
Abstract
Background
Ring chromosome 15 has been associated in previous studies with different clinical characteristic such as cardiac problems, digit and musculoskeletal abnormalities, and mental and motor problems among others. Only 97 clinical cases of ring chromosome 15 syndrome have been reported since 1966 and a common phenotype for these patients has not been established.
Case presentation
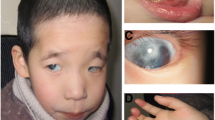
The present case report describes a 15-month-old girl from the Amazon region of Ecuador, of Mestizo ancestry, who after cytogenetic tests showed a 46,XX,r(15) karyotype in more than 70% of metaphases observed. Her parents were healthy and non-related. The pregnancy was complicated and was positive for intrauterine growth retardation. Her birth weight was 1950 g, her length was 43.5 cm, and she had a head circumference of 29.3. In addition to postnatal growth delay, she had scant frontal hair, small eyes, hypertelorism, low-set of ears, flattened nasal bridge, anteverted nostrils, down-turned mouth, three café au lait spots, and delayed dentition.
Conclusions
Despite the frequency of some phenotypes expressed in the different clinical cases reviewed and the present case, a common phenotype for patients with ring 15 could not be determined and it is restricted to the region of the chromosome lost during the ring formation.
Similar content being viewed by others

Background
Rings are aberrant structures that arise from random events during cell replication, which yield a circular chromosome [1]. They have been identified for all human chromosomes [2]. Ring formation often results from terminal breaks in both chromosome arms, followed by a fusion event of the broken ends [1, 3]. As a result, genetic material may or may not be lost, leading to different types of rings [2]. Rings also occur due to the fusion of subtelomeric sequences like telomere–telomere fusion. This fusion might lead to complete ring formation with no significant loss of genetic material, as seen in patients with normal phenotype [1].
Due to the circular nature of the affected chromosomes, they tend to be unstable during cell division [4]. Sister chromatid exchanges due to cell mistakes during mitosis can result in chromosomal abnormalities that produce dicentric rings, interlocked rings, or ring loss leading to monosomy or mosaicism [4, 5]. Furthermore, transmission of unstable rings can lead to de novo ring formations in the next generation [2, 3, 6].
Ring chromosome 15 has been previously described in the literature. The first case reported [7], described a patient with café au lait macules, strabismus, and diminished mental and motor development. Further clinical features ranged from cardiac problems to digit and musculoskeletal abnormalities. Dysmorphic features, such as growth retardation, triangular faces, eye abnormalities, and developmental abnormalities are common features defined in ring 15 syndrome, which in this case report is abbreviated to r(15) syndrome [8, 9]. Since the first case report of a patient with ring chromosome 15 syndrome in 1966, only 97 clinical cases have been published in the literature to the date. In addition, it has been suggested that the clinical phenotype is correlated with the amount of deletions and genomic imbalances during the ring formation and the instability of the latter [1, 5, 10, 11].
This report describes the traits, demographic details, cytogenetic analysis, molecular diagnosis, and clinical manifestations of a baby girl with ring chromosome 15 and compares this case with other cases previously reported in the literature. The aim is to identify a common phenotype of ring chromosome 15 syndrome and improve our understanding of this genetic disorder.
Case presentation
Our patient came from Coca city in the Amazon region of Ecuador; according to the ancestry profile of the Ecuadorian population [12] she is a Mestizo: 63.1% Native American, 30.3% European, and 6.6% African ancestry. The first contact with our patient was in April 2016 at 11-months old, she was the second child of non-consanguineous healthy parents. Her father was 32-years old and her mother was 31 at the time of birth. Our patient was born at 38 weeks of gestation from a complicated pregnancy. Intrauterine growth retardation, hypermature placenta, and low amniotic fluid were detected during the fifth month of pregnancy. The infant was delivered by cesarean C-section. Her birth weight was 1950 g (<third centile), her length was 43.5 cm (<third centile), and she had a head circumference (HC) of 29.3 (<third centile). The Appearance, Pulse, Grimace, Activity, and Respiration (APGAR) scores were 6–8. She required gastric probing, phototherapy, and thermo-cradle care for 10 days after birth.
