
Abstract
Cigarettes are well-recognized risk factors responsible for the emergence of a variety of pathologic conditions affecting both the airways and the lungs. Smoking-related lung diseases can be classified as chronic obstructive pulmonary disease (COPD) and several types of interstitial diseases, such as pulmonary Langerhans cell histiocytosis, bronchiolitis, desquamative interstitial pneumonitis, acute eosinophilic pneumonia, and interstitial fibrosing lung diseases. The evidence of combined lower lung fibrosis and predominant upper lung emphysema is renowned as a distinct clinical entity, named combined pulmonary fibrosis and emphysema. Although computerized tomography permits an adequate classification and distinction of these diseases, the clinical, imaging, and histological features often overlap and coexist in a single patient. Therefore, a combined radiologic and pathologic approach, in the appropriate clinical setting, is useful for best comprehension and distinction of these entities. Our goals are to describe the imaging features in smoking-related lung diseases and how the pathological manifestations translate on high-resolution computerized tomography.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Key points
-
COPD is one of the most common smoke-related causes of death, depicted by the spirometric evidence of irreversible and usually progressive airflow limitation. It includes chronic bronchitis and emphysema. The peripheral and smaller airways are also affected in both number and caliber.
-
Changes in smoking habits are known inducers of the development of acute eosinophilic pneumonia (AEP): initiation of smoking habit, resumption after interruption, and increased frequency of smoking.
-
Pulmonary Langerhans cell histiocytosis (PLCH) is an uncommon disease virtually exclusive of smokers. Over time, the cellular nodules are replaced by polymorphic fibrotic scars associated with distorted and enlarged air spaces.
-
Besides the difference of imaging findings, desquamative interstitial pneumonia (DIP) and respiratory bronchiolitis/respiratory bronchiolitis-interstitial lung disease (RB/RB-ILD) are a spectrum of the same pathologic event, characterized by the excess of macrophages in the distal airways.
-
Fibrosis is also a common radiological feature ranging from sparse fibrosis along the alveolar walls, termed AEF, to a pattern of diffuse interstitial fibrosis, which can represent usual interstitial pneumonia (UIP) in some cases.
Introduction
Cigarettes are a noxious mixture containing around 5000 chemicals and considered as one of the most important causes of chemically mediated disorders in humans. Both direct toxicity and induced immune-mediated response lead to both reversible and irreversible damage from central airways to most distal airways and lung parenchyma [1,2,3].
The most common smoking-related causes of death include numerous types of cancer, particularly lung cancer, and chronic obstructive pulmonary disease (COPD). Adding to these disorders, cigarettes are a well-known etiologic factor linked with the development of some types of interstitial lung disease (ILD), namely AEP, DIP, RB-ILD, and PLCH. Smoking is also responsible for the development of fibrotic lung disease [4,5,6,7]. These lung diseases related to smoking are a spectrum of the same pathologic process. Pathologists usually find a mixture of histopathological patterns and a single diagnosis is often difficult to make [5]. Although the majority of the smokers have a certain degree of inflammatory changes in the airways, just a subgroup of individuals develops clinically relevant respiratory disease. Both genetic and exogenous triggers, such as allergens or infections, may be implicated in the development of the disease [7].
In this article, we describe and illustrate the characteristic clinical features, imaging findings, and pathologic findings of diffuse lung diseases related to smoking, encompassing COPD and ILDs. We emphasize the need of a multidisciplinary approach (clinical, radiological, and pathological) for better comprehension and distinction of these entities.
Discussion
Chronic obstructive pulmonary disease
COPD is depicted by the spirometric evidence of irreversible and usually progressive airflow limitation. The disease comprehends distinct however overlapping obstructive disorders, such as chronic bronchitis, emphysema, and also affecting the distal airways of the lung, with both reductions in the number and the caliber. Bronchiolitis is the earliest lesion in COPD, with narrowing and loss of terminal bronchioles preceding emphysematous changes [3, 8]. Emphysema results from permanent enlargement and wall destruction of the airways distal to the surviving terminal bronchioles, progressing in severe cases to coalescence of destroyed lobules [8, 9]. Centrilobular and panlobular emphysema have clinical significance, often associated with increased dyspnea and poorer functional capacity. Additionally, centrilobular emphysema is related to smoking habits, and panlobular emphysema is associated with low body mass index (BMI). Paraseptal emphysema is more common in men and is frequently of little physiologic significance, except for the development of pneumothorax secondary to the presence of paraseptal bleb/bulla [10]. Visual CT evaluation is considered the clinical gold standard for the assessment and characterization of centrilobular and panlobular emphysema, also demonstrated to be valid regarding the pathologic assessments [10,11,12,13,14].
Chronic bronchitis consists in the presence of inflammation in the large airways. Clinically, patients present with chronic cough and sputum during at least 3 months per year in two consecutive years [15]. Chronic bronchitis is triggered and sustained by the activation of an abnormal immune response due to long-term cigarette smoking, causing overproduction of mucus from goblet cells, thickening, and fibrosis of the bronchial walls (Fig. 1). The condition promotes a further reduction of the caliber of the airways, predisposing expiratory collapse. Imaging depicts wall thickening of the large airways, and endobronchial mucus plugging may also be depicted (Fig. 2) [15, 16]. Bronchial wall thickening is consistently associated with a decline in FEV1 and with the risk of acute exacerbation and hospitalization [16,17,18,19].

