
Abstract
Background
Growing evidence suggests that not only cerebrovascular disease but also Alzheimer’s disease (AD) pathological process itself cause cerebral white matter degeneration, resulting in white matter hyperintensities (WMHs). Some preclinical evidence also indicates that white matter degeneration may precede or affect the development of AD pathology. This study aimed to clarify the direction of influence between in vivo AD pathologies, particularly beta-amyloid (Aβ) and tau deposition, and WMHs through longitudinal approach.
Methods
Total 282 older adults including cognitively normal and cognitively impaired individuals were recruited from the Korean Brain Aging Study for the Early Diagnosis and Prediction of Alzheimer’s Disease (KBASE) cohort. The participants underwent comprehensive clinical and neuropsychological assessment, [11C] Pittsburgh Compound B PET for measuring Aβ deposition, [18F] AV-1451 PET for measuring tau deposition, and MRI scans with fluid-attenuated inversion recovery image for measuring WMH volume. The relationships between Aβ or tau deposition and WMH volume were examined using multiple linear regression analysis. In this analysis, baseline Aβ or tau were used as independent variables, and change of WMH volume over 2 years was used as dependent variable to examine the effect of AD pathology on increase of WMH volume. Additionally, we set baseline WMH volume as independent variable and longitudinal change of Aβ or tau deposition for 2 years as dependent variables to investigate whether WMH volume could precede AD pathologies.
Results
Baseline Aβ deposition, but not tau deposition, had significant positive association with longitudinal change of WMH volume over 2 years. Baseline WMH volume was not related with any of longitudinal change of Aβ or tau deposition for 2 years. We also found a significant interaction effect between baseline Aβ deposition and sex on longitudinal change of WMH volume. Subsequent subgroup analyses showed that high baseline Aβ deposition was associated with increase of WMH volume over 2 years in female, but not in male.
Conclusions
Our findings suggest that Aβ deposition accelerates cerebral WMHs, particularly in female, whereas white matter degeneration appears not influence on longitudinal Aβ increase. The results also did not support any direction of influence between tau deposition and WMHs.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Introduction
Alzheimer’s disease (AD) and cerebrovascular disease (CVD) commonly co-occur [1] and have additive effects on cognitive decline or onset of dementia [2, 3]. White matter hyperintensities (WMHs) on T2-weighted magnetic resonance image (MRI) are frequently found in AD dementia as well as CVD. Since WMHs have been related to demyelination and axonal loss caused by chronic ischemia due to cerebral small vessel disease, WMHs in individuals with AD dementia are commonly considered as a marker of comorbid CVD [4].
However, growing evidence suggests that not only CVD but also AD pathological process itself cause cerebral white matter degeneration, resulting in WMHs. Several postmortem studies demonstrated that both beta-amyloid protein (Aβ) deposition and intraneuronal aggregation of phosphorylated tau protein (p-tau) were associated with white matter degeneration via disorganizing myelin architecture [5,6,7]. A couple of preclinical studies also showed that AD transgenic mice exhibited oligodendrocyte dysfunction and axonal degeneration, suggesting that white matter degeneration may be an important pathophysiological feature of AD [8, 9].
Meanwhile, some preclinical evidence indicated that white matter degeneration may precede or affect the development of AD pathology. An experimental study reported that myelin basic proteins regulated Aβ deposition by binding Aβ and inhibiting formation Aβ fibril and their decline related to the accumulation of Aβ [10]. Other preclinical studies also showed that ischemic axonal injury was associated with increase of tau phosphorylation and neurofibrillary tangle [11, 12].
The relationships between in vivo AD biomarkers and WMHs on MRI were also demonstrated in living human by several cross-sectional studies. Lower cerebrospinal fluid (CSF) Aβ1−42, but not with CSF total tau (t-tau) or p-tau, was associated with higher WMH volume [13, 14]. Similarly, higher Aβ, but not tau, deposition on positron emission tomography (PET) was associated with greater WMH burden [15, 16]. Unlike CSF tau or tau on PET, increased plasma tau concentrations were associated with higher WMH volume [17].