At 15-months old after a physical examination, she showed a weight of 6000 g (<third centile), length of 63 cm (<third centile), and HC of 40 cm (third centile). In addition to postnatal growth delay, she had scant frontal hair, small eyes, hypertelorism, low-set of ears, flattened nasal bridge, anteverted nostrils, down-turned mouth, three café au lait spots, and delayed dentition. Developmentally, she showed normal psychomotor progress. She held up her head at 3 months, sat down at 6 months, and walked with support at 15 months. Bone age was determined by bone densitometry.
A Giemsa trypsin banding (GTG) technique performed on our patient, at 450-band resolution, showed 73 out of 100 metaphases with a 46,XX,r(15) karyotype (Fig. 1). The parental karyotypes were normal. In addition, 750 ng of patient’s deoxyribonucleic acid (DNA) was used for hybridization in the Affymetrix 750K Array (Affymetrix, Santa Clara, CA, USA). The arrays were processed in the Fluidics Affymetrix 450 Station and scanned with GeneChip™ 3000 (Affymetrix, Santa Clara, CA, USA).

Karyotype of the patient from the present study showing a 46,XX,r(15)
The arrays analysis revealed two copy number loss in the r(15)(q26.2q26.3) chromosomal region of 5,927,199 kbp, encompassing genes NR2F2 to OR4F13P, and in the 15q26.3 chromosomal region of 3,459,976 kbp, encompassing genes FAM169B to OR4F13P (Table 1). The analysis also revealed additional abnormalities in chromosomes 14 and X. In chromosome 14, our patient had a three-copy gain of genetic information at 369,716 kbp in the 14q32.33 chromosomal region, including genes KIAA0125, ADAM6. While on chromosome X, there was a copy number loss of the genes CT45A1 to CT45A6 in the 119,101kbp region of Xq26.3 and a three-copy number gain of the genes spindlin family member 4 (SPIN4) and LOC92249 in the 347,978kbp region of Xq11.1 (Table 2).
As a common phenotype for r(15) syndrome is not clearly described, we compared our patient case with 97 clinical cases of ring chromosome 15 found in the literature. In all 98 cases the average age at diagnosis was 10.85 years in females and 12.21 in males. The average maternal and paternal age at the time of birth ranged between 20 and 42 years (29.73%) and 22–46 (31.59%) years, respectively. Furthermore, the average weight and height for patients was 2252 g and 43.94 cm for females and 2345 g and 46.29 cm for males.
Among all phenotypic features, the following six were the most common in all 98 cases: growth retardation (76.53%), microcephaly (39.8%), clinodactyly (33.67%), triangular faces (32.65%), brachymesophalangy (32.65%), and low weight (42.86%). Other abnormalities for instance café au lait macules (25.51%), and psychological and behavioral abnormalities, such as developmental delay (27.55%), mental deficit (40.82%), and language deficit (27.55%) were also present (Table 3).
Discussion
Ring chromosome 15 syndrome is a rare genetic disorder, which is still not completely understood. Ring chromosomes are formed due to a loss of the distal fraction of the chromosome, followed by a fusion event to the terminal region of the short arm of the same chromosome [1, 5]. The size and position of the lost fragment determines the phenotype of the patients. As the nature of the chromosome is unstable and depending on what genes have been lost, the spectrum of clinical features can vary from patient to patient [1, 4].
Copy number variations (CNVs) are one of the most referred causes of human disease pathogenesis because they often include important and functional DNA sequences. It has been reported that gene expression variability is highly related to CNVs larger than 40 kb, which is consistent with the number of copies lost in this study [13]. Geneticists are no longer associating diseases with common genetic variants inherited through different generations, but with large and rare structural variants of recent origin (like ring formations). Large and rare structural variants of recent origin are responsible for serious conditions like autism, schizophrenia, and intellectual disability; intellectual disability is one of the most common phenotypes found in the different clinical cases analyzed [14].
The clinical manifestations of the case described in this study fit well with previous ones reported in literature. In most of the cases, there is the presence of invariable size rings with a minimal loss of genetic material. We identified that there is a common breakpoint in most: the genomic region 15q26. Duplication, deletions, and gains in this region could be associated with different phenotypic manifestations as the region harbors different genes [15]. For instance, insulin-like growth factor 1 receptor (IGF1R) is involved in normal growth and development [16]. Mutations and changes in this gene, such as abnormal number of structures of receptor, can lead to different pathologies associated with growth deficit, such as Prader–Willi syndrome and Silver–Russell syndrome [17, 18]. The loss of one copy of the gene can also lead to abnormal head and body size. Other genes such as synemin (SYNM) and tetratricopeptide repeat domain 23 (TTC23), which play a role in the assembly of intermediate filaments in the Z-ring, are shared among previously described cases. Abnormalities in these genes can lead to musculoskeletal disorders, such as delayed bone age, digit and finger clinodactyly, and brachydactyly [19].
In addition, with the array analysis we found two other chromosomes involved in loss and gain of genetic material: chromosome 14 and chromosome X. Genes identified to be part of the region lost, Xq26.3, included several members of the family cancer/testis antigen family 45 (CT45), such as CT45A1–CT45A6 associated with oncogenic function [20]. On the other hand, gain of genetic material on region Xq11.1 involved genes SPIN4 and LOC92249; losses and mutations on these genes have been associated with periodontitis phenotype [21].
The literature review for phenotype comparison (Table 3) also led to interesting findings; the different phenotypes of all 98 reviewed cases were categorized. We reported the frequency of shared phenotypes and found that growth retardation, microcephaly, and a low weight were the main characteristics in cases with r(15) (Fig. 2). Figure 3 also shows phenotypes shared among the clinical cases in the literature with mental deficit, brachymesophalangy, clinodactyly, and triangular face as the dominant traits. Finally, the most shared phenotypes between all the cases were selected and compared (Fig. 4) with the aim of finding a common trait among the patients; we found that growth retardation was present in almost 80% of the cases. However, due to the low statistical significance of the frequency of the phenotype expressed by all cases, a common phenotype for patients with ring chromosome 15 could not be established and is highly restricted to the region of chromosome 15 lost due to the ring formation. It has been suggested that the clinical phenotype of patients with ring chromosome depends on two main features: the first is related to telomeric deletions responsible for the ring formation, and the second is directly related to the amount of DNA lost and the functional genes involved in this region [5].

Shared phenotype frequencies among 97 clinical cases and the present case

Shared phenotype frequencies among the 97 reviewed clinical cases

Frequencies of the most common phenotype expressed by 97 clinical cases and the patient of this study
Conclusions
Ring chromosome 15 syndrome shows a heterogeneous phenotype which is dependent on the region and the genes involved in the break, although there are some traits like the ones shown in Figs. 2 and 3 that have more than 40% frequency in the individual cases reviewed. Those traits can be suspected to be the shared phenotype of r(15) syndrome. Cases that show a breakpoint on genomic region 15q26 show common dysmorphic features, such as musculoskeletal abnormalities and growth retardation. Furthermore, cytogenetic and molecular approaches could facilitate the association of phenotypic and genotypic correlations. Further investigation and description of patients’ cases could provide an insight to the genetic aberrations involved in ring chromosome 15 disorders, in order to offer more information on the genes and the genomic regions affected.
Abbreviations
- APGAR:
-
Appearance, Pulse, Grimace, Activity, and Respiration
- CNV:
-
Copy number variation
- CT45 :
-
Cancer/testis antigen family 45
- GTG:
-
Giemsa trypsin banding
- HC:
-
Head circumference
- IGF1R :
-
Insulin-like growth factor 1 receptor
- r(15):
-
Ring 15
- SPIN4 :
-
Spindlin family member 4
- SYNM :
-
Synemin
- TTC23 :
-
Tetratricopeptide repeat domain 23
References
Yip M-Y. Autosomal ring chromosomes in human genetic disorders. Transl Pediatr. 2015;4:164–74. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26835370%5Cn. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4729093
Rajesh H, Freckmann M-L, Chapman M. Azoospermia and paternal autosomal ring chromosomes: case report and literature review. Reprod BioMed Online. 2011;23:466–70.
Habib AGK, Sugiura K, Ueno M. Chromosome passenger complex is required for the survival of cells with ring chromosomes in fission yeast. PLoS One. 2018;13:e0190523. https://doi.org/10.1371/journal.pone.0190523.
Sodré CP, Guilherme RS, VFA M, Brunoni D, Juliano Y, Andrade JAD, et al. Ring chromosome instability evaluation in six patients with autosomal rings. Genet Mol Res. 2010;9:134–43.
Paz-y-Mino C, Benitez J, Ayuso C, Sanchez-Cascos A. Ring chromosome 6: clinical and cytogenetic behaviour. Am J Med Genet. 1990;35:481–3.
Eid MM, El-Bassyouni HT, Eid OM, Hamad SA, Elgerzawy A, Zaki MS, et al. Ring chromosome 15: expanding the phenotype. Genet Couns. 2013;24:417–25.
Jacobsen P. A ring chromosome in the 13–15 group associated with microcephalic dwarfism, mental retardation and emotional immaturity. Hereditas. 1966;55:188–91.
Fryns J, Timmermans J, D’Hondt F. Ring chromosome 15 syndrome. Hum Genet. 1979;51:43–8.
Fujimaki W, Baba K, Tatara K, Umezu R, Kusakawa S, Mashima Y. Ring chromosome 15 in a mother and her children. Hum Genet. 1987;76:302.
Guilherme RS, Ayres Meloni VF, Kim CA, Pellegrino R, Takeno SS, Spinner NB, et al. Mechanisms of ring chromosome formation, ring instability and clinical consequences. BMC Med Genet. 2011;12:171.
Daber RD, Conlin LK, Leonard LD, Canevini MP, Vignoli A, Hosain S, et al. Ring chromosome 20. Eur J Med Genet. 2012;55:381–7.
Zambrano AK, Gaviria A, Vela M, Cobos S, Leone PE, Gruezo C, et al. Ancestry characterization of Ecuador’s Highland mestizo population using autosomal AIM-INDELs. Forensic Sci Int Genet Suppl Ser. 2017;6:e477–8.
Ionita-Laza I, Rogers AJ, Lange C, Raby BA, Lee C. Genetic Association Analysis of Copy Number Variation (CNVs) in Human Disease Pathogenesis. Genomics. 2009;93:22–6.
Eichler E. Copy Number Variation and Human Disease. Nat Educ. 2008;1:1.
Szabó A, Czakó M, Hadzsiev K, Duga B, Bánfai Z, Komlósi K, et al. Small supernumerary marker chromosome 15 and a ring chromosome 15 associated with a 15q26.3 deletion excluding the IGF1R gene. Am J Med Genet Part A. 2018;176:443–9.
Riedemann J, Macaulay VM. IGF1R signalling and its inhibition. Endocr Relat Cancer. 2006;13(Suppl 1):S33–43.
Roback EW, Barakat AJ, Dev VG, Mbikay M, Chretien M, Butler MG. An infant with deletion of the distal long arm of chromosome 15 (q26.1----qter) and loss of insulin-like growth factor 1 receptor gene. Am J Med Genet. 1991;38:74–9.
Puchalska-Niedbał L, Zajączek S, Petriczko E, Kulik U. Ophthalmic treatment and vision care of a patient with rare ring chromosome 15: a case report. Case Rep Pediatr. 2014;2014:285132. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4065758&tool=pmcentrez&rendertype=abstract
Titeux M, Brocheriou V, Xue Z, Gao J, Pellissier JF, Guicheney P, et al. Human synemin gene generates splice variants encoding two distinct intermediate filament proteins. Eur J Biochem. 2001;268:6435–48.
Shang B, Gao A, Pan Y, Zhang G, Tu J, Zhou Y, et al. CT45A1 acts as a new proto-oncogene to trigger tumorigenesis and cancer metastasis. Cell Death Dis. 2014;5:e1285.
Teumer A, Holtfreter B, Völker U, Petersmann A, Nauck M, Biffar R, et al. Genome-wide association study of chronic periodontitis in a general German population. J Clin Periodontol. 2013;40:977–85.
Fujita H, Matsumoto H. Ring chromosome 15; 46,XX,r(15) (p11q26) in a girl. Jinrui Idengaku Zasshi. 1978;23:233–7.
Yunis E, Leibovici M, Quintero L. Ring (15) chromosome. Hum Genet. 1981;57:207–9.
Butler MG, Fogo AB, Fuchs DA, Collins FS, Dev VG, Phillips JA III. Brief clinical report and review: Two patients with ring chromosome 15 syndrome. Am J Med Genet. 1988;29:149–54.
de Jong G, Rossouw RA, Retief AE. Ring chromosome 15 in a patient with features of Fryns’ syndrome. J Med Genet. 1989;26:469–70.
Smith A, den Dulk G, Viersbach R, Michas J. Ring chromosome 15 and 15qs+ mosaic: Clinical and cytogenetic behaviour spanning 29 years. Am J Med Genet. 1991;40:460–3.