Chronic bronchitis. a and b Low-power hematoxylin and eosin stain (H&E) shows an increased number of goblet cells (arrow in a) and hyperplasia of submucosal glands (arrow in b). Hypersecretion of mucus from goblet cells is also visualized

Bronchial wall thickening in a 79-year-old heavy smoker man. a and b Axial images from a chest CT show thickening of the bronchial walls (arrows). Bronchial wall thickening is an important predictor factor of FEV1 and of the risk of acute exacerbation
Centrilobular emphysema (CLE) results from an abnormal dilatation of the airspaces distal to the terminal bronchioles. This subtype of emphysema is strongly related to cigarette smoking with higher airway inflammatory cell content and usually with upper lung predominance [10]. CLE originates from the destruction and dilation of bronchioles, with further coalescence of several primary lesions (Fig. 3). On imaging, small well or poorly demarcated regions of low attenuation can be depicted, surrounded by areas of the normal lung [3, 8,9,10]. Fleischner Society’s guideline scoring of CLE [20] characterizes emphysema as trace when involving less than 0.5% of a lung region, mild when concerning 0.5–5%, and moderate if more than 5% (Fig. 4). Severe emphysema is classified as confluent (coalescence of centrilobular lucencies) or advanced destructive emphysema (ADE) if the expansion of the entire secondary lobule, distortion of the pulmonary architecture, and splaying or decreased caliber of vessels are present (Fig. 5).

Centrilobular emphysema. Low-power hematoxylin and eosin (H&E) stain shows emphysematous spaces surrounding the terminal bronchioles (arrow; arrowhead in normal parenchyma). Abnormally large airspaces are the result of the destruction of alveolar walls

Subtypes of CLE according to the Fleischner Society guidelines on HRCT. Axial images in 3 smokers show a trace CLE, with minimal centrilobular lucencies occupying less than 0.5% of a lung zone (arrows); b mild CLE, defined as scattered centrilobular lucencies affecting 0.5% to 5% of a lung zone; and c moderate CLE, with numerous centrilobular lucencies occupying more than 5% of any lung zone

Subtypes of severe CLE according to the Fleischner Society guidelines on HRCT. a Axial CT image depicts confluent CLE with coalescent centrilobular lucencies without significant distortion of the pulmonary architecture. b Axial and c coronal images show ADE, with panlobular lucencies, distortion of the underlying pulmonary architecture, and hyperexpansion of the lung
Panlobular emphysema (PLE) like ADE indicates destruction across the lobule. This type of emphysema presents in a younger age group (30–44 years of age). The findings are lower lung predominant in about two-thirds of the individuals (Fig. 6) [3, 8,9,10, 20]. It is commonly linked to a mutation in the alpha 1-antitrypsin gene, in which Z allele accounts for approximately 95% of clinically recognized cases, causing unopposed action of neutrophil elastase with consequent destruction of lung parenchyma [21].

PLE in a 51-year-old man with alpha-1 antitrypsin deficiency. a Axial and b coronal CT images show hyperinflation and lower lobe predominant emphysema, involving the entire secondary pulmonary lobules. Bronquial wall thickening is also depicted (arrows)
Paraseptal emphysema (PSE) is secondary to emphysematous changes of the distal acinus, adjacent to the visceral pleura, including fissures. PSE is also scored as mild (up to 1 cm juxtapleural well-demarcated lucencies) or substantial (cyst-like lucencies or bullae greater than 1 cm adjacent to the pleura) (Fig. 7). Bullae are a risk factor for spontaneous pneumothorax, and they can also be large enough to cause severe compression and reduction of the pulmonary function of the remaining lung (Fig. 8) [20].

Subtypes of PSE according to the Fleischner Society guidelines on HRCT. a Axial CT shows mild PSE (white arrows) with up to 1 cm juxtapleural well-demarcated lucencies. b Axial CT depicts substantial PSE, defined as cyst-like lucencies or bullae greater than 1 cm adjacent to the pleura. The patient had spontaneous pneumothorax and black arrow depicts a chest drain. Subcutaneous emphysema is also visible in the right thoracic wall

Bullous emphysema in a 55-year-old man. a Posteroanterior (PA) radiograph shows lucencies in the superior half of the right hemithorax and in the upper left hemithorax. b Axial CT shows upper lobe predominant bullae (asterisks) in the subpleural surface of the right lung. Moderate CLE is also present (arrow)
Acute eosinophilic pneumonia
AEP is an uncommon condition that occurs most frequently in males between the second and forth decades of life. Approximately two-thirds of patients have smoking habits [22,23,24,25]. Changes in smoking habits are known inducers of the development of AEP: initiation of smoking habit, resumption after interruption, and increased frequency of smoking [26,27,28]. The clinical presentation is generally unspecific, letting the disease be frequently misdiagnosed as other most common entities such as community-acquired pneumonia. The clinical symptoms are acute, with duration of the respiratory illness of less than a month, and characterized by moderate fever, cough, dyspnea, pleuritic pain, malaise, myalgia, and night sweats. Acute respiratory failure is frequent and mechanical ventilation is often required [22, 25]. Therefore, the disease is severe, and most patients fulfill diagnostic criteria for acute lung injury (ALI) or acute respiratory distress syndrome (ARDS). The key to diagnosis is evidence of eosinophilia in the bronchoalveolar lavage (BAL), with more than 25% of eosinophils on the cell count. Nevertheless, blood eosinophils are frequently at normal levels at the beginning of the clinical presentation and may rise after a few days. BAL is sterile with no bacterial growth during the disease course [22, 23, 25].
Lung biopsy is usually not needed to meet the diagnosis. It shows alveolar and interstitial eosinophilia and alveolar damage. Additional features include nonnecrotizing perivascular inflammation, eosinophilic abscesses, interstitial lymphocytes, organizing fibrinous exudate in the alveoli, type II pneumocyte hyperplasia, and also involvement of the airway [22, 29].
Imaging findings are predominant in the lower lungs, showing diffuse consolidations, ground-glass opacities (GGO), ill-defined centrilobular nodules, smooth septal thickening, and unilateral or bilateral pleural effusion (Fig. 9). The radiologic differential includes infection, fluid overload, ALI/ARDS, hypersensitivity to drugs, and pulmonary hemorrhage [7, 24, 25, 29, 30].

