
Abstract
Smoking-related lung abnormalities are now an increasing public health concern. According to the findings of large-cohort studies, approximately 8% of smokers have interstitial lung abnormalities, which are associated with a relatively high risk of all-cause mortality. We reviewed the radiological and pathological findings of smoking-related interstitial lung diseases, such as respiratory bronchiolitis-interstitial lung disease, desquamative interstitial pneumonia, and airspace enlargement with fibrosis. We have also discussed the histological basis of unclassifiable interstitial pneumonia in smokers, which exhibits airway-centered cystic lesions with fibrosis. A variety of radiological findings coexist in the lungs of a smoker. This overlapping of multiple pathological conditions might cause the radiological patterns of diseases to become unclassifiable. Therefore, diagnosis should be performed not on the basis of a single radiological finding, but in a comprehensive manner, by including clinical symptoms and disease behavior. Among interstitial abnormalities in smokers, the usual interstitial pneumonia (UIP) pattern is correlated with a worse prognosis than others. Basal-predominant subpleural reticulation is a clue for accurate diagnosis of UIP, which can be achieved by computer-aided quantitative analysis.
Similar content being viewed by others


Introduction
A variety of pathological and physiological abnormalities exist in the lungs of smokers and ex-smokers, including emphysema and various interstitial lung diseases (ILDs). Traditional smoking-related lung diseases include Langerhans cell histiocytosis (LCH), respiratory bronchiolitis-ILD (RB-ILD), and desquamative interstitial pneumonia (DIP) [1]. Smoking is a risk factor for chronic fibrosing interstitial pneumonias — including usual interstitial pneumonia (UIP) and non-specific interstitial pneumonia (NSIP) [2] — as well as unclassifiable idiopathic interstitial pneumonias (unclassifiable IIPs) [3]. High-resolution computed tomography (HRCT) can demonstrate these abnormalities before the appearance of characteristic clinical symptoms [4]. A prospective, large-scale, cohort study demonstrated that approximately 8% of smokers have interstitial lung abnormalities (ILAs) [5]. These abnormalities are associated with a relatively high risk of all-cause mortality [6], and some progress into pulmonary fibrosis [7]. Araki et al. analyzed serial CT images of 1867 participants in the Framingham Heart Study and observed the development of ILAs, including de novo UIP-pattern fibrosis [7]. In the background of these findings, smoking-related lung abnormalities are now considered an increasing public health concern.
In this review, we discuss HRCT findings observed in the lungs of smokers and identify characteristics that could differentiate interstitial lung abnormalities from idiopathic pulmonary fibrosis (IPF), which has a worse prognosis than the former.
Smoking-related lung diseases
Table 1 summarizes the abnormalities observed in the lungs of smokers. We stress that many pathological and radiological findings coexist in the lungs of a smoker; for example, emphysema can coexist with interstitial fibrosis, which appears to be at the opposite end of the disease spectrum [8]. Overlapping of multiple pathological conditions might cause the radiological patterns of diseases to become unclassifiable. Therefore, diagnosis should be performed not on the basis of a single radiological finding, but in a comprehensive manner, by including clinical symptoms and disease behavior.
According to the official statement of the American Thoracic Society and European Respiratory Society (ATS/ERS) [9], RB-ILD and DIP are categorized as smoking-related IIPs. These two diseases are thought to be different end-points of a similar pathological condition [10]; however, they differ in terms of clinical presentation, imaging findings, and response to therapy and, therefore, remain classified separately.
Langerhans cell histiocytosis
LCH is an uncommon disease characterized by histiocyte proliferation. Patients with LCH are usually young adults, in their third or fourth decade of life. There is strong association between smoking and LCH — over 90% of patients with LCH have a history of smoking [11].
The mechanism of LCH pathogenesis is unclear, although there is some evidence that the characteristic proliferation of Langerhans cells represents an abnormal immune reaction, possibly directed at the components of cigarette smoke [12, 13]. A very small percentage of smokers develop LCH, and a genetic background to the disease is also speculated [14, 15]. The diagnostic histological features of LCH are follows: (1) nodular interstitial infiltrates of Langerhans cells; (2) peribronchiolar location and stellate shape; (3) adjacent traction emphysema; and (4) RB with intra-alveolar macrophage accumulation (DIP-like reaction) (Fig. 1).

A 33-year-old male smoker with LCH. HRCT images of the a left upper and b left lower lobes. c Histological specimen (H & E staining; original magnification, × 600). While HRCT images show upper-lobe predominant, poorly defined, numerous stellate nodules, cysts, and irregular lines (a, b), Histology shows the area of accumulation of Langerhans cells, which have cerebriform vesicular nuclei with longitudinal grooves and palely eosinophilic cytoplasm. Arrows reveal representative cells (c)
On HRCT images of LCH, nodules (Fig. 1) and cysts (Fig. 2) are frequently observed with upper- and middle-zone predominance of distribution (Fig. 1). While nodules are usually less than 10 mm in diameter, they may also be as large as several centimeters in size and exhibit cavitation [2]. Serial HRCT findings have demonstrated the transformation of large nodules into thick-walled cysts, which later transform into thin-walled cysts [16]. Only coalescent cysts can be observed on follow-up CT images of LCH (Fig. 2) [17]. Emphysema may be identifiable on HRCT images at any stage of LCH. However, differentiation between coalescent cysts and de novo emphysema becomes increasingly challenging with disease progression.