Horigome Y, Kondo I, Kuwajima K, Suzuki T. Familial occurrence of ring chromosome 15. Clin Genet. 1992;41:178–80.
Tamura T, Tohma T, Ohta T, Soejima H, Harada N, Abe K, et al. Ring chromosome 15 involving deletion of the insulin-like growth factor 1 receptor gene in a patient with features of Silver-Russell syndrome. Clin Dysmorphol. 1993;2:106–13.
Matsuishi T, Yamada Y, Endo K, Sakai H, Fukushima Y. Ring chromosome 15 syndrome in an adult female. J Intellect Disabil Res. 1996;40:478–81.
de Lacerda L, Carvalho JA, Stannard B, Werner H, Boguszewski MC, Sandrini R, et al. In vitro and in vivo responses to short-term recombinant human insulin-like growth factor-1 (IGF-I) in a severely growth-retarded girl with ring chromosome 15 and deletion of a single allele for the type 1 IGF receptor gene. Clin Endocrinol (Oxf). 1999;51:541–50.
Werner M, Ben-Neriah Z, Silverstein S, Lerer I, Dagan Y, Abeliovich D. A patient with Prader-Willi syndrome and a supernumerary marker chromosome r(15)(q11.1-13p11.1)pat and maternal heterodisomy. Am J Med Genet A. 2004;129:176.
Glass IA, Rauen KA, Chen E, Parkes J, Alberston DG, Pinkel D, et al. Ring chromosome 15: Characterization by array CGH. Hum Genet. 2006;118:611–7.
Klaassens M, Galjaard D, Scott H, Brüggenwirth D, Van Opstal MV, Fox R, et al. Prenatal detection and outcome of congenital diaphragmatic hernia (CDH) associated with deletion of chromosome 15q26: two patients and review of the literature. Am J Med Genet A. 2007;143A:2106–12.
Boente MDC, Bazan C, Montanari D. Cutis tricolor parvimaculata in two patients with ring chromosome 15 syndrome. Pediatr Dermatol. 2011;28:670–3.
Guilherme RS, Meloni Vde F, Takeno SS, Pellegrino R, Brunoni D, Kulikowski LD, et al. Twenty-year cytogenetic and molecular follow-up of a patient with ring chromosome 15: a case report. J Med Case Rep. 2012;6:283.
Britto ISW, Regina Silva Herbest S, Tedesco GD, Drummond CL, Bussamra LCS, Araujo Júnior E, et al. Prenatal Diagnosis of a Fetus with Ring Chromosomal 15 by Two- and Three-Dimensional Ultrasonography. Case Rep Obstet Gynecol. 2014;2014:1–4. Available from: http://www.hindawi.com/journals/criog/2014/495702/
Gediya SG, Poswal L, Sanadhya A, Goyal S. Ring chromosome 15 presenting as short stature, intellectual disability and café-au-lait spots. Int J Res Med Sci. 2016;4(11) https://doi.org/10.18203/2320%2D6012.ijrms20163825. Available from: http://www.msjonline.org/index.php/ijrms/article/view/344
Cannarella R, Mattina T, Condorelli RA, Mongioi LM, Pandini G, La Vignera S, et al. Chromosome 15 structural abnormalities: effect on IGF1R gene expression and function. Endocr Connect. 2017;6:528–39.
Kalantari H, Karimi H, Almadani SN, Fakhri M, Mokhtari P, Gourabi H, et al. Fecundity in an infertile man with r(15) – a challenge to the current paradigm. Reprod BioMed Online. 2018;36:210–8.
Rumenić L, Joksimović I, Anaf M. Ring chromosome 15 in child with a minor dysmorphism of phenotype. Hum Genet. 1976;33:187–8.
Pfeiffer RA, Dhadial R, Lenz W. 46,XX/46,XX,r(15) mosaiciam: report of a case. J. Med. Genet. 1977;14:63–5.
Otto J, Back E, Fürste HO, Abel M, Böhm N, Pringsheim W. Dysplastic features, growth retardation, malrotation of the gut, and fatal ventricular septal defect in a 4-month-old girl with ring chromosome 15. Eur J Pediatr. 1984;142:229–31.
Wilson GN, Sauder SE, Bush M, Beitins IZ. Phenotypic delineation of ring chromosome 15 and Russell-Silver syndromes. J Med Genet. 1985;22:233–6.
Borghgraef M, Fryns J-P, Berghe H. Psychological findings in three children with ring 15 chromosome. J Ment Defic Res. 1988;32(Pt 4):337–47.
Kitatani M, Takahashi H, Ozaki M, Okino E, Maruoka T. A case of ring chromosome 15 accompanied by almost normal intelligence. Hum Genet. 1990;85:138–9.
Nuutinen M, Kouvalainen K, Knip M. Good growth response to growth hormone treatment in the ring chromosome 15 syndrome. J Med Genet. 1995;32:486–7.
Tümer Z, Harboe TL, Blennow E, Kalscheuer VM, Tommerup N, Brøndum-Nielsen K. Molecular cytogenetic characterization of ring chromosome 15 in three unrelated patients. Am J Med Genet A. 2004;130A:340–4.
Pinson L, Perrin A, Plouzennec C, Parent P, Metz C, Collet M, et al. Detection of an unexpected subtelomeric 15q26.2 → qter deletion in a little girl: Clinical and cytogenetic studies. Am J Med Genet. 2005;138 A:160–5.
Emberger J, Rossi D, Jean R, Bonnet H, Dumas R. Etude d’une observation de chromosome du groupe 13–15 en anneau (46,XY,15r). Hum Genet. 1971;11:295–9.
Forabosco A, Dutrillaux B, Vassoler G, Lejeune J. Ring 15 chromosome: r(15). Identification by controlled denaturation. Ann Genet. 1972;15:267–70.
Stoll C, Juif JG, Luckel JC, Lausecker C. Ring chromosome 15:r(15). Identification by R banding. Hum Genet. 1975;27:259–62.
László J, Gaál M, Bósze P. Chromosome studies in ovarian hypoplasia. Clin Genet. 1976;9:61–70.
Peoples R, Milatovich A, Francke U. Hemizygosity at the insulin-like growth factor I receptor (IGF1R) locus and growth failure in the ring chromosome 15 syndrome. Cytogenet Cell Genet. 1995;70:228–34.
Rogan PK, Seip JR, Driscoll DJ, Papenhausen PR, Johnson VP, Raskin S, et al. Distinct 15q genotypes in Russell-Silver and ring 15 syndromes. Am J Med Genet. 1996;62:10–5.
Rujirabanjerd S, Suwannarat W, Sripo T, Dissaneevate P, Permsirivanich W, Limprasert P. De novo subtelomeric deletion of 15q associated with satellite translocation in a child with developmental delay and severe growth retardation. Am J Med Genet Part A. 2007;143:271–6.
Hitayezu J, Uwineza A, Murorunkwere S, Ndinkabandi J, Mutesa L. A case of Rwandan patient with ring chromosome 15 syndrome. Rwanda Med J. 2013;70:27–8.
Tewari S, Lubna N, Shah R, Al-Rikabi ABH, Shah K, Sheth J, et al. Molecular characterization and evaluation of complex rearrangements in a case of ring chromosome 15. Mol Cytogenet. 2017;10:38.
Nishikawa K, Itoi F, Nagahara M, Jose M, Matsunaga A, Ueda J, et al. The normality of sperm in an infertile man with ring chromosome 15: a case report. J Assist Reprod Genet. 2018;35:251–6.
Kousseff BG. Ring chromosome 15 and failure to thrive. Am J Dis Child. 1980;134:798–9.
Kosztolanyi G, Pap M. Severe growth failure associated with atrophic intestinal mucosa and ring chromosome 15. Acta Paediatr Scand. 1986;75:326–31.
Tan SJ, Chen CH, Chen CP, Chen CW, Chen CY, Hwang KS. Prenatal diagnosis of mosaic ring chromosome 15 with abnormal maternal serum Down syndrome screening and Dandy-Walker malformation. Taiwan J Obstet Gynecol. 2012;51:109–11.
Quinonez SC, Gelehrter TD, Uhlmann WR. A Marfan syndrome-like phenotype caused by a neocentromeric supernumerary ring chromosome 15. Am J Med Genet Part A. 2017;173:268–73.
Zou YS, McGrann PS, Uphoff TS, Van Dyke DL. A case of mosaic supernumerary ring chromosome 15 with two copies of the segment 15p11.1-q14. Am J Med Genet Part A. 2006;140:1663–8.
Xu F, Zou CC, Liang L, Huang XM, Shao YN. Ring chromosome 15 syndrome: Case report and literature review. Hong Kong J. Paediatr. 2011;16:175–9.
Meinecke P, Koske-Westphal T. Ring chromosome 15 in a male adult with radial defects. Evaluation of the phenotype. Clin Genet. 1980;18:428–33.
Teplitz RL, Miller D, Hansson KM, Rundall TS. A human ring D chromosome associated with multiple congenital abnormalities. J Pediatr. 1967;70:936–41.
Hatem E, Meriam BR, Walid D, Adenen M, Moez G, Ali S. Molecular characterization of a ring chromosome 15 in a fetus with intra uterine growth retardation and diaphragmatic hernia. Prenat Diagn. 2007;27:471–4.
Manolakos E, Vetro A, Kitmirides S, Papoulidis I, Kosyakova N, Mrasek K, et al. Prenatal diagnosis of a fetus with ring chromosome 15 characterized by array-CGH. Prenat Diagn. 2009;29:884–8.
Schroer RJ, Phelan MC, Michaelis RC, Crawford EC, Skinner SA, Cuccaro M, et al. Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet. 1998;76:327–36.
Neri G, Ricci R, Pelino A, Bova R, Tedeschi B, Serra A. A boy with ring chromosome 15 derived from a t(15q;15q) Robertsonian translocation in the mother: Cytogenetic and biochemical findings. Am J Med Genet. 1983;14:307–14.
Schmid M, Henrichs I, Nestler H, Knörr-Gärtner H, Teller WM, Krone W. Analysis of banding patterns and mosaic configurations in a case of ring chromosome 15. Hum Genet. 1978;41:289–99.
Ledbetter DH, Riccardi VM, Au WW, Wilson DP, Holmquist GP. Ring chromosome 15: Phenotype, Ag-NOR analysis, secondary aneuploidy, and associated chromosome instability. Cytogenet Genome Res. 1980;27:111–22.
Li MM, Nimmakayalu MA, Mercer D, Andersson HC, Emanuel BS. Characterization of a Cryptic 3.3 Mb Deletion in a Patient With a “Balanced t(15;22) Translocation” Using High Density Oligo Array CGH and Gene Expression Arrays. Am. J. Med. Genet. A. 2008;146A:368–75.
Liu YH, Chang SD, Chen FP. Increased fetal nuchal fold leading to prenatal diagnosis of ring chromosome 15. Prenat Diagn. 2001;21:1031–3.
Wang Q, Wu W, Xu Z, Luo F, Zhou Q, Li P, et al. Copy number changes and methylation patterns in an isodicentric and a ring chromosome of 15q11-q13: Report of two cases and review of literature. Mol Cytogenet. 2015;8:198–203.
Moreau N, Teyssier M. Ring chromosome 15: report of a case in an infertile man. Clin Genet. 1982;21:272–9.
Hernando C, Grao P, Santos M, García M, De la Maza L, Egozcue J, et al. Caracterización del cromosoma 15 en anillo presente en 2 pacientes con gran retraso de crecimiento mediante hibridación genómica comparada. An Pediatría. 2005;62:607–27. Elsevier; [cited 2018 Apr 26] Available from: http://linkinghub.elsevier.com/retrieve/pii/S1695403305701302
Acknowledgements
We like to thank Universidad UTE for their unconditional support in the research process to our laboratory.
Funding
Funding for this research was completely managed by the institutions affiliated.
Availability of data and materials
All data used in this study are available from the corresponding author on request.
Author information
Authors and Affiliations
Contributions
JGA and APYM sent the patient and made physical and endocrinology analysis. CPYM is the responsible M.D. of the patient. IAC, VY, FV, and PEL contributed to the literature review, cytogenetic analysis, and wrote the manuscript. JMH and JLG made the genetic arrays analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The present work was approved by the Bioethics committee from “Universidad de las Américas” with approval number 2015-0702.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian(s) for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Paz-y-Miño, C., Guevara-Aguirre, J., Paz-y-Miño, A. et al. Ring chromosome 15 – cytogenetics and mapping arrays: a case report and review of the literature. J Med Case Reports 12, 340 (2018). https://doi.org/10.1186/s13256-018-1879-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-018-1879-5