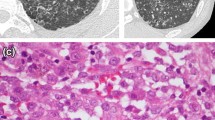
Radiologic-pathologic correlation of AEP. a Posteroanterior (PA) radiograph shows bilateral ground-glass opacities and consolidation in the mid-left lung zone. A small left pleural effusion is present. b Axial CT shows peripheral lower lobe consolidations and ground-glass opacities, mainly on the left. c Transthoracic lung biopsy showing eosinophils (black arrow) infiltrating the interstitium and the alveolar spaces; edema (green arrow) and reactive pneumocytes are seen; no necrotizing vasculitis is observed (H&E, × 400)
Treatment with steroids achieves an excellent response, with resolution within days of the immunologic process [22, 23].
Pulmonary Langerhans cell histiocytosis
PLCH is an unusual respiratory disease found most commonly in young adults between the third and fourth decades of life. It is exceedingly rare in children and occurs in most cases as a part of disseminated LCH secondary to an abnormal immune response. Over 95% of PLCH patients are smokers and it is estimated that the disease affects approximately 3–4% of smokers. PLCH affects both genders equally [31,32,33]. Patients usually report fatigue, weight loss, exertional dyspnea, and non-productive cough. Pneumothorax may be the first sign in 15–20% of patients. Although, approximately 20% of patients reported no symptoms at the time of disease detection [33].
Histopathologic findings reveal bronchiole-centered, stellate nodules, containing Langerhans cells, and interspersed with other inflammatory cells (lymphocytes, macrophages, monocytes). Langerhans cells are quite large cells, containing pale cytoplasms and convoluted nucleus, which resemble coffee grains. They are immunoreactive with CD1a and S-100. Along the disease course, cellular nodules evolve from mixed cellular and fibrotic nodules to entirely polymorphic scars associated with enlarged and distorted air spaces [32,33,34,35,36,37,38].
Imaging reveals upper and middle lung predominance of nodules and cysts with variable wall thickness and irregular margins. The disease characteristically spares the costophrenic sulci, extreme apices, and part of the middle lobe and lingula. Imaging findings have a typical progression over time from a predominant nodular pattern to diffusely distributed cysts with bizarre shapes (Fig. 10). The later disease stage is associated with significant emphysematous areas either related to PLCH scars or usual emphysema due to smoking (Fig. 11). GGOs are a frequent imaging finding and may be associated with the presence of other smoke-related diseases, for example, RB and DIP [6, 7, 24, 29, 30, 33, 38]. Smoking cessation is a fundamental key in the treatment process of PLCH. After smoking cessation, symptoms and radiologic alterations may partially regress or stabilize in more than half of the patients (Fig. 12) [34, 39,40,41,42,43,44,45,46]. However, one third to a half of the patients may show respiratory failure and disease progression, despite smoking cessation (Fig. 13) [47].

Pulmonary Langerhans cell histiocytosis in a 40-year-old man. a Axial CT image shows upper lobe predominance of nodules and cysts of varying wall thickness and irregular margins. Arrow depicts a stellate cellular nodule. b Transthoracic lung biopsy of the nodule highlighted in a shows a nodular aggregate of cells (lymphocytes, eosinophils, plasma cells, and Langerhans cells) centered in bronchioles (arrow) and extending to the interstitium (H&E, × 200). c CD1a immunostaining highlights the Langerhans cells (CD1a, × 100)

PLCH. a Coronal and b axial CT images in a 30-year-old man show diffuse upper lobe predominant lucencies with basilar sparing. Imaging findings were thought to represent severe confluent emphysema. c Small cysts (asterisk) with nodular aggregates of Langerhans cells in their walls, with a stellate appearance and centered in bronchioles (arrows) (H&E, × 40). d The cells in the nodules have vesicular nuclei and a pale cytoplasm and sometimes with a reniform appearance; some eosinophils are present (H&E, × 400). e CD1a immunostaining highlights the Langerhans cells

PLCH. Regression of the imaging findings over time (2-year interval), after smoking cessation. a Axial CT image showing cysts with different shapes and sizes diffusely distributed in both lungs. A few cellular and mixed nodules are also present. b Axial CT image at 2-year interval showing almost complete resolution of the imagiological findings

PLCH. Progression of the imaging findings over time (10-year interval), despite smoking cessation, in a 30-year-old man. a Axial CT image showing diffuse bizarre cysts with walls of varying thickness. b Axial CT image at 10-year interval showing progression of the number and size of the cysts in both lungs
PLCH should be differentiated from lymphangioleiomyomatosis (LAM), in which cysts tend to have more regular and rounded shapes and more uniformly distribution through the lung. Other differentials should also be considered, such as pulmonary metastasis, sarcoidosis, Birt-Hogg-Dubé syndrome, and infections (Fig. 14) [32, 35, 48, 49].

Differential diagnosis of PLCH. a Sagittal and b axial CT images show a 50-year-old woman with LAM. There are multiple rounded-shaped cysts, with a relatively uniform distribution with no zonal predilection and affecting the lung bases. c Coronal and d axial CT images depict multiple cysts with a lower zone distribution, in a 35-year-old male with Birt-Hogg-Dubé syndrome. The patient had a history of recurrent pneumothoraces
Respiratory bronchiolitis and desquamative interstitial pneumonia
RB-ILD and DIP are moderately uncommon diseases related to smoking. On clinical presentation, patients usually complain about insidious dyspnea and cough over the course of weeks to months [5,6,7, 50,51,52,53,54]. The clinical disease course of RB-ILD and DIP tends to be stable in most of the patients. However, the rates of impairment are worse in those with DIP, in which diffusing capacity for carbon monoxide (DLco) may be severely reduced. As a consequence, deaths can occur in patients with DIP, but there are no described related deaths in those with RB-ILD [5, 50, 51]. RB is a classic histological marker of smoking, encountered in the lungs of nearly all active smokers. However, patients with RB are fundamentally asymptomatic, and the disease does not portend any clinical significance. RB and RB-ILD are differentiated by each other by the presence of respiratory symptoms and abnormal pulmonary function tests in the last one [5,6,7, 50, 51].
Histopathologic examinations of RB and DIP show an excess number of pigmented or smokers’ macrophages involving the distal airways and peribronchiolar airspaces. However, DIP presents with pigmented macrophages diffusely filling the alveoli, with associated thickening of the septa secondary to the presence of inflammatory cells. On DIP findings, the degree of interstitial fibrosis is usually mild and more severe than in RB. Both entities represent a severity spectrum of the same pathologic event, with an excess number of macrophages filling the distal airways and alveoli secondary to an immune-mediated response due to smoking [5, 24, 29, 51, 55, 56].
On imaging, RB/RB-ILD shows upper lung predominance of the findings, characterized by the evidence of low attenuation centriacinar nodules, GGOs, bronchial wall thickening, few thickened interlobular septa, and also lobular air-trapping, especially depicted on expiratory CT. These findings are generally associated with emphysema (Fig. 15). The radiological findings in DIP are lower lobe predominant and characterized by GGOs and reticular opacities interposed with relatively normal lung zones, forming a mosaic attenuation. Small cystic spaces may be depicted in the areas of GGO, representing dilated alveolar ducts or centrilobular emphysema. The distribution of findings is more often subpleural but may also be random or diffuse (Fig. 16) [6, 7, 24, 29, 30, 54, 56]. Associated findings of RB and emphysema are frequently visualized. The evidence of areas of mild pulmonary fibrosis in DIP, characterized by increased septal lines, traction bronchiectasis, and lung distortion, is associated with disease progression in some individuals, despite smoking cessation and steroids [53].

RB-ILD. a Axial CT image depicts diffuse, ill-defined centrilobular nodules. Due to deteriorating symptoms, the patient underwent lung biopsy. b Excess numbers of alveolar macrophages (arrows) with light brown granules filling the cytoplasm, in a predominant centriacinar location; mild interstitial lymphocytic infiltrate (H&E, × 100)

DIP in a 58-year-old heavy-smoker man. a Posteroanterior (PA) radiograph shows reticular opacities and ground-glass attenuation in both lungs, predominantly in the lower lung zones. b Axial CT image shows ground-glass opacities with cystic changes in the lower and mid lobes and lingula. Images show well-defined areas of sparing, creating a mosaic attenuation. Mild areas of reticulation and bronchiectasis are also present, signifying fibrosis. c Large numbers of alveolar macrophages with light brown granules filling the cytoplasm (arrow); mild interstitial lymphocytic infiltrate and mild fibrosis (arrowhead) (H&E, × 200)
Some differential diagnoses are important to be considered in association with patient’s complete clinical history, including NSIP, hypersensitivity pneumonitis, and atypical infections, including pneumocystosis (Figs. 17 and 18) [7, 24, 29, 50].

Differential diagnosis of RB-ILD. a Axial CT image depicts subacute hypersensitivity pneumonitis (HP) in a 71-year-old woman with a long history of exposition to moldy hay (farmer’s lung). Imaging findings of HP are similar to RB-ILD, which also appears as centrilobular nodules predominantly in the upper lobes (arrow) and patchy areas of ground-glass attenuation. A clinical history is crucial to make the differential diagnosis. b Sarcoidosis in a 37-year-old man. Axial CT image shows diffuse areas of nodularity with peribronchial distribution and also in relation to the subpleural region (arrow). There are also some surrounding ground-glass opacities

Differential diagnosis of DIP. a Axial and b coronal CT images depict NSIP in a female patient with systemic sclerosis. CT findings show bilateral and asymmetrical ground-glass opacities, with a lower lobe predominance and immediate subpleural sparing. c Axial and d coronal CT images depict pneumocystosis in an immunocompromised patient with HIV. Imaging findings show bilateral ground-glass opacities, predominantly in the lower lobes
Interstitial fibrosis
Airspace enlargement with fibrosis/smoking-related interstitial fibrosis
Cigarette smoke leads to alveolar wall fibrosis that increases with time and intensity of exposure, called AEF, also termed as SRIF. Pulmonary fibrosis severity ranges from sparse fibrosis in the alveolar walls to diffuse fibrosis, with dense, paucicellular, eosinophilic collagen that has a waxy quality on pathologic examination. Hypertrophic smooth muscle bundles can also be depicted and perhaps predominate. AEF is also accompanied by features of emphysema and RB [57,58,59]. The fibrosis is confined in the subpleural and peribronchiolar interstitium, with relative preservation of the lung architecture 60. It is important to mention that AEF is an incidental histologic or radiological finding characterized by interstitial fibrosis exceeding the fine fibrosis often seen in emphysema alone. Most patients have stable disease and good survival time [60, 61]. Therefore, patients with progressively worsening exertional dyspnea and cough might have AEF accompanied by chronic interstitial pneumonia such as UIP or nonspecific interstitial pneumonia (NSIP). In these cases, lung biopsy is crucial for definitive diagnosis [62].
Radiological features of AEF are subpleural sparing thin-walled cysts (TWCs), associated with reticular and ground-glass opacities. Imaging distinction between the typical honeycombing present in idiopathic pulmonary fibrosis (IPF) and TWCs related to smoking may be confusing. The cysts in AEF appear as thin-walled cysts (less than 1 mm) predominantly distributed in the upper lobes and upper and middle portion of the lower lobes, slightly distant from pleura, affecting deeper lung parenchyma (Fig. 19a) [63,64,65].

AEF and DIP combined with fibrotic NSIP pattern. a Axial CT image through the upper lobes shows CLE and PLE, associated with reticulation (black arrows) and discrete patchy ground-glass opacities (white arrows). The areas of emphysema within the areas of GGO appear to have more well-defined walls, a finding termed as AEF. b Axial CT at the lower lobes shows GGO with scattered cystic changes, mild reticulation, and bronchiectasis (orange arrow), signifying underlying fibrosis. c Lung transbronchial criobiopsy with uniform thickening of alveolar septa by collagen deposition (black arrows), mild associated inflammation, emphysema, and respiratory bronchiolitis (white arrow) (H&E, × 100)
NSIP/UIP
The progression of fibrosis leads to increasing lower-lobe predominant GGOs, traction bronchiectasis, and reticulation, in a pattern compatible with NSIP on imaging examination (Fig. 19) [5, 24, 61, 66]. These changes are often accompanied by other smoking-related findings, such as emphysema, DIP, and RB. In AEF, emphysema may appear better-demarcated secondary to the presence of fibrosis in the alveolar walls [5, 63,64,65].
IPF is a progressive chronic fibrosing lung disease with unknown etiology and pathologically described by a pattern consistent with UIP [67]. It represents the most common but also the most severe type of ILD, affecting most frequently males over the age of 65 years. The median survival time after the diagnosis ranges from 2 to 4 years. Cigarette smoking is considered a probable risk factor for the development of IPF, with an odds ratio ranging from 1.6 to 2.9 [50, 67,68,69]. Cigarette smoking is also linked to lower survival time, in relation to non-smoker IPF patients [70, 71]. Imaging features are often bilateral and asymmetric and include peripheral and basal predominant traction bronchiectasis, reticular opacities, and honeycombing, with minimal GGOs. Pulmonary volumes are typically low. The findings are spatially and temporally heterogeneous, with areas of varying disease extent and severity adjacent to regions of a more normal lung. The evidence of any of the following features should prompt an alternative diagnosis: fibrosis predominantly located in the upper or mid lungs, extensive GGOs, peribronchovascular distribution, diffuse cysts or nodules, predominant consolidation, and presence of air trapping with substantial mosaic attenuation [72].
Combined pulmonary fibrosis and emphysema
The disease termed CPFE is a severe respiratory condition integrating imaging features of both pulmonary fibrosis and emphysema. Patients present with similar symptoms to IPF and emphysema; however, they have relatively preserved spirometry, as a result of the combined obstructive disease in distal airways and restrictive fibrosis. The clue for the diagnosis is the evidence of the physiologic decline in diffusing capacity and characteristic CT features [73,74,75,76,77,78,79].
The histological and radiological patterns of interstitial fibrosis described in CPFE are most commonly UIP and, in a minority of reported cases, NSIP [50]. CPFE is estimated to arise in about 8% of patients diagnosed with IPF [80]. CPFE syndrome is found more frequently in heavy-smoker males and portends a tendency to appear in a slightly older age group in relation to IPF alone (mean age of 65–70 years) 77-79. CPFE confers a median survival time nearly double that of IPF (approximately 6.1 years) [81]; however, CPFE portends a higher risk of development of pulmonary hypertension (ranging from 50% to 90%) [82, 83] and may also be associated with a greater chance of lung cancer, with consequent lower survival time [84]. Imaging features are emphysema (both CLE and PSE) in the upper lobes associated with interstitial fibrosis in the lower lobes [31, 50, 76,77,78,79,80,81,82, 85, 86]. Ryerson et al. [80] reported that CLE should involve a minimum of 10% of the lung volume to allow the diagnosis. Bulky cystic lesions with thick walls predominantly located in the upper lobes are also apparent, which may correspond to emphysema with fibrosis or AEF (Fig. 20) [85, 86].

Combined pulmonary fibrosis and emphysema (CPFE). a Posteroanterior (PA) radiograph shows right volume loss, diffuse bilateral reticular opacities, and GGO. There are lucencies in the upper lobes (white arrow), signifying bullous emphysema. b Axial and c coronal CT images show centrilobular, paraseptal, and bullous emphysema predominantly in the upper lobes. d Axial image through the lower lobes shows peripheral predominant fibrosis with reticulation, traction bronchiectasis, and honeycombing (black arrow), in a UIP pattern
Conclusion
Diffuse smoking-related lung diseases exemplify a wide clinicopathologic manifestation secondary to the same process of lung injury. Histologic findings frequently overlap in a single patient and as a consequence mixed patterns of the disease may be depicted on HRCT. The multidisciplinary approach by an integrated clinical, radiological, and pathological study is useful for the best comprehension and distinction of these entities.
Availability of data and materials
All data and materials presented were from our hospital and daily practice.
Abbreviations
- AEF:
-
Airspace enlargement with fibrosis
- AEP:
-
Acute eosinophilic pneumonia
- ALI:
-
Acute lung injury
- ARDS:
-
Acute respiratory distress syndrome
- BAL:
-
Bronchoalveolar lavage
- BMI:
-
Body mass index
- CLE:
-
Centrilobular emphysema
- COPD:
-
Chronic obstructive pulmonary disease
- CPFE:
-
Combined pulmonary fibrosis and emphysema
- DIP:
-
Desquamative interstitial pneumonia
- DLco:
-
Diffusing capacity for carbon monoxide
- GGO:
-
Ground-glass opacity
- HRCT:
-
High-resolution computerized tomography
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- LAM:
-
Lymphangioleiomyomatosis
- NSIP:
-
Nonspecific interstitial pneumonia
- PLCH:
-
Pulmonary Langerhans cell histiocytosis
- PLE:
-
Panlobular emphysema
- PSE:
-
Paraseptal emphysema
- RB:
-
Respiratory bronchiolitis
- SRIF:
-
Smoking-related interstitial fibrosis
- TWC:
-
Thin-walled cyst
- UIP:
-
Usual interstitial pneumonia
References
Talhout R, Schulz T, Florek E et al (2011) Hazardous compounds in tobacco smoke. Int J Environ Res Public Health. 8(2):613–628
Fowles J, Dybing E (2003) Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tob Control. 12:424–430
Celli BR, MacNee W, Force AET (2004) Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 23:932–946
Ezzati M, Lopez AD (2003) Estimates of global mortality attributable to smoking in 2000. Lancet. 362:847–852
Konopka KE, Myers JL (2018) A review of smoking-related interstitial fibrosis, respiratory bronchiolitis, and desquamative interstitial pneumonia: overlapping histology and confusing terminology. Arch Pathol Lab Med. 142(10):1177–1118
Walsh SL, Nair A, Desai SR (2015) Interstitial lung disease related to smoking: imaging considerations. Curr Opin Pulm Med. 21:407–416
Vassallo R, Ryu JH (2012) Smoking-related interstitial lung diseases. Clin Chest Med. 33:165–178
Viegi G, Pistelli F, Sherrill DL et al (2007) Definition, epidemiology and natural history of COPD. Eur Respir J. 30(5):993–1013
The definition of emphysema (1985) Report of a National Heart, Lung, and Blood Institute, Division of Lung Diseases workshop. Am Rev Respir Dis 132:182–185
Smith BM, Austin JH, Newell JD Jr et al (2014) Pulmonary emphysema subtypes on computed tomography: the MESA COPD study. Am J Med 127(1):94.e7–94e23
McDonough JE, Yuan R, Suzuki M et al (2011) Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 365(17):1567–1575
Diaz AA, Valim C, Yamashiro T et al (2010) Airway count and emphysema assessed by chest CT imaging predicts clinical outcome in smokers. Chest. 138:880–887
Kuwano K, Matsuba K, Ikeda T et al (1990) The diagnosis of mild emphysema. Correlation of computed tomography and pathology scores. Am Rev Respir Dis. 141:169–178
Spouge D, Mayo JR, Cardoso W et al (1993) Panacinar emphysema: CT and pathologic findings. J Comput Assist Tomogr. 17:710–713
MacNee W, Tuder RM (2009) New paradigms in the pathogenesis of chronic obstructive pulmonary disease I. Proc Am Thorac Soc. 6(6):527–531
Kim V, Criner GJ (2013) Chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 187(3):228–237
Vestbo J, Prescott E, Lange P (1996) Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Copenhagen City Heart Study Group. Am J Respir Crit Care Med. 153:1530–1535
Guerra S, Sherrill DL, Venker C et al (2009) Chronic bronchitis before age 50 years predicts incident airflow limitation and mortality risk. Thorax. 64:894–900
Sherman CB, Xu X, Speizer FE et al (1992) Longitudinal lung function decline in subjects with respiratory symptoms. Am Rev Respir Dis. 146:855–859
Lynch DA, Austin JH, Hogg JC et al (2015) CT-definable subtypes of chronic obstructive pulmonary disease: a statement of the Fleischner Society. Radiology. 277(1):192–205
Stoller JK, Aboussouan LS (2012) A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 185:246–259
De Giacomi F, Vassallo R, Yi ES et al (2018) Acute eosinophilic pneumonia: causes, diagnosis and management. Am J Respir Crit Care Med. 197(6):728–736
Sohn JW (2013) Acute eosinophilic pneumonia. Tuberc Respir Dis. 74:51–55
Kligerman S, Franks T, Galvin J (2016) Clinical-radiologic-pathologic correlation of smoking-related diffuse parenchymal lung disease. Radiol Clin North Am. 54(6):1047–1063
Cottin V, Cordier JF (2012) Eosinophilic lung diseases. Immunol Allergy Clin N Am. 32(4):557–586
Uchiyama H, Suda T, Nakamura Y et al (2008) Alterations in smoking habits are associated with acute eosinophilic pneumonia. Chest. 133(5):1174–1180
Philit F, Etienne-Mastroianni B, Parrot A et al (2002) Idiopathic acute eosinophilic pneumonia: a study of 22 patients. Am J Respir Crit Care Med. 166:1235–1239
Shorr AF, Scoville SL, Cersovsky SB et al (2004) Acute eosinophilic pneumonia among US military personnel deployed in or near Iraq. JAMA. 292:2997–3005
Galvin JR, Franks TJ (2009) Smoking-related lung disease. J Thorac Imaging. 24:274–284
Attili AK, Kazerooni EA, Gross BH et al (2008) Smoking-related interstitial lung disease: radiologic–clinical–pathologic correlation. Radiographics. 28:1383–1396
Bano S, Chaudhary V, Narula MK et al (2014) Pulmonary Langerhans cell histiocytosis in children: a spectrum of radiologic findings. Eur J Radiol. 83(1):47–56
Radzikowska E (2017) Pulmonary Langerhans’ cell histiocytosis in adults. Adv Respir Med. 85:277–289
Vassallo R, Harari S, Tazi A (2017) Current understanding and management of pulmonary Langerhans cell histiocytosis. Thorax. 72(10):937–945
Girschikofsky M, Arico M, Castillo D et al (2013) Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis. 8:72
Harari S, Torre O, Cassandro R et al (2012) Bronchoscopic diagnosis of Langerhans cell histiocytosis and lymphangioleiomyomatosis. Respir Med. 106(9):1286–1292
Picarsic J, Jaffe R (2015) Nosology and pathology of Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 29:799–823
Torre O, Elia D, Caminati A, Harari S (2017) New insights in lymphangioleiomyomatosis and pulmonary Langerhans cell histiocytosis. Eur Respir Rev 26(145). https://www.ncbi.nlm.nih.gov/pubmed/28954765
Rao RN, Goodman LR, Tomashefski JF Jr (2008) Smoking-related interstitial lung disease. Ann Diagn Pathol. 12:445–457
Tazi A (2006) Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J. 27(6):1272–1285
Tazi A, de Margerie C, Naccache JM et al (2015) The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis.
Aricò M, Girschikofsky M, Généreau T et al (2003) Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer 39(16):2341–8. https://www.ncbi.nlm.nih.gov/pubmed/14556926
Vassallo R, Ryu JH, Schroeder DR et al (2002) Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med. 346(7):484–490
Stockschlaeder M, Sucker C (2006) Adult Langerhans cell histiocytosis. Eur J Haematol. 76(5):363–368
Elia D, Torre O, Cassandro R et al (2015) Pulmonary Langerhans cell histiocytosis: a comprehensive analysis of 40 patients and literature review. Eur J Intern Med. 26(5):351–356
Schönfeld N, Dirks K, Costabel U et al (2012) A prospective clinical multicentre study on adult pulmonary Langerhans’ cell histiocytosis. Sarcoidosis Vasc Diffuse Lung Dis. 29(2):132–138
Negrin-Dastis S, Butenda D, Dorzee J et al (2007) Complete disappearance of lung abnormalities on high-resolution computed tomography: a case of histiocytosis X. Can Respir J. 14(4):235–237
Tazi A, Marc K, Dominique S et al (2012) Serial computed tomography and lung function testing in pulmonary Langerhans’ cell histiocytosis. Eur Respir J. 40:905–912
Castoldi MC, Verrioli A, De Juli E et al (2014) Pulmonary Langerhans cell histiocytosis: the many faces of presentation at initial CT scan. Insights Imaging. 5:483–492
Richards JC, Lynch DA, Chung JH (2015) Cystic and nodular lung disease. Clin Chest Med. 36:299–312
Madan R, Matalon S, Vivero M (2016) Spectrum of smoking-related lung diseases: imaging review and update. J Thorac Imaging. 31(2):78–91
Ryu JH, Myers JL, Capizzi SA, Douglas WW, Vassallo R, Decker PA (2005) Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest. 127:178–184
Moon J, du Bois RM, Colby TV et al (1999) Clinical significance of respiratory bronchiolitis on open lung biopsy and its relationship to smoking related interstitial lung disease. Thorax. 54:1009–1014
King TE Jr (1993) Respiratory bronchiolitis-associated interstitial lung disease. Clin Chest Med. 14:693–698
Park JS, Brown KK, Tuder RM et al (2002) Respiratory bronchiolitis-associated interstitial lung disease: radiologic features with clinical and pathologic correlation. J Comput Assist Tomogr l26:13–20
Craig PJ, Wells AU, Doffman S et al (2004) Desquamative interstitial pneumonia, respiratory bronchiolitis and their relationship to smoking. Histopathology. 45(3):275–282
Liebow AA, Steer A, Billingsley JG (1965) Desquamative interstitial pneumonia. Am J Med. 39:369–404
Reddy TL, Mayo J, Churg A (2013) Respiratory bronchiolitis with fibrosis: high resolution computed tomography findings and correlation with pathology. Ann Am Thorac Soc. 10(6):590–601
Katzenstein AL (2012) Smoking-related interstitial fibrosis (SRIF), pathogenesis and treatment of usual interstitial pneumonia (UIP), and transbronchial biopsy in UIP. Mod Pathol. 25(Suppl 1):S68–S78
Katzenstein AL (2013) Smoking-related interstitial fibrosis (SRIF): pathologic findings and distinction from other chronic fibrosing lung diseases. J Clin Pathol. 66(10):882–887
Katzenstein AL, Mukhopadhay S, Zanardi C, Dexter E (2010) Clinically occult interstitial fibrosis in smokers: classification and significance of a surprisingly common finding in lobectomy specimens. Hum Pathol. 41(3):316–325
Yousem SA (2006) Respiratory bronchiolitis-associated interstitial lung disease with fibrosis is a lesion distinct from fibrotic nonspecific interstitial pneumonia: a proposal. Mod Pathol. 19(11):1474–1479
Kawabata Y, Hoshi E, Murai K et al (2008) Smoking-related changes in the background lung specimens resected for lung cancer: a semiquantitative study with correlation to postoperative course. Histopathology. 53(6):707–714
Yamada T, Nakanishi Y, Homma T et al (2013) Airspace enlargement with fibrosis shows characteristic histology and immunohistology different from usual interstitial pneumonia, nonspecific interstitial pneumonia and centrilobular emphysema. Pathol Int. 63(4):206–213
Watanabe Y, Kawabata Y, Kanauchi T et al (2015) Multiple, thin-walled cysts are one of the HRCT features of airspace enlargement with fibrosis. Eur J Radiol. 84(5):986–992
Franks TJ, Galvin JR (2015) Smoking-related “interstitial” lung disease. Arch Pathol Lab Med. 139(8):974–977
Marten K, Milne D, Antoniou K et al (2009) Non-specific interstitial pneumonia in cigarette smokers: a CT study. Eur Radiol. 19(7):1679–1685
Raghu G, Collard HR, Egan JJ et al (2011) An official ATS/ERS/ JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 183:788–824
Baumgartner KB, Samet JM, Stidley CA et al (1997) Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 155:242–248
Ley B, Collard HR, King TE (2011) Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 183:43140
Richeldi L, Collard HR, Jones MG (2017) Idiopathic pulmonary fibrosis. Lancet. 389:1941–1952
Antoniou KM, Hansell DM, Rubens MB et al (2008) Idiopathic pulmonary fibrosis: outcome in relation to smoking status. Am J Respir Crit Care Med. 177:190–194
Lynch DA, Sverzellati N, Travis WD et al (2018) Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 6(2):138–153
Kitaguchi Y, Fujimoto K, Hanaoka M et al (2010) Clinical characteristics of combined pulmonary fibrosis and emphysema. Respirology. 15:265–271
Grubstein A, Bendayan D, Schactman I et al (2005) Concomitant upper-lobe bullous emphysema, lower-lobe interstitial fibrosis and pulmonary hypertension in heavy smokers: report of eight cases and review of the literature. Respir Med. 99:948–954
Mura M, Zompatori M, Pacilli AM et al (2006) The presence of emphysema further impairs physiologic function in patients with idiopathic pulmonary fibrosis. Respir Care. 51:257–265
Jankowich MD, Polsky M, Klein M et al (2008) Heterogeneity in combined pulmonary fibrosis and emphysema. Respiration. 75:411–417
Cottin V, Cordier JF (2009) The syndrome of combined pulmonary fibrosis and emphysema. Chest. 136(1):1–2
Mitchell PD, Das JP, Murphy DJ et al (2015) Idiopathic pulmonary fibrosis with emphysema: evidence of synergy among emphysema and idiopathic pulmonary fibrosis in smokers. Respir Care. 60:259–268
Zhang L, Zhang C, Dong F et al (2016) Combined pulmonary fibrosis and emphysema: a retrospective analysis of clinical characteristics, treatment and prognosis. BMC Pulm Med. 16:137
Ryerson CJ, Hartman T, Elicker BM et al (2013) Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest. 144(1):234–240
Cottin V, Nunes H, Brillet PY et al (2005) Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 26:586–593
Mejía M, Carrillo G, Rojas-Serrano J et al (2009) Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 136:10–15
Cottin V, Le Pavec J, Prévot G et al (2010) Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 35:105–111
Usui K, Tanai C, Tanaka Y et al (2011) The prevalence of pulmonary fibrosis combined with emphysema in patients with lung cancer. Respirology. 16:326–331
Matsuoka S, Yamashiro T, Matsushita S et al (2015) Morphological disease progression of combined pulmonary fibrosis and emphysema: comparison with emphysema alone and pulmonary fibrosis alone. J Comput Assist Tomogr. 39:153–159
Wright JL, Tazelaar HD, Churg A (2011) Fibrosis with emphysema. Histopathology. 58:517–524
Acknowledgements
None
Funding
The authors state that this work has received any funding.
Author information
Authors and Affiliations
Contributions
All authors contributed to the collections of cases and development/content of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sousa, C., Rodrigues, M., Carvalho, A. et al. Diffuse smoking-related lung diseases: insights from a radiologic-pathologic correlation. Insights Imaging 10, 73 (2019). https://doi.org/10.1186/s13244-019-0765-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13244-019-0765-z