However, limited information is available regarding the direction of causal relationship between AD pathology and WMHs in living human brain. Only a couple of longitudinal studies have investigated a causal relationship between WMH and Aβ using CSF or imaging data, with mixed results in both directions [18,19,20]. Moreover, no longitudinal study investigated the effect of baseline tau deposition on WMH change or the effect of baseline WMHs on tau deposition change yet.
Therefore, this study aimed to investigate the direction of relationship between in vivo AD pathologies, particularly Aβ and tau deposition, and WMHs through a longitudinal approach in older adults with diverse cognitive spectrum including normal cognition, mild cognitive impairment (MCI) and AD dementia.
Methods
Participants
This study is a part of the Korean Brain Aging Study for Early Diagnosis and Prediction of Alzheimer’s Disease (KBASE), which is an ongoing prospective, longitudinal cohort study that began in 2014 [21]. Total 282 older adults aging from 55 to 90 years including cognitively normal (CN), MCI and AD dementia individuals were recruited from the KBASE cohort. CN individuals were defined as individuals without MCI or dementia and with clinical dementia rating (CDR) global score of 0. MCI was defined as having CDR score of 0.5 and meeting core clinical criteria for diagnosis of MCI on the National Institute on Aging and Alzheimer’s Association (NIA-AA) guideline [22]. AD dementia was defined as meeting the criteria for dementia on the Diagnostic and Statistical Manual of Mental Disorders 4th edition (DSM-IV-TR) [23] and the NIA-AA criteria for probable AD [24]. All participants with AD dementia had CDR score of 0.5 or 1. The exclusion criteria were the following: (1) presence of a major psychiatric illness; (2) significant neurological or medical condition or comorbidity that could affect mental functioning; (3) contraindications for an MRI scan (e.g., pacemaker or claustrophobia); (4) illiteracy; (5) presence of significant visual/hearing difficulties and/or severe communication or behavioral problems that would make clinical examinations or brain scans difficult; (6) pregnancy or lactation; and, (7) use of an investigational drug. Additional comprehensive participant details were previously provided [21].
Clinical and neuropsychological assessments
All participants were given standardized clinical assessments by trained board-certificated psychiatrists based on the KBASE clinical assessment protocol which incorporated the Korean version of the Consortium to Establish a Registry for Alzheimer’s Disease Assessment (CERAD-K) clinical assessment [25]. Vascular risk factor scores (VRS) were calculated as the total number of vascular risk factors including hypertension, diabetes mellitus, coronary heart disease, hyperlipidemia, cerebrovascular accident and transient ischemic attacks [26]. All participants were also administered a comprehensive neuropsychological assessment battery by clinical neuropsychologists or trained psychometrists in accordance with a standardized protocol which incorporated the CERAD-K neuropsychological battery [27]. Details on information of clinical and neuropsychological assessments were described previously [21].
Apolipoprotein E ε4 genotyping and coding
Genomic DNA was extracted from whole blood samples and apolipoprotein E genotyping was performed as described previously [28]. Apolipoprotein ε4 (APOE ε4) positivity was coded as positive if there was at least one ε4 allele and coded as negative if there was no ε4 allele.
Measurement of cerebral Aβ deposition
All participants underwent simultaneous three-dimensional (3D) [11C] Pittsburg compound B (PiB)-PET and 3D T1-weighted MRI using the 3.0T Biograph mMR (PET-MR) scanner (Siemens, Washington DC, USA) at both the initial assessment and 2-year follow-up (Fig. 1). The image processing was conducted utilizing SPM8. An automatic anatomic labeling algorithm and a region combining method were applied to identify specific regions of interests (ROIs) [29, 30]. To quantify the extent of cerebral Aβ deposition, the uptake value of [11C] PiB was extracted from ROIs encompassing the frontal, lateral parietal, posterior cingulate-precuneus, and lateral temporal regions. The calculation of the voxel-weighted mean standardized uptake value ratio (SUVR) for these ROIs was achieved by dividing the mean [11C] PiB uptake value of these ROIs by the corresponding mean uptake values of cerebrum white matter, cerebellar white matter, pons and inferior cerebellar gray matter regions [31,32,33]. More detailed information about the methodology used for measuring cerebral Aβ deposition can be found in a prior publication [21].

Datasets for analysis. Dataset for analysis of association between Aβ or Tau deposition and WMH volume. Aβ = Beta-amyloid, WMH = White matter hyperintensity. A = Aβ-PET, M = MRI scan, T = Tau-PET
Measurement of cerebral tau deposition
A subgroup of participants (n = 150) underwent [18F] AV1451-PET scans using Biograph Truepoint 40 PET/CT scanner (Siemens, Washington DC, USA). While all the other neuroimaging scans were performed at baseline and then at 2-year follow-up, [18F] AV-1451 PET imaging was first administered at 2-year follow-up time and then repeated 2 years (i.e., 4-year follow-up time) (Fig. 1). Images were coregistered and resliced into T1-weighted MRI for further processing. The computation of the weighted mean SUVR was carried out in native space, incorporating partial volume correction, and normalized by the mean hemispheric cerebrum white matter uptake. This calculation was employed to compute composite ROIs that combined a set of AD-signature regions including entorhinal, amygdala, parahippocampal, fusiform, inferior temporal, and middle temporal ROIs [34]. Further elaboration on the methodology utilized for quantifying cerebral tau deposition can be found in a previous publication [35].
Measurement of WMHs
Each participant underwent simultaneous 3D fluid-attenuated inversion-recovery (FLAIR) images using the 3.0T Biograph mMR (PET-MR) scanner (Siemens, Washington DC, USA). FLAIR images were obtained at baseline, 2-year follow-up, and 4-year follow-up (Fig. 1). FLAIR images were acquired in sagittal plane, with the following parameter settings: TR, 5000ms; TE, 173ms; echo spacing, 3.46ms; FOV, 250 mm; matrix size, 256 × 256; slice thickness, 1.0 mm. To calculate the volume of WMH on FLAIR images, a previously validated automated approach was employed [36]. The procedure can be briefly summarized as follows: spatial coregistration of T1 and FLAIR images; fusion of T1 and FLAIR images; segmentation of T1; attainment of transformation parameters; deformation and obtainment of the white matter mask; obtainment of FLAIR within the white matter mask; intensity normalization of the masked FLAIR; nomination of candidate WMH with a designated threshold; creation of a junction map; and elimination of the junction. Two modifications were made to adapt for the current data. First, a threshold value of 70, more suitable for our specific data, was applied instead of the original reference value of 65. Second, given the exclusion of participants with acute cerebral infarcts, diffusion-weighted imaging was not integrated into the procedure. WMH candidate images were utilized to extract WMH volumes based on lobar ROIs in the native space for each participant. WMH volumes were log-transformed to reduce skewness (Supplementary Figs. 1 and 2).
Statistical analysis
To investigate the relationship of AD pathology on WMH volume change over 2 years, we tested multiple linear regression model with brain Aβ or tau deposition on initial PET scan as an independent variable and longitudinal change of WMH volume over next 2 years as a dependent variable controlling age, sex, APOE ε4 positivity, VRS, CDR-Sum of Boxes (SOB) score, and baseline WMH volume as covariates. We also analyzed the regression model with baseline WMH volume as an independent variable and longitudinal change of Aβ or tau deposition for 2 years as a dependent variable controlling age, sex, APOE ε4 positivity, VRS, CDR-SOB, and baseline Aβ or tau deposition as covariates. The longitudinal change of Aβ, tau, or WMH volume was calculated by subtracting baseline value from the value at follow-up, which was represented as delta (∆) (Fig. 1). Additionally, we performed exploratory analyses including interaction term as an additional independent variable in the multiple regression model to investigate the moderating effects of age, sex, APOE ε4 positivity, VRS, CDR-SOB, and baseline value of neuroimaging marker on the relationships which showed statistical significance by any analysis described above. When a significant interaction effect was found, subsequent subgroup analyses were performed. All analyses were performed using R Statistical Software (v4.1.2; R Core Team 2021).
Results
Participant characteristics
Table 1 shows the characteristics of the participants. To examine the relationship between baseline Aβ deposition and change of WMH volume over 2 years and vice versa, overall 282 participants were included in the analyses (Fig. 1). For the association between initial tau deposition and change of WMH volume over next 2 years, 117 participants were included in the analysis (Fig. 1). Among them, 66 participants (56.4%), who received the second Tau-PET images at 4-year follow-up, were included in the analysis to investigate the relationship between baseline WMH volume and change of tau deposition over 2 years (Fig. 1).
Longitudinal relationship between cerebral Aβ deposition and WMH volume
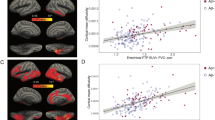
Higher baseline cerebral Aβ deposition was associated greater increase of WMH volume for 2 years (β = 0.238, p = 0.009; Table 2; Fig. 2A). In contrast, there was no significant relationship between baseline WMH volume and change of Aβ deposition over 2 years (β = 0.008, p = 0.502; Table 2; Fig. 2C).

Bidirectional relation between Aβ or Tau deposition and WMH volume. A, Relation between Aβ and change of WMH for 2 years. Partial regression plot consisted of change of WMH for 2 years as dependent variable, and baseline Aβ as independent variable, adjusted for age, sex, APOE ε4 positivity, Vascular risk score, CDR sum of box and baseline WMH. B, Relation between tau and change of WMH for 2 years. Partial regression plot consisted of change of WMH for 2 years as dependent variable, and baseline tau as independent variable, adjusted for age, sex, APOE ε4 positivity, Vascular risk score, CDR sum of box and baseline WMH. C, Relation between WMH and change of Aβ for 2 years. Partial regression plot consisted of change of Aβ for 2 years as dependent variable, and baseline WMH as independent variable, adjusted for age, sex, APOE ε4 positivity, Vascular risk score, CDR sum of box and baseline Aβ. D, Relation between WMH and change of tau for 2 years. Partial regression plot consisted of change of tau for 2 years as dependent variable, and baseline WMH as independent variable, adjusted for age, sex, APOE ε4 positivity, Vascular risk score, CDR sum of box and baseline tau
Longitudinal relationship between cerebral tau deposition and WMH volume
Baseline tau deposition was not related with the 2-year change of WMH volume (β = -0.232, p = 0.128; Table 2; Fig. 2B). Baseline WMH volume was also not associated with change in tau deposition over 2 years (β = 0.054, p = 0.503; Table 2; Fig. 2D).
Moderation on the relationship of cerebral Aβ deposition with WMH volume change
We explored the moderating effects of age, sex, APOE ε4 positivity, VRS, CDR-SOB, or baseline WMH volume on the relationship between baseline Aβ deposition and change of WMH volume over 2 years which showed statistical significance. There was a significant baseline Aβ deposition × sex interaction effect on longitudinal change of WMH volume (β = -0.350, p = 0.011; Table 3; Fig. 3A). Subsequent subgroup analyses showed that higher baseline Aβ deposition was associated with greater increase of WMH volume over 2 years in female, but not in male (female: β = 0.427, p = 0.001; male: β = -0.049, p = 0.696; Fig. 3). In contrast, we did not find any significant interaction between Aβ and any other variables mentioned above (Table 3). While there was no significant association between baseline tau and WMH volume change, we further analyzed the moderating effects of age, sex, APOE ε4 positivity, VRS, CDR-SOB, or baseline WMH volume on the relationship between baseline tau deposition and change of WMH volume for comparison purposes. However, we did not find any significant moderating effects (Supplementary Table 2).

Moderating effect of sex on association between Aβ and WMH volume. A, Effect of sex on association between baseline Aβ and change of WMH volume for 2 years. Partial regression plot adjusted for age, APOE ε4 positivity, Vascular risk score, CDR sum of box and baseline WMH. Values in a box indicate coefficient and p-value of interaction term between baseline Aβ and sex. B, Relation between Aβ and change of WMH for 2 years in male. Partial regression plot consisted of change of WMH for 2 years as dependent variable, and baseline Aβ as independent variable, adjusted for age, APOE ε4 positivity, Vascular risk score, CDR sum of box and baseline WMH. C, Relation between Aβ and change of WMH for 2 years in female. Partial regression plot consisted of change of WMH for 2 years as dependent variable, and baseline Aβ as independent variable, adjusted for age, APOE ε4 positivity, Vascular risk score, CDR sum of box and baseline WMH.
Discussion
In the present study, we found that higher baseline cerebral Aβ deposition, but not tau deposition, was associated with greater increase of WMH volume over 2 years in older adults with diverse cognitive spectrum including normal cognition, MCI and AD dementia. In contrast, WMH volume at baseline did not show any relationship with the change of brain Aβ or tau deposition over 2 years.
Regarding the longitudinal relationship between Aβ and WMH volume, our findings suggest that baseline cerebral Aβ deposition may affect increase of WMH volume, whereas baseline WMH volume may not facilitate or precede the change of Aβ deposition. These observations were similar to a previous report which showed that lower baseline CSF Aβ1−42 was associated with increase of WMH volume over one year, while baseline WMH volume was not associated with change of CSF Aβ1−42 in individuals including CN, MCI and AD dementia [18]. In contrast, a study based on individuals with normal cognition reported that there was no relationship between baseline CSF Aβ1−42 and change of WMH volume [20]. Individuals with normal cognition, who possibly have a relatively low Aβ accumulation and small WMH volume change, might make it difficult to detect the relationship of Aβ with the increase of WMH volume. Additionally, in contrast to our result, another study which focused only on the influence of WMHs on Aβ change demonstrated that baseline WMH volume was significantly related with increase of cerebral Aβ deposition [19]. Although the study was conducted for cognitively unimpaired participants, they were followed up for relatively longer period (mean: 4.4 years; range from 1 to 8 years) compared to our study, which might have it possible to detect the relationship.
In regard of longitudinal tau-WMH relationship, we found no significant results for any direction of relationship, which is generally in line with previous reports based on cross-sectional analysis [13, 15]. In contrast, some diffusion tensor imaging (DTI) studies demonstrated a significant association between higher brain tau deposition and decreased white matter microstructural integrity as measured by fractional anisotropy (FA) or mean diffusivity (MD) [37,38,39]. Given these results, cerebral tau deposition may cause subtle or microstructural white matter damages that can be detected by FA or MD on DTI, but not by WMHs on T2 weighted MRI.
To date, the mechanism underlying the effect of Aβ on white matter injury (WMI) expressed as WMHs has not been clearly identified. Some evidence indicates that Aβ-induced death of oligodendrocytes and neuroinflammation may be primary mechanisms accounting for the effect of Aβ on white matter degeneration, demyelination in particular. Oligodendrocytes play an essential role in repairing impaired myelin sheath by remyelination. Cytotoxic effect of increasing Aβ can induce the death of oligodendrocytes, resulting in remyelination failure of damaged axons [40]. Increase of Aβ can also promote neuroinflammatory environment by fostering release of cytokine from astrocytes [41]. Neuroinflammation induced by Aβ can directly cause demyelination [42] and indirectly contribute to demyelination by interfering with an efficient remyelination of oligodendrocytes [43].
Additional exploratory analyses showed that the relationship between Aβ deposition and increase of WMH volume was significant only in female, but not in male. Such female predominant relationship may relate to drastic decrease in estrogen and progesterone during menopause and perimenopause [44]. The estrogen and progestogen can affect the production and activity of immune cells, and thus can be neuroprotective against AD and CVD pathogenesis [45, 46]. Weakened immune system resulted from decrease of these hormones can induce more vulnerable environment in which the toxicity of Aβ have a greater impact on brain in female. Some previous studies reported that effects of Aβ on neurodegeneration or cognitive decline were significant only in female, not male [47, 48]. A recent study also reported that WMH volume increases more rapidly in postmenopausal women compared to premenopausal women and men of the same age range [49].
The current study is unique in that it specifically investigated the directionality of the relationship between AD pathology and WMHs through longitudinal approaches. Nevertheless, it still had some limitations. First, sample size for the analysis of the relationship between cerebral tau deposition and WMH volume was relatively small, which might contribute to the non-significant results by reducing statistical power. Second, the 2-year follow-up period for longitudinal analysis also might not be sufficient to confirm some relationship between AD biomarkers and WMHs; for example, the relationship of baseline WMH volume with the change of brain Aβ or tau over 2 years. Further studies with a longer follow-up period are needed.
Conclusions
The present findings in older adults with diverse cognitive spectrum suggested that baseline Aβ deposition contribute to the increase of WMHs over 2 years, especially in female, whereas WMIs appear not influence on longitudinal Aβ increase. The results also did not support any direction of influence between tau deposition and WMHs.
Data availability
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AD:
-
Alzheimer’s disease
- CVD:
-
Cerebrovascular disease
- WMH:
-
White matter hyperintensity
- WMI:
-
White matter injury
- Aβ:
-
Beta-amyloid protein
- p-tau:
-
Phosphorylated tau protein
- t-tau:
-
Total tau protein
- CSF:
-
Cerebrospinal fluid
- MRI:
-
Magnetic resonance image
- PET:
-
Positron emission tomography
- FLAIR:
-
Fluid-attenuated inversion-recovery
- DTI:
-
Diffusion tensor imaging
- FA:
-
Fractional anisotropy
- MD:
-
Mean diffusivity
- SUVR:
-
Standardized uptake value ratio
- ROI:
-
Regions of interest
- CN:
-
Cognitively normal
- MCI:
-
Mild cognitive impairment
- CDR:
-
Clinical dementia rating
- SOB:
-
Sum of Boxes
- VRS:
-
Vascular risk factor scores
- APOE ε4:
-
Apolipoprotein ε4
- KBASE:
-
Korean Brain Aging Study for Early Diagnosis and Prediction of Alzheimer’s Disease
- NIA-AA:
-
National Institute on Aging and Alzheimer’s Association
- DSM-IV-TR:
-
Diagnostic and Statistical Manual of Mental Disorders 4th edition
- CERAD-K:
-
Korean version of the Consortium to Establish a Registry for Alzheimer’s Disease Assessment
References
Love S, Miners JS. Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathol. 2016;131(5):645–58.
Koncz R, Sachdev PS. Are the brain’s vascular and Alzheimer pathologies additive or interactive? Curr Opin Psychiatry. 2018;31(2):147–52.
Roseborough A, Ramirez J, Black SE, Edwards JD. Associations between amyloid beta and white matter hyperintensities: a systematic review. Alzheimers Dement. 2017;13(10):1154–67.
Prins ND, Scheltens P. White matter hyperintensities, cognitive impairment and dementia: an update. Nat Rev Neurol. 2015;11(3):157–65.
McAleese KE, Firbank M, Dey M, Colloby SJ, Walker L, Johnson M, et al. Cortical tau load is associated with white matter hyperintensities. Acta Neuropathol Commun. 2015;3:60.
McAleese KE, Walker L, Graham S, Moya ELJ, Johnson M, Erskine D, et al. Parietal white matