tomography (CT) scan showed multiple osteoblastic bone lesions scattered throughout his spine, his pelvis and to a lesser extent his ribs, but no lymph adenopathy was identified (Fig. 1a). A positon emission tomography (PET)-CT scan confirmed the presence of the lesions identified in the CT scan and revealed further bone lesions in his pelvis (the right ischium, the pubic area, the left acetabulum, the left part of the sacrum, and the right and left iliac wing), in his sternum with a maximum standardized uptake value (SUVmax) of 4.8, in his right (SUVmax = 5.2) and left humerus (SUVmax = 4.3), and in his right scapula (SUVmax = 4.8). No soft tissue lesions (Virchow’s node included) were found, confirming the exclusive involvement of bones (Fig. 1). The analysis of lymph acquired through fine-needle aspiration of his left supraclavicular lymph node excluded lymph node metastasis. A brain MRI showed local disease progression with an increase in the volume of the right temporoparietal tumor, which had spread to the left temporal region and the superior sagittal sinus. Cytological and biochemical analysis of cerebrospinal fluid excluded carcinomatosis meningitis. Immunohistochemical analyses were carried out on a bone marrow sample from his left iliac crest. A tumoral proliferation of ovoid cells was observed in the medullary cavity. Cells were hyperchromic, had pale cytoplasm and irregular nuclei, and mitosis was occurring: this morphological aspect perfectly reflected the diagnostic for the initial anaplastic oligodendroglioma brain tumor. Immunostaining was positive for glial fibrillary acidic protein (GFAP) and the mutated form of IDH1, and therefore excluded any diagnosis other than oligodendroglioma metastasis (Fig. 2). The second-line PCV chemotherapy, initiated following the third subtotal resection 4 months before the current presentation, was continued with two additional cycles in the absence of a real therapeutic alternative and at our patient’s request. It was stopped due to the worsening of his clinical status with positional vertigo, nausea, vomiting, and headache compatible with carcinomatous meningitis, followed by status epilepticus and lethargy; he died after 4 months after the diagnosis of metastases.

A computed tomography scan of the pelvis shows multiple osteoblastic bone lesions (a). Positon emission tomography-computed tomography scan demonstrates the hypermetabolic nature of these spinal bone lesions (b) located beyond the axial skeleton, shown here on the right scapula (c) and the left humerus (d)

Photomicrographs (magnification × 20) of a hematoxylin and eosin-stained section of metastatic oligodendroglial cells in the bone specimen (a), oval tumor cells with pale cytoplasm and irregular cell nuclei with positive Olig2 immunostaining (b), and the mutated form (R132H) of IDH1 (c)
Discussion
Oligodendrogliomas are rare tumors, and represent 5 to 20% of gliomas. Like other glioma tumors, their evolution is characterized by local relapses in the vast majority of cases, and the occurrence of cases of metastasis at sites located outside the CNS is extremely rare. Among these tumors, oligodendrogliomas present the lowest risk of extracerebral metastasis [1]. Different hypotheses have been proposed to explain this phenomenon, which could be due to the anatomical properties of the CNS (absence of a lymphatic system, presence of a neuromeningeal barrier or an environment that is not hostile enough to allow the selection of clones that can metastasize outside the CNS) or could also be explained by a patient history in which death occurred before metastases could spread [1, 44]. However, several publications described how malignant gliomas could bypass the possible aforementioned mechanisms to metastasize outside the CNS. Many factors associated with metastatic relapse outside the CNS have been described, such as repeated intracranial surgery, the presence of a shunt, or prolonged survival [1, 35, 39, 44, 45]. The four intracranial surgeries could explain the metastatic evolution described in this case study. Although there is currently no published evidence, repeated craniotomies may give tumor cells access to the cerebral parenchyma venous pathway, the dura mater, or the scalp, thus bypassing the blood–brain barrier.
Another mechanism that is described is the direct impact of the tumor on the meninges, which increases the permeability of the blood–brain barrier and could therefore facilitate the hematogenous dissemination of tumor cells [31]. Finally, bone flap infection, as observed in this case, could also provide a means of metastatic spread that has not been reported in the literature to date. Here, tumor cells may access vascular pathways following local inflammation, during which vasoactive factors could lead to vasodilation and increased permeability, causing vasogenic edema. However, like the aforementioned mechanisms, the validity of this theory cannot be verified. The delayed occurrence of this metastatic spread may result from a selection of clones following multimodal treatment (RT and chemotherapy), which may make them more aggressive [37, 46]. Tumor cells may also be diffused via the cephalo-rachidian liquid, where they were present in 14% of patients in a series from the 1980s [46]. Our patient presented an oligodendroglioma with 1p/19q codeletion which corresponded to a specific tumor. This molecular change is observed in 30 to 65% of anaplastic oligodendrogliomas [47,48,49]. These tumors are characterized by a high sensitivity to RT and chemotherapy, and longer survival with and without treatment. The mutation of IDH1 or IDH2 genes is observed in all anaplastic oligodendrogliomas with 1p/19q codeletion, and this mutation concerns the IDH1 gene in the present case study. Only five other cases of metastasis outside the CNS in oligodendroglioma with 1p/19q codeletion have been described in the literature, and no correlation was found between this chromosomal signature and the metastatic relapse [9, 10, 39,40,41]. The 10-year prolonged survival of patients with this tumor sub-type, compared to 2 years for tumors without this molecular alteration, has been highlighted by previous publications as a possible explanation for the metastatic evolution observed in one of the rare cases reported, in which a 15-year interval occurred between the baseline diagnosis and the metastatic relapse. [40, 50]. Our case does not confirm this hypothesis because metastatic relapse occurred after a particularly short period of 16 months. This case is also singular in terms of metastatic distribution: whereas bone locations with medullary sites appear to be the most frequently reported metastatic sites for oligodendrogliomas (less than 20 cases reported in the literature), only a few descriptions of similar disseminated bone metastases have been reported in the rachis and beyond the axial skeleton [36, 37, 41].
A last explanation could be found in the genetic alteration. Some studies have shown that gliomas result from the accumulation of several distinct chromosomal alterations such as loss of heterozygosity (LOH) on the short arm of chromosome 1 (1p), the long arm of chromosome 19 (19q), and the long arm of chromosome 10 (10q). These acquired genomic alterations in tumor cells have an important role in formulating the prognosis of patients with gliomas, in addition to the histological classification, because some correlate with the clinical outcome. Recently, a correlation between the loss of 10q and anaplastic glial tumor and overall survival was demonstrated [51]. Of note, 10q is the site of PTEN encoded on position 10q23 [52]. Today, PTEN is considered a tumor suppressor and metabolic regulator [53]. A whole genome analysis on prostate cancer-derived cells showed that PTEN loss was robustly associated with the bone metastasis-derived PC3 prostate cancer cell lines but not with the lymph node metastasis-derived LNCaP prostate cancer cell lines [54]. Furthermore, PTEN loss was the worst prognostic factor of gastric cancer that developed bone metastasis and was a factor of bone tumor invasion and tumor cell spread in bone [55, 56].
Conclusion
Metastases from oligodendrogliomas are extremely rare. The novelty of this case lies in the early occurrence of metastatic evolution in a sub-type of oligodendroglioma that habitually has a good prognosis, the singular distribution of metastases, and the different mechanisms that may play a role in tumor cell diffusion.
References
Liwnicz BH, Rubinstein LJ. The pathways of extraneural spread in metastasizing gliomas. Hum Pathol. 1979;10(4):453–67.
Koeller KK, Rushing EJ. From the archives of the AFIP: Oligodendroglioma and its variants: radiologic-pathologic correlation. Radiogr Rev Publ Radiol Soc N Am Inc. 2005;25(6):1669–88.
Stupp R, Brada M, van den Bent MJ, Tonn J-C, Pentheroudakis G, ESMO Guidelines Working Group. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):iii93–101.
Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66(20):9852–61.
Choon A, Roepke JE. Importance of immunohistochemical staining in metastatic anaplastic oligodendroglioma. Arch Pathol Lab Med. 2004;128(4):489–90.
Lee C-C, Jiang J-S, Chen ET, Yokoo H, Pan Y-H, Tsai M-D. Cytologic diagnosis of a metastatic oligodendroglioma in a pleural effusion. A case report. Acta Cytol. 2006;50(5):542–4.
Verma N, Nolan C, Hirano M, Young RJ. Intramedullary spinal cord and leptomeningeal metastases from intracranial low-grade oligodendroglioma. Clin Imaging. 2014;38(4):505–7.
Liu H, Lu X, Thakral B. A rare presentation of anaplastic oligodendroglioma metastatic to bone marrow, mimicking blast cells. Br J Haematol. 2016;174(3):344.
Demeulenaere M, Duerinck J, Du Four S, Fostier K, Michotte A, Neyns B. Bone Marrow Metastases from a 1p/19q Co-deleted Oligodendroglioma - A Case Report. Anticancer Res. 2016;36(8):4145–9.
Krijnen JLM, Fleischeur REM, van Berkel M, Westenend PJ. Metastatic oligodendroglioma: a case report and incidence in The Netherlands. Clin Neuropathol. 2010;29(3):141–6.
Garner J, Morcos Y, Bari M. Extradural cord compression due to metastatic oligodendroglioma. J Neuro-Oncol. 2002;58(1):71–5.
Anand M, Kumar R, Jain P, Gupta R, Ghosal N, Sharma A, et al. Metastatic anaplastic oligodendroglioma simulating acute leukemia. A case report. Acta Cytol. 2003;47(3):467–9.
Can B, Akpolat I, Meydan D, Üner A, Kandemir B, Söylemezoğlu F. Fine-needle aspiration cytology of metastatic oligodendroglioma: case report and literature review. Acta Cytol. 2012;56(1):97–103.
Pectasides D, Halikia A, Visvikis A, Bountouroglou N, Tzikas I, Iakovidou I, et al. Successful management of extracranial metastases from cerebral high-grade oligodendroglioma: report of a case with literature review. J BUON. 2002 Sep;7(3):273–6.
Han SR, Yoon SW, Yee GT, Choi CY, Lee DJ, Sohn MJ, et al. Extraneural metastases of anaplastic oligodendroglioma. J Clin Neurosci. 2008;15(8):946–9.
Natelson SE, Dyer ML, Harp DL. Delayed CSF seeding of benign oligodendroglioma. South Med J. 1992;85(10):1011–2.
Steininger H, von Streitberg U. Oligodendroglioma with cervical lymph node metastasis. Pathologe. 1993;14(6):386–90.
Schröder R, Lorenzen J, Ostertag H, Ortmann M, Hansmann ML. Extraneural metastasis of brain and spinal cord tumors. Report of 2 cases. Pathologe. 1995 May;16(3):223–9.
Gerrard GE, Bond MG, Jack AS. Bone marrow infiltration by a parietal lobe grade III oligodendroglioma. Clin Oncol. 1995;7(5):321–2.
Monzani V, Rovellini A, Masini B, Cappricci E, Miserocchi G. Metastatic oligodendroglioma. Case report. J Neurosurg Sci. 1996;40(3–4):239–41.
Ng H-K, Sun DTF, Poon W-S. Anaplastic oligodendroglioma with drop metastasis to the spinal cord. Clin Neurol Neurosurg. 2002;104(4):383–6.
McBryde C, Hamid N, Mitchell R. Anaplastic oligodendroglioma with metastasis to the spinal cord. Br J Neurosurg. 2003;17(4):364–6.
Ng WH, Lim TCC, Tan KK. Disseminated spread of recurrent oligodendroglioma (WHO Grade II). J Clin Neurosci. 2006;13(5):602–7.
Ozişik PA, Işikay I, Oruçkaptan H, Söylemezoğlu F, Ozcan OE. Unusual massive spinal metastasis of an intracranial oligodendroglioma. Turk Neurosurg. 2008;18(3):276–80.
Volavšek M, Lamovec J, Popović M. Extraneural metastases of anaplastic oligodendroglial tumors. Pathol Res Pract. 2009;205(7):502–7.
Noshita N, Mashiyama S, Fukawa O, Asano S, Watanabe M, Tominaga T. Extracranial metastasis of anaplastic oligodendroglioma with 1p19q loss of heterozygosity--case report. Neurol Med Chir (Tokyo). 2010;50(2):161–4.
Oshiro S, Komatsu F, Tsugu H, Nabeshima K, Abe H, Ohkawa M, et al. A case of intramedullary cervical metastasis from cerebellar anaplastic oligodendroglioma without typical MR appearance for CSF dissemination. No Shinkei Geka. 2010;38(3):279–85.
Wu Y, Liu B, Qu L, Tao H. Extracranial skeletal metastasis in anaplastic oligodendroglioma: case report and review of the literature. J Int Med Res. 2011;39(3):960–7.
Cordiano V, Miserocchi F, Storti M. Bone marrow metastases from anaplastic oligodendroglioma presenting with pancytopenia and hypogammaglobulinemia: a case report. Tumori. 2011;97(6):808–11.
Elefante A, Peca C, Del Basso De Caro ML, Russo C, Formicola F, Mariniello G, et al. Symptomatic spinal cord metastasis from cerebral oligodendroglioma. Neurol Sci. 2012;33(3):609–13.
Mazza E, Belli C, Terreni M, Doglioni C, Losio C, Cantore M, et al. Breast metastases from oligodendroglioma: an unusual extraneural spread in two young women and a review of the literature. Crit Rev Oncol Hematol. 2013;88(3):564–72.
Zustovich F, Della Puppa A, Scienza R, Anselmi P, Furlan C, Cartei G. Metastatic oligodendrogliomas: a review of the literature and case report. Acta Neurochir. 2008;150(7):699–702. discussion 702-703
Uzuka T, Kakita A, Inenaga C, Takahashi H, Tanaka R, Takahashi H. Frontal anaplastic oligodendroglioma showing multi-organ metastases after a long clinical course. Case report. Neurol Med Chir (Tokyo). 2007;47(4):174–7.
Giordana MT, Ghimenti C, Leonardo E, Balteri I, Iudicello M, Duò D. Molecular genetic study of a metastatic oligodendroglioma. J Neuro-Oncol. 2004;66(3):265–71.
Kim JG, Park CO, Hyun DK, Ha YS. Spinal epidural metastasis of cerebral oligodendroglioma. Yonsei Med J. 2003;44(2):340–6.
Maloney PR, Yamaki VN, Kumar R, Johnson D, Hunt C, Jentoft ME, et al. Osteosclerosis Secondary to Metastatic Oligodendroglioma. Rare Tumors. 2017;9(1):6837.
Kural C, Pusat S, Sentürk T, Seçer Hİ, Izci Y. Extracranial metastases of anaplastic oligodendroglioma. J Clin Neurosci. 2011;18(1):136–8.
Carlsen JG, Tietze A, Lassen YA, Rosendal F. Paraplegia due to drop metastases from anaplastic oligodendroglioma. Br J Neurosurg. 2012;26(1):94–5.
Greene J, Cadoo K, Ti J, O’Donnell N, Allcutt D, Farrell M, et al. 1p19q co-deleted oligodendroglioma metastatic to bone. Clin Neuropathol. 2013;32(2):139–41.
Merrell R, Nabors LB, Perry A, Palmer CA. 1p/19q chromosome deletions in metastatic oligodendroglioma. J Neuro-Oncol. 2006;80(2):203–7.
Li G, Zhang Z, Zhang J, Jin T, Liang H, Gong L, et al. Occipital anaplastic oligodendroglioma with multiple organ metastases after a short clinical course: a case report and literature review. Diagn Pathol. 2014;9:17.
Morrison T, Bilbao JM, Yang G, Perry JR. Bony metastases of anaplastic oligodendroglioma respond to temozolomide. Can J Neurol Sci. 2004;31(1):102–8.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med. 2005;352(10):987–96.
Al-Ali F, Hendon AJ, Liepman MK, Wisniewski JL, Krinock MJ, Beckman K. Oligodendroglioma Metastatic to Bone Marrow. Am J Neuroradiol. 2005;26(9):2410–4.
Houston SC, Crocker IR, Brat DJ, Olson JJ. Extraneural metastatic glioblastoma after interstitial brachytherapy. Int J Radiat Oncol Biol Phys. 2000;48(3):831–6.
Macdonald DR, O’Brien RA, Gilbert JJ, Cairncross JG. Metastatic anaplastic oligodendroglioma. Neurology. 1989;39(12):1593–6.
Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473–9.
Idbaih A, Dalmasso C, Kouwenhoven M, Jeuken J, Carpentier C, Gorlia T, et al. Genomic aberrations associated with outcome in anaplastic oligodendroglial tumors treated within the EORTC phase III trial 26951. J Neuro-Oncol. 2011;103(2):221–30.
Intergroup Radiation Therapy Oncology Group Trial 9402, Cairncross G, Berkey B, Shaw E, Jenkins R, Scheithauer B, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24(18):2707–14.
Smith JS, Perry A, Borell TJ, Lee HK, O’Fallon J, Hosek SM, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol Off J Am Soc Clin Oncol. 2000;18(3):636–45.
Ramirez C, Bowman C, Maurage C-A, Dubois F, Blond S, Porchet N, et al. Loss of 1p, 19q, and 10q heterozygosity prospectively predicts prognosis of oligodendroglial tumors—towards individualized tumor treatment? Neuro Oncol. 2010;12(5):490–9.
Pulido R, Baker SJ, Barata JT, Carracedo A, Cid VJ, Chin-Sang ID, et al. A unified nomenclature and amino acid numbering for human PTEN. Sci Signal. 2014;7(332):pe15.
Chen C-Y, Chen J, He L, Stiles BL. PTEN: Tumor Suppressor and Metabolic Regulator. Front Endocrinol. 2018;9:338.
Seim I, Jeffery PL, Thomas PB, Nelson CC, Chopin LK. Whole-Genome Sequence of the Metastatic PC3 and LNCaP Human Prostate Cancer Cell Lines. G3. 2017;7(6):1731–41.
Kim C, Lee C-K, Chon HJ, Kim JH, Park HS, Heo SJ, et al. PTEN loss and level of HER2 amplification is associated with trastuzumab resistance and prognosis in HER2-positive gastric cancer. Oncotarget. 2017;8(69):113494–501.
Xi Y, Chen Y. Oncogenic and Therapeutic Targeting of PTEN Loss in Bone Malignancies. J Cell Biochem. 2015;116(9):1837–47. [cited 2019 Feb 26]. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcb.25159
Acknowledgements
Not applicable.
Availability of data and materials
All data and materials related to this report are accessible at any time upon request.
Author information
Authors and Affiliations
Contributions
MB, GN, RS followed the patient clinically. MB, GN and RS wrote the case report. MPC and KB contributed to the medical diagnosis and staging. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient's next of kin for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Burgy, M., Chenard, MP., Noël, G. et al. Bone metastases from a 1p/19q codeleted and IDH1-mutant anaplastic oligodendroglioma: a case report. J Med Case Reports 13, 202 (2019). https://doi.org/10.1186/s13256-019-2061-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-019-2061-4