Follow-up CT findings of a 33-year-old male smoker with LCH. HRCT findings at initial presentation (a), and (b) 10-year follow-up. Nodules and cysts are found mainly in the upper lobe (a). After cessation of smoking, the nodules have disappeared, and the air space has enlarged (b)
Differential diagnoses of LCH include diffuse cystic diseases such as lymphangiomyomatosis (LAM) and Birt–Hogg–Dubé disease (BHD) [18]. LAM is an uncommon cystic lung disease caused by infiltration of the lungs with smooth muscle cells originating from an unknown source, spreading through blood and the lymphatic system, and containing growth-activating mutations in tuberous sclerosis genes [19]. On HRCT images, cysts of LAM exhibit diffuse distribution. In a patient with typical HRCT findings of LAM, a serum vascular endothelial growth factor D level > 800 pg/ml is also diagnostic for the disease [20]. BHD is a rare, autosomal-dominant disorder characterized by the development of hair-follicle tumors, renal neoplasms, and pulmonary cysts [21]. Cystic lung lesions in BHD are typically seen predominantly in the lung base and paraseptal locations [22].
Respiratory bronchiolitis-interstitial lung disease
RB is a common histological lesion seen in smokers. The majority of cases of RB are asymptomatic. It is characterized by pigmented macrophage accumulation within bronchioles (Fig. 3) [13].

A 43-year-old male smoker with RB-ILD. HRCT images of the a right upper and b right lower lobes. c Histological specimen (H & E staining; original magnification, × 10). In HRCT images, bronchial wall thickening (a, arrow), centrilobular nodules, GGO, and cysts (a, b, circles) are seen mainly in the upper lobe (a, b). In histological specimens, RB (c, arrow) and centriacinar emphysema are observed
RB-ILD — a clinical diagnosis made in heavy smokers — is extensive enough to cause symptoms and show evidence of ILD. Myers et al. described six patients with clinical, radiological, and physiological manifestations of restrictive chronic ILD, but with only RB observed upon open biopsy [23]. Histological findings of RB-ILD include RB and peribronchiolar fibrosis as well as peribronchiolar macrophage distribution. Some patients show widespread macrophage distribution, overlapping with DIP [24].
In current smokers and ex-smokers with RB, chest radiographic appearance of the lungs is often normal, and any abnormalities tend to be limited and non-specific [25]. On HRCT images, centrilobular nodules and ground-glass opacities (GGO) are predominant abnormalities in RB (Fig. 3) [9, 26]. In the largest HRCT series of patients with a clinicopathological diagnosis of RB-ILD (n = 21), Park et al. found the most frequent HRCT features to be central bronchial wall thickening, centrilobular nodules, and areas of GGO [27]. Although a few other studies had reported the upper lung zones to be predominantly affected [26], Park et al. did not report any zonal predominance. Remy-Jardin et al. reported ill-defined ground-glass micronodules with upper-lobe predominance in 27% of current smokers and none of the never-smokers in their study; they also found that emphysema and GGOs increase with smoking and that these CT findings were partially reversible in patients who stopped smoking [4]. These findings suggest that RB-ILD is characterized by micronodules and GGOs, with extensive macrophage accumulation in respiratory bronchioles, alveolar ducts, and alveoli.
In clinical practice, RB-ILD is being increasingly diagnosed without surgical lung biopsy in smokers with these HRCT findings as well as in cases where histological findings of bronchoalveolar lavage demonstrate smoker’s macrophages and absence of lymphocytosis, which are potentially suggestive of hypersensitivity pneumonitis (HP) [9, 28]. The differential diagnosis of RB-ILD on the basis of HRCT findings is HP, which presents with centrilobular nodules and GGOs [9]. In RB-ILD, partial regression occurs with smoking cessation, with or without treatment [28].
Desquamative interstitial pneumonia
DIP is an uncommon condition observed in the fourth or fifth decade of life. Histological hallmarks of DIP include prominent intra-alveolar accumulation of macrophages, hyperplasia of type II pneumocytes, and, more variably, diffuse alveolar septal thickening (Fig. 4) [10]. Relative to RB-ILD, DIP presents with a greater extent of interstitial fibrosis and more prominent lymphoid follicles and eosinophilic infiltration [1].

DIP in a 52-year-old male smoker. Histological specimens (H & E staining) of the upper lobe (a, b) (original magnification, × 5 and × 20, respectively). c, d Initial and e, f 11-year follow-up HRCT findings. Right upper-lobe biopsy findings reveal diffuse, sheet-like, intra-alveolar macrophage accumulation (a, b). Pigmented macrophages (occasionally multinucleated) and eosinophil infiltration are noted. Mild interstitial fibrosis is also present (b). Initial HRCT images show widespread GGO in the middle and lower zones, with peripheral or patchy distribution. Cystic spaces are also noted (c, d). In follow-up CT images, the GGOs have disappeared after steroid therapy and cessation of smoking. However, emphysema-like cysts remain (e, f)
One of the HRCT findings of DIP is widespread GGOs in the middle and lower zones, with peripheral or patchy distribution (Fig. 4) [2]. Centrilobular nodules are also observed in DIP, which probably represent coexisting RB. Cystic spaces are also noted. Akira et al. suggested that these cysts probably correspond to dilated alveolar ducts or pulmonary cysts [29]. In follow-up CT findings of histologically proven DIP, GGOs were reported to have disappeared, with only the emphysema remaining; this suggests that some of the cysts serve as foci of emphysema (Fig. 4). A typical honeycomb pattern is not observed in biopsy-proven DIP.
Relationship between non-specific interstitial pneumonia and smoking
Idiopathic NSIP is now recognized as one of the IIPs [30]. It is well known that some patients with NSIP exhibit autoimmune features although they do not satisfy any of the diagnostic criteria for autoimmune diseases [31]. Upon excluding patients with NSIP exhibiting autoimmune features, a non-negligible proportion of residual cases of biopsy-proven fibrosing NSIP (fNSIP) are found to include smokers and ex-smokers (Fig. 5) [32]. Some authors have proposed that smoking is closely related to NSIP-pattern fibrosis [33, 34].

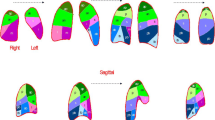
A 57-year-old man with fNSIP. a Initial and b 5-year follow-up HRCT findings. c 3D images reconstructed from CT images by GHNC. In the GHNC image: pink = normal (N); dark blue = emphysema (E); light green = GGO (G); light blue = reticulation (R); yellow = honeycomb (H); dark green = trachea and bronchi; and orange = vessels. In the initial CT image, diffuse reticulation with traction bronchiectasis is noted, predominantly in the lower lobe. While reticulation shows peripheral predominance, the subpleural region is relatively spared (a). In follow-up CT images, the reticulation has remained, and traction bronchiectasis is seen to have progressed (b). In the 3D GHNC image viewed from the posterior left direction, the lung is covered in a light-blue R-pattern, which corresponds to relatively homogenous fibrosis. Regions exhibiting a yellow H-pattern are few (c)
It is easy to confuse DIP with fNSIP in smokers. Smokers with fNSIP frequently present with the DIP pattern [35]. If alveolar macrophages were to be excluded, the biopsy findings of DIP would resemble those of fNSIP (Fig. 4) [10]. Overlapping of DIP and NSIP is a common phenomenon [35]. Such cases of overlap might exhibit progression of fibrosis (Fig. 6). In our experience, progression to severe fibrosis is rare in biopsy-proven typical DIP. However, differentiating among fNSIP, DIP, and an overlapping pattern of DIP and NSIP (DIP–NSIP) on the basis of initial HRCT findings is challenging; this requires clinicians to observe disease behavior, including treatment response.

A 40-year-old male smoker exhibiting overlapping NSIP and DIP upon surgical biopsy. a Initial and b 5-year follow-up HRCT findings. In the initial CT image, diffuse reticulation with traction bronchiectasis is noted along bronchovascular bundles, predominantly in the lower lobe (a). In the follow-up CT image, reticulation is seen to have increased (b)
Overlapping of NSIP and UIP is also common among smokers (Fig. 7). In a study by Kambouchner et al., an overlapping pattern of NSIP and UIP (NSIP–UIP) was observed in 26% of patients with biopsy-proven NSIP; two-thirds of these patients were smokers [35]. Jin et al. reported a 9.7% (86 of 884 participants of the National Lung Screening Trial population evaluated at a single site) prevalence of ILAs in their study; it is very interesting that 37% of patients with fibrotic ILAs in this study exhibited disease progression after 2 years of follow-up, and all these patients exhibited NSIP–UIP [3]. Distinguishing between UIP and NSIP in smokers is challenging [36]; NSIP-pattern fibrosis, especially peribronchiolar fibrosis, is commonly observed in the end-stage of IPF/UIP [37]. We believe that fNSIP is one of the common histological components of smoking-related lung diseases.

A 73-year-old male ex-smoker exhibiting overlapping NSIP and UIP upon surgical biopsy. HRCT images of the upper (a), middle (b), and lower (c) lungs show GGOs with peripheral-predominant, patchy consolidation along bronchovascular bundles (b, arrow). At the same time, subpleural reticulation is noted at the lung base, with small dots and short lines in the dorsal subpleural regions of the upper lobe, indicating UIP-pattern fibrosis (a, c, arrow heads)
Usual interstitial pneumonia in smokers
Smoking is strongly associated with IPF [9]. The pattern of UIP fibrosis also overlaps with emphysema and classic smoking-related IIPs. It is peculiar that emphysema and fibrosis — diseases on opposite ends of the spectrum — coexist in the same set of lungs.
Figure 8 shows the schema of coexistence of UIP-pattern fibrosis and centrilobular emphysema. Typical centrilobular emphysema is observed in the centriacinar part with respiratory bronchiolitis. On the other hand, UIP-pattern fibrosis is distributed in the perilobular (periacinar) area, and it is characterized by alveolar collapse [38] and decreased lung volume [39]. The predominant locations of centrilobular emphysema and UIP are microscopically different; therefore, emphysema and UIP-pattern fibrosis can coexist and are simply superimposed in the same lobule (Fig. 9) [40]. Rogliani et al. discussed matrix metalloproteinases (MMPs), a family of structurally related enzymes capable of degrading all components of the extracellular matrix and basement membranes; the authors found MMPs to be expressed at relatively high levels in fibroblastic foci in both UIP and emphysema/UIP [41]. Cysts with small dots are frequently seen on HRCT images of smokers. We believe that some of these cysts indicate destruction of lung tissue by emphysema and that the small dots are remnants of vessels and bronchi (Fig. 9).

Superimposed CT images of emphysema and UIP-pattern fibrosis. Scheme of centriacinar (a) and periacinar (c) distribution and their superimposition (b) in CT images of a normal ex vivo lung. CT images of d centrilobular emphysema; e IPF/UIP in a smoker; and f periacinar fibrosis in a non-smoker with IPF. e and f are multiplanar reconstruction images, almost parallel to the pleura. In the non-smoker with IPF, periacinar fibrosis is noted as a polygonal structure with traction bronchiectasis (f). In the smoker with IPF, polygonal structure and centriacinar emphysema are observed, and the lungs exhibit a macroscopic honeycomb cystic pattern (e)

A 66-year-old male smoker with IPF/UIP. a Histological specimen (H & E staining; original magnification, × 2). HRCT images of the right lower lobe at (b) initial presentation and (c) 7-year follow-up. In the histological specimen, emphysema and perilobular UIP-pattern fibrosis are noted (a). Several cysts are noted in the subpleural region in the initial CT image (b). In the follow-up CT image, macroscopic honeycombing pattern is seen to have increased in volume (c). Arrows indicate cysts with small dot-like appearance (b, c)
Honeycombing is an important histological and radiological finding for diagnosis of IPF. However, the histological honeycomb is not equal to the honeycomb pattern observed on HRCT images. Most honeycomb cysts that pathologists usually observe are less than 1 mm in diameter, sometimes filled with mucus; these cannot be visualized on HRCT images, especially in early UIP (Fig. 10) [42]. Staats et al. compared the histological and HRCT findings of 69 explants with UIP and concluded that HRCT findings typical of UIP and the HRCT honeycomb pattern correlate best with histological findings of bronchiolectasis [43]. Yamauchi et al. reported that typical macroscopic honeycomb cysts are not observed in IPF in never-smokers, who only exhibit peripheral collapse and traction bronchiectasis in their follow-up evaluation despite exhibiting honeycombing in histological specimens [44]. In smokers, fibroses of all types — including UIP-pattern (Fig. 10) and collagenous-type (Fig. 11) fibrosis — are superimposed with emphysema; both exhibit a multiple-cyst pattern resembling a honeycomb on HRCT images. This is one of the reasons that emphysema with fibrosis resembles IPF/UIP. We discuss this matter in the following section.

A 70-year-old male never-smoker with IPF. a, b Initial and c 4-year follow-up HRCT findings. d 3D-GHNC image of the initial CT scan. e Histological specimens (H & E staining; original magnification, × 1). Initial HRCT findings show subpleural reticulation in the lung base (b, arrow), with small dots and short lines in subpleural regions of the upper lobe (a, arrow). After 4 years, although the reticulation has increased, there are no honeycomb cysts (c). The peripheral vessels are closer to the pleura, which indicates alveolar collapse (c, arrow). In the 3D-GHNC image (left posterior view), a yellow H-pattern — indicative of fibrosis — covers the lung surface, including the dorsal lung base. Perilobular fibroses are seen at junctions between the pleura and interlobular septum (we have termed these points “pleuro-septal junctions”) (e, arrows). These subpleural fibroses correspond to small opacities resembling thorns, located beneath the pleura on CT images (a, b arrows). The histological specimen shows microscopic honeycombing (e, black circles); these mucus-filled small cysts cannot be detected on CT images

Histological specimen showing AEF (H & E staining; original magnification, × 2). It shows ropey, collagen-type fibrosis with background of centriacinar emphysema
Airspace enlargement with fibrosis and combined pulmonary fibrosis and emphysema
Emphysema is characterized by permanently enlarged airspaces distal to the terminal bronchiole along with destruction of alveolar walls; absence of “obvious fibrosis” was historically regarded as an additional criterion [45]. This is still the standard definition of emphysema. However, microscopic fibrosis is a fairly common finding in centrilobular emphysema, and it is extremely common in paraseptal emphysema [40]. On the basis of biochemical and histochemical findings, Wright et al. demonstrated increased collagen concentration in the lesions of centrilobular emphysema; the authors described fibrosis as an integral part of the emphysematous process [40].
Several authors have proposed names for the pathological findings of fibrosis coexisting with emphysema. While Katzenstein et al. proposed smoking-related interstitial fibrosis [46], Yousem and Reddy et al. described the condition as RB with fibrosis [47, 48]. Wright et al. described fibrosis coexisting with emphysema as “emphysema with fibrosis” [40]. The term “airspace enlargement with fibrosis” (AEF) was a proposed by Yousem [47] and Kawabata et al. [49]. Kawabata et al. confirmed that these findings were observed more frequently in heavy smokers than in other patients [49]. We believe that AEF is a common phrase for representing the histological findings of fibrosis coexisting with emphysema.
In histological specimens, AEF is characterized by the following: (1) fibrous interstitium with structural remodeling; (2) emphysematous change; (3) frequent bronchiolocentric location; and (4) absence of fibroblastic foci (Fig. 11). Kawabata et al. reported the absence of acute exacerbation after surgery in patients with AEF [49]. Katzenstein et al. reported that most patients with smoking-related interstitial fibrosis are asymptomatic or only mildly symptomatic and that the clinical course of the disease is stable in most cases [46]. Therefore, AEF needs to be distinguished from UIP and other IIPs [9, 50].
Several studies have reported on the radiological findings of AEF. While AEF is grossly visible as variously sized thin-walled cysts in histological specimens [49], it presents as thin-walled cysts on HRCT images. In our experience, HRCT patterns in patients with histologically proven AEF are variable and include cystic lesions in reticulations (Fig. 12) and thin-walled cysts (Fig. 13). In some cases, it is difficult to distinguish between AEF and UIP on CT images (Fig. 14) [51].

A 79-year-old male ex-smoker with lung cancer and histologically proven AEF. a Initial and b 5-year follow-up HRCT findings. Reticulation of the left lower lobe has not changed after 5 years from initial presentation. Cystic space in the reticulation has slightly increased at the 5-year follow-up (b)

A 66-year-old male ex-smoker with lung cancer and histologically proven AEF. HRCT images of the upper (a) and lower (b) lobes show upper-lobe emphysema and multiple thin-walled cysts in the lower lobe

A 73-year-old male ex-smoker with histologically proven IPF/UIP. HRCT images show subpleural reticulation in the upper (a) and lower (b) lobes. Multiple cysts are also noted in these reticulations. Some of the cysts with small dots (arrows) might represent superimposed emphysema and UIP-pattern fibrosis (see Fig. 9 and text)
Combined pulmonary fibrosis and emphysema (CPFE), proposed by Cottin et al. is a clinical manifestation reflecting the coexisting patterns of interstitial fibrosing and emphysema [52]. The type of fibrosis in CPFE has yet to be defined; currently, any type of fibrosis, including AEF, NSIP, and UIP, may be included. Cottin et al. reported on 61 patients (all smokers) with low carbon monoxide transfer coefficients; 47 and 55% of these patients exhibited pulmonary hypertension at diagnosis and during follow-up, respectively, and all patients exhibited coexistence of emphysema with upper-zone predominant parenchymal lung disease suggestive of pulmonary fibrosis of the lower lobes on CT images (Fig. 15). Ryerson et al. defined CPFE as ≥ 10% emphysema on HRCT images; in their study, although 8% of patients with IPF met this criterion, there was no significant difference in mortality between patients with IPF with and without CPFE [53]. These findings suggest that fibrosis type might play a key role in patient survival.

A 60-year-old male ex-smoker exhibiting combined pulmonary emphysema and fibrosis, with pulmonary hypertension. (a) A chest radiograph and (b, c) HRCT images of the upper and lower lung lobes. These images show upper-lobe emphysema and reticulation in the lower lobe (a–c)
Unclassifiable IIP in smokers
The ATS/ERS statement proposed an “unclassifiable” category of IIPs, acknowledging that final diagnosis might not be achieved even after lengthy multidisciplinary diagnosis [9]. Overlapping of different histological patterns is common among smokers. Therefore, smokers and ex-smokers with DIP–NSIP and NSIP–UIP may be easily classified as having unclassifiable IIP. Besides such cases of overlap, unclassifiable IIPs include new entities or unusual variants of recognized entities not adequately characterized by the current classification (e.g., a variant of organizing pneumonia with supervening fibrosis) [9]. Ryerson et al. retrospectively reviewed 1370 cases of ILD and diagnosed unclassifiable IIP in 132 cases (10%), of which 64% involved smokers or ex-smokers; the authors also reported that unclassifiable IIP exhibits slow progression and longer patient survival than IPF [54].
Among smokers and ex-smokers, we recognized a considerable number of cases of unclassifiable IIP with peculiar pathological changes that cannot be explained by the ATS/ERS classification. All of these cases involved smokers, and the histopathological specimens exhibited airway-centered cystic lesions with fibrosis, accompanied by bronchiolar metaplasia, mucostasis, and fibroblastic foci (Figs. 16, 17, 18). On HRCT images, cysts were noted in reticulations, usually apart from the pleura (Fig. 16). Cystic dilatation of peripheral bronchi was frequently seen on multiplanar reconstruction images (Fig. 17). On serial HRCT images, the airway-centered cysts were seen to slowly progress, finally creating large airspaces and causing lung parenchyma destruction in several cases (Fig. 18). These patients exhibited symptoms of dyspnea on exertion. In pulmonary function tests, many of these patients exhibited nearly normal levels of forced vital capacity and forced expiratory volume in 1 s, although the carbon monoxide diffusion capacity of the lungs was low. Despite the slow progression of symptoms, lung cancer and coronary arterial diseases frequently occur in patients with unclassifiable IIP.

A 69-year-old male ex-smoker (49 pack-years) with unclassifiable interstitial pneumonia. a Initial and b 4- and c 12-year follow-up HRCT findings of the right lung. d Histological specimen (H & E staining, original magnification, × 5) of the left lower lobe biopsied after the second CT examination. GGOs are noted along bronchovascular bundles in the initial CT image (a). Small cysts are noted in GGOs on the 4-year follow-up CT image (b). On the 12-year follow-up CT image, these cysts are seen to have enlarged and increased in number (c); however, subpleural regions are spared. The histological specimen shows airway-centered cystic lesions with fibrosis, accompanied by bronchiolar metaplasia, mucostasis, and fibroblastic focus, next to the intrapulmonary lymph node (d, arrow)

A 50-year-old male ex-smoker with unclassifiable interstitial pneumonia. a Initial and b 7- and c 10-year follow-up HRCT images. d, e Coronal reconstructed images of the initial and 7-year follow-up CT scans. Over the follow-up period, the reticulation is seen to have gradually increased, while the normal lung is seen to have decreased in area. Cysts in the reticulation are seen to have enlarged. In coronal reconstruction images, cysts directly involving the bronchus are also seen to have increased in size (d, e, arrows)

A 55-year-old male current smoker with unclassifiable interstitial pneumonia. a Initial and b 5-year follow-up HRCT images. In the initial pulmonary function test, the %predicted carbon monoxide diffusion capacity of the lungs was 36.0%. Initial CT images show thin-walled multiple cysts at the lung base; a large number of GGOs are also noted. In the follow-up CT image, these cysts are seen to have enlarged, which appears like the destruction of lung parenchyma
Several authors have already discussed airway-centered lesions. Fukuoka et al. mainly mentioned bronchial metaplasia in diffuse lung diseases [55]. Churg et al. proposed an ILD pathologically characterized by small airway-centered interstitial fibroses and metaplastic bronchiolar epithelium extending around and often linking fibrotic and sometimes heavily muscularized bronchioles [56]. Airway-centered interstitial fibrosis is also observed in non-smoking patients [57,58,59]. The findings of these small retrospective series suggest that ILDs mainly affect airways.
Some authors have described unclassifiable IIPs with a cystic pattern on HRCT images in smokers. Matsuoka et al. also demonstrated enlargement of low-attenuation areas on serial HRCT images in patients with emphysema and fibrosis [60]. In a study by Inomata et al., thick-walled cystic lesions — lesions involving one or more acini, with dense wall fibrosis and occasional fibroblastic foci surrounded by honeycombing and normal alveoli — were confirmed by postmortem examination in patients exhibiting CPFE-pattern findings on CT images [61]. Emphysematous destruction and enlargement of membranous and respiratory bronchioles with fibrosis were observed in these thick-walled cystic lesions.
On the basis of these findings, we believe that some smokers show unclassifiable IIP with airway-centered cystic lesions with fibrosis. The existence of fibroblastic foci and progression of cystic destruction are different from AEF, which does not show marked progression.
Differential diagnosis of smoking-related interstitial lung diseases
There are many kinds of smoking-related lung diseases, including emphysema and IPF. Among patients with these conditions, those with pathologically proven AEF show the best prognosis and require no treatment, while patients with IPF/UIP exhibit the worst prognosis and require early diagnosis and appropriate intervention with anti-fibrotic drugs. In cases of overlap, such as combined emphysema and UIP and NSIP–UIP, patient management depends on the extent of UIP-pattern fibrosis [53]. Therefore, detection of UIP-pattern fibrosis is important.
We believe that subpleural abnormalities are important for detection of UIP-pattern fibrosis on HRCT images. Small opacities resembling thorns, located beneath the pleura, are characteristic of UIP [62, 63], corresponding to fibrosis at the pleuroseptal junction (Fig. 10). Multiplanar reconstruction images show irregular polygonal structures with traction bronchiectasis in the subpleural area, corresponding to perilobular collapse and fibrosis (Fig. 8) [32].
Computer-based analysis is helpful for detecting subpleural fibrosis. For example, a Gaussian histogram normalized correlation (GHNC) system can segment the lungs along the lines of predesigned samples on the basis of CT attenuation values and local histogram findings [64,65,66,67]. In the GHNC system, an H-pattern is recognized on the basis of a sample of honeycomb lungs with fibrosis and traction bronchiectasis. Upon direct comparison between GHNC and surgical biopsy findings in a previous study, H-pattern volume ratios at biopsy sites were found to be significantly correlated with histologically identified honeycombing (r = 0.355; p < 0.001), subpleural collapse (r = 0.410; p < 0.001), and heterogeneity (r = 0.484; p < 0.001) [32]. Moreover, since the lung surface is widely covered by H-pattern lesions, computer-aided analysis of three-dimensional (3D) images can clearly demonstrate subpleural fibrosis (Fig. 19). Disease progression is the most important point for distinguishing between patients with IPF/UIP and stable patients with pathologically proven AEF. In clinical practice, however, prompt diagnosis is required, for example, before surgery. We believe that computer-aided 3D image analysis is useful for diagnosis in such cases.

Comparison of 3D-GHNC images of AEF and IPF. a, b Chest radiographs and c, d 3D-GHNC images. a, c A patient with adenocarcinoma and pathologically proven AEF (the same patient as in Fig. 12), who was alive 5 years after surgery. b, d A 58-year-old male ex-smoker with squamous cell carcinoma and IPF/UIP, who died from acute exacerbation after surgery for lung cancer. Chest radiographs and 3D-GHNC images both show lower lung volume in the second subject (b, d), relative to that in the first subject (a, c). In the 3D-GHNC image of the second subject, almost the entire lung is covered in a yellow H-pattern, which indicates advanced subpleural fibrosis
Conclusion
We have reviewed the radiological and pathological findings of smoking-related ILDs, including LCH, RB-ILD, DIP, and AEF. We have also discussed the characteristics of IPF/UIP, NSIP, and unclassifiable IIP in smokers. Many histological patterns of ILDs coexist in the lungs of smokers, which makes it difficult or impossible to classify them as a specific disease. Management approaches should be based on the most probable diagnosis after multidisciplinary evaluation and consideration of expected disease behavior. Since UIP-pattern fibrosis exhibits the worst prognosis of all ILDs, clinicians should pay close attention to subpleural fibrosis with alveolar collapse indicative of UIP.
Abbreviations
- 3D:
-
Three-dimensional
- AEF:
-
Airspace enlargement with fibrosis
- CPFE:
-
Combined pulmonary fibrosis and emphysema
- CT:
-
Computed tomography
- DIP:
-
Desquamative interstitial pneumonia
- fNSIP:
-
Fibrosing non-specific interstitial pneumonia
- HRCT:
-
High-resolution computed tomography
- H&E:
-
Hematoxylin–eosin
- IIP:
-
Idiopathic interstitial pneumonia
- ILD:
-
Interstitial lung diseases
- IPF:
-
Idiopathic pulmonary fibrosis
- GGO:
-
Ground-glass opacity
- LCH:
-
Langerhans cell histiocytosis
- NSIP:
-
Non-specific interstitial pneumonia
- RB-ILD:
-
Respiratory bronchiolitis-interstitial lung diseases
- UIP:
-
Usual interstitial pneumonia
References
Craig PJ, Wells AU, Doffman S, Rassl D, Colby TV, Hansell DM, et al. Desquamative interstitial pneumonia, respiratory bronchiolitis and their relationship to smoking. Histopathology. 2004;45:275–82.
Nair A, Hansell DM. High-resolution computed tomography features of smoking-related interstitial lung disease. Semin Ultrasound CT MR. 2014;35:59–71.
Jin GY, Lynch D, Chawla A, Garg K, Tammemagi MC, Sahin H, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology. 2013;268:563–71.
Remy-Jardin M, Edme JL, Boulenguez C, Remy J, Mastora I, Sobaszek A. Longitudinal follow-up study of smoker’s lung with thin-section CT in correlation with pulmonary function tests. Radiology. 2002;222:261–70.
Washko GR, Hunninghake GM, Fernandez LE, Nishino M, Okajima Y, Yamashiro T, et al. Lung volumes and emphysema in smokers with interstitial lung abnormalities. N Engl J Med. 2011;364:897–906.
Putman RK, Hatabu H, Araki T, Gudmundsson G, Gao W, Nishino M, et al. Association between interstitial lung abnormalities and all-cause mortality. JAMA. 2016;315:672–81.
Araki T, Putman RK, Hatabu H, Gao W, Dupuis J, Latourelle JC, et al. Development and progression of interstitial lung abnormalities in the Framingham Heart study. Am J Respir Crit Care Med. 2016;194:1514–22.
Madan R, Matalon S, Vivero M. Spectrum of smoking-related lung diseases: imaging review and update. J Thorac Imaging. 2016;31:78–91.
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–48.
Tazelaar HD, Wright JL, Churg A. Desquamative interstitial pneumonia. Histopathology. 2011;58:509–16.
Abbott GF, Rosado-de-Christenson ML, Franks TJ, Frazier AA, Galvin JR. From the archives of the AFIP: pulmonary Langerhans cell histiocytosis. Radiographics. 2004;24:821–41.
Katzenstein AL. Langerhans cell histiocytosis. In: Katzenstein AL, editor. Katzenstein and Askin’s surgical pathology of non-neoplastic lung disease. 4th ed. Philadelphia: Saunders Elsevier; 2006. p. 417–25.
Crotty Alexander LE, Shin S, Hwang JH. Inflammatory diseases of the lung induced by conventional cigarette smoke: a review. Chest. 2015;148:1307–22.
Mourah S, How-Kit A, Meignin V, Gossot D, Lorillon G, Bugnet E, et al. Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J. 2016;47:1785–96.
Kamionek M, Ahmadi Moghaddam P, Sakhdari A, Kovach AE, Welch M, Meng X, et al. Mutually exclusive extracellular signal-regulated kinase pathway mutations are present in different stages of multi-focal pulmonary Langerhans cell histiocytosis supporting clonal nature of the disease. Histopathology. 2016;69:499–509.
Brauner MW, Grenier P, Mouelhi MM, Mompoint D, Lenoir S. Pulmonary histiocytosis X: evaluation with high-resolution CT. Radiology. 1989;172:255–8.
Kim HJ, Lee KS, Johkoh T, Tomiyama N, Lee HY, Han J, et al. Pulmonary Langerhans cell histiocytosis in adults: high-resolution CT—pathology comparisons and evolutional changes at CT. Eur Radiol. 2011;21:1406–15.
Raoof S, Bondalapati P, Vydyula R, Ryu JH, Gupta N, Raoof S, et al. Cystic lung diseases: algorithmic approach. Chest. 2016;150:945–65.
Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. Diffuse cystic lung disease. Part I. Am J Respir Crit Care Med. 2015;191:1354–66.
Young L, Lee HS, Inoue Y, Moss J, Singer LG, Strange C, et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med. 2013;1:445–52.
Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. Diffuse cystic lung disease. Part II. Am J Respir Crit Care Med. 2015;192:17–29.
Kumasaka T, Hayashi T, Mitani K, Kataoka H, Kikkawa M, Tobino K, et al. Characterization of pulmonary cysts in Birt-Hogg-Dubé syndrome: histopathological and morphometric analysis of 229 pulmonary cysts from 50 unrelated patients. Histopathology. 2014;65:100–10.
Myers JL, Veal CF Jr, Shin MS, Katzenstein AL. Respiratory bronchiolitis causing interstitial lung disease. A clinicopathologic study of six cases. Am Rev Respir Dis. 1987;135:880–4.
Moon J, du Bois RM, Colby TV, Hansell DM, Nicholson AG. Clinical significance of respiratory bronchiolitis on open lung biopsy and its relationship to smoking related interstitial lung disease. Thorax. 1999;54:1009–14.
Wells AU, Nicholson AG, Hansell DM. Challenges in pulmonary fibrosis. 4: Smoking-induced diffuse interstitial lung diseases. Thorax. 2007;62:904–10.
Heyneman LE, Ward S, Lynch DA, Remy-Jardin M, Johkoh T, Müller NL. Respiratory bronchiolitis, respiratory bronchiolitis-associated interstitial lung disease, and desquamative interstitial pneumonia: different entities or part of the spectrum of the same disease process? AJR Am J Roentgenol. 1999;173:1617–22.
Park JS, Brown KK, Tuder RM, Hale VA, King TE Jr, Lynch DA. Respiratory bronchiolitis-associated interstitial lung disease: radiologic features with clinical and pathologic correlation. J Comput Assist Tomogr. 2002;26:13–20.
Sieminska A, Kuziemski K. Respiratory bronchiolitis-interstitial lung disease. Orphanet J Rare Dis. 2014;9:106.
Akira M, Yamamoto S, Hara H, Sakatani M, Ueda E. Serial computed tomographic evaluation in desquamative interstitial pneumonia. Thorax. 1997;52:333–7.
Travis WD, Hunninghake G, King TE Jr, Lynch DA, Colby TV, Galvin JR, et al. Idiopathic nonspecific interstitial pneumonia: report of an American Thoracic Society project. Am J Respir Crit Care Med. 2008;177:1338–47.
Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46:976–87.
Iwasawa T, Takemura T, Okudera K, Gotoh T, Iwao Y, Kitamura H, et al. The importance of subpleural fibrosis in the prognosis of patients with idiopathic interstitial pneumonias. Eur J Radiol. 2017;90:106–13.
Marten K, Milne D, Antoniou KM, Nicholson AG, Tennant RC, Hansel TT, et al. Non-specific interstitial pneumonia in cigarette smokers: a CT study. Eur Radiol. 2009;19:1679–85.
Shinohara T, Kadota N, Hino H, Naruse K, Ohtsuki Y, Ogushi F. Improvement in idiopathic nonspecific interstitial pneumonia after smoking cessation. Respir Med Case Rep. 2014;14:7–9.
Kambouchner M, Levy P, Nicholson AG, Schubel K, Magois E, Feuillet S, et al. Prognostic relevance of histological variants in nonspecific interstitial pneumonia. Histopathology. 2014;65:549–60.
Akira M, Inoue Y, Kitaichi M, Yamamoto S, Arai T, Toyokawa K. Usual interstitial pneumonia and nonspecific interstitial pneumonia with and without concurrent emphysema: thin-section CT findings. Radiology. 2009;251:271–9.
Rabeyrin M, Thivolet F, Ferretti GR, Chalabreysse L, Jankowski A, Cottin V, et al. Usual interstitial pneumonia end-stage features from explants with radiologic and pathological correlations. Ann Diagn Pathol. 2015;19:269–76.
Galvin JR, Frazier AA, Franks TJ. Collaborative radiologic and histopathologic assessment of fibrotic lung disease. Radiology. 2010;255:692–706.
Iwasawa T, Ogura T, Sakai F, Kanauchi T, Komagata T, Baba T, et al. CT analysis of the effect of pirfenidone in patients with idiopathic pulmonary fibrosis. Eur J Radiol. 2014;83:32–8.
Wright JL, Tazelaar HD, Churg A. Fibrosis with emphysema. Histopathology. 2011;58:517–24.
Rogliani P, Mura M, Mattia P, Ferlosio A, Farinelli G, Mariotta S, et al. HRCT and histopathological evaluation of fibrosis and tissue destruction in IPF associated with pulmonary emphysema. Respir Med. 2008;102:1753–61.
Akira M. Radiographic differentiation of advanced fibrocystic lung diseases. Ann Am Thorac Soc. 2017;14:432–40.
Staats P, Kligerman S, Todd N, Tavora F, Xu L, Burke A. A comparative study of honeycombing on high resolution computed tomography with histologic lung remodeling in explants with usual interstitial pneumonia. Pathol Res Pract. 2015;211:55–61.
Yamauchi H, Bando M, Baba T, Kataoka K, Yamada Y, Yamamoto H, et al. Clinical course and changes in high-resolution computed tomography findings in patients with idiopathic pulmonary fibrosis without honeycombing. PLoS One. 2016;11:e0166168.
Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology. 2008;246:697–722.
Katzenstein AL, Mukhopadhyay S, Zanardi C, Dexter E. Clinically occult interstitial fibrosis in smokers: classification and significance of a surprisingly common finding in lobectomy specimens. Hum Pathol. 2010;41:316–25.
Yousem SA. Respiratory bronchiolitis-associated interstitial lung disease with fibrosis is a lesion distinct from fibrotic nonspecific interstitial pneumonia: a proposal. Mod Pathol. 2006;19:1474–9.
Reddy TL, Mayo J, Churg A. Respiratory bronchiolitis with fibrosis: high-resolution computed tomography findings and correlation with pathology. Ann Am Thorac Soc. 2013;10:590–601.
Kawabata Y, Hoshi E, Murai K, Ikeya T, Takahashi N, Saitou Y, et al. Smoking-related changes in the background lung of specimens resected for lung cancer: a semiquantitative study with correlation to postoperative course. Histopathology. 2008;53:707–14.
Katzenstein AL. Smoking-related interstitial fibrosis (SRIF), pathogenesis and treatment of usual interstitial pneumonia (UIP), and transbronchial biopsy in UIP. Mod Pathol. 2012;25(Suppl 1):S68–78.
Watanabe Y, Kawabata Y, Kanauchi T, Hoshi E, Kurashima K, Koyama S, et al. Multiple, thin-walled cysts are one of the HRCT features of airspace enlargement with fibrosis. Eur J Radiol. 2015;84:986–92.
Cottin V, Nunes H, Brillet PY, Delaval P, Devouassoux G, Tillie-Leblond I, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26:586–93.
Ryerson CJ, Hartman T, Elicker BM, Ley B, Lee JS, Abbritti M, et al. Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest. 2013;144:234–40.
Ryerson CJ, Urbania TH, Richeldi L, Mooney JJ, Lee JS, Jones KD, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J. 2013;42:750–7.
Fukuoka J, Franks TJ, Colby TV, Flaherty KR, Galvin JR, Hayden D, et al. Peribronchiolar metaplasia: a common histologic lesion in diffuse lung disease and a rare cause of interstitial lung disease: clinicopathologic features of 15 cases. Am J Surg Pathol. 2005;29:948–54.
Churg A, Myers J, Suarez T, Gaxiola M, Estrada A, Mejia M, et al. Airway-centered interstitial fibrosis: a distinct form of aggressive diffuse lung disease. Am J Surg Pathol. 2004;28:62–8.
Smith ML. Update on pulmonary fibrosis: not all fibrosis is created equally. Arch Pathol Lab Med. 2016;140:221–9.
Kuranishi LT, Leslie KO, Ferreira RG, Coletta EA, Storrer KM, Soares MR, et al. Airway-centered interstitial fibrosis: etiology, clinical findings and prognosis. Respir Res. 2015;16:55.
Pradere P, Gauvain C, Danel C, Debray MP, Borie R, Plantier L, et al. Airway-centered fibroelastosis: a distinct entity. Chest. 2016;149:767–74.
Matsuoka S, Yamashiro T, Matsushita S, Fujikawa A, Kotoku A, Yagihashi K, et al. Morphological disease progression of combined pulmonary fibrosis and emphysema: comparison with emphysema alone and pulmonary fibrosis alone. J Comput Assist Tomogr. 2015;39:153–9.
Inomata M, Ikushima S, Awano N, Kondoh K, Satake K, Masuo M, et al. An autopsy study of combined pulmonary fibrosis and emphysema: correlations among clinical, radiological, and pathological features. BMC Pulm Med. 2014;14:104.
Sumikawa H, Johkoh T, Fujimoto K, Arakawa H, Colby TV, Fukuoka J, et al. Pathologically proved nonspecific interstitial pneumonia: CT pattern analysis as compared with usual interstitial pneumonia CT pattern. Radiology. 2014;272:549–56.
Hunninghake GW, Lynch DA, Galvin JR, Gross BH, Müller N, Schwartz DA, et al. Radiologic findings are strongly associated with a pathologic diagnosis of usual interstitial pneumonia. Chest. 2003;124:1215–23.
Asakura A, Gotoh T, Iwasawa T, Saito H, Akasaka H. Classification system of the CT images with nonspecific interstitial pneumonia. J Inst Image Electron Eng Jpn. 2004;33:180–8.
Iwao Y, Gotoh T, Kagei S, Iwasawa T, Tsuzuki MSG. Integrated lung field segmentation of injured region with anatomical structure analysis by failure–recovery algorithm from chest CT images. Biomed Signal Process Control. 2014;12:28–38.
Iwasawa T, Kanauchi T, Hoshi T, Ogura T, Baba T, Gotoh T, et al. Multicenter study of quantitative computed tomography analysis using a computer-aided three-dimensional system in patients with idiopathic pulmonary fibrosis. Jpn J Radiol. 2016;34:16–27.
Iwasawa T, Iwao Y, Takemura T, Okudera K, Gotoh T, Baba T, et al. Extraction of the subpleural lung region for detecting interstitial lung disease on computed tomography images. Jpn J Radiol. 2017;35:681–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Tae Iwasawa received a research grant for this study (KAKENHI 15K09901).
Conflict of interest
Tae Iwasawa received a consulting fee from Ono pharmaceutical Co. Ltd. Takashi Ogura received a research grant from North East Japan Study Group and lecture fees from Shionogi & Co., Ltd. & Berlinger Ingelheim Co., Ltd. Tamiko Takemura has no conflicts of interest.
Ethnical statement
This article does not contain any studies with human participants or animal performed by any of authors.
About this article

Cite this article
Iwasawa, T., Takemura, T. & Ogura, T. Smoking-related lung abnormalities on computed tomography images: comparison with pathological findings. Jpn J Radiol 36, 165–180 (2018). https://doi.org/10.1007/s11604-017-0713-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11604-017-0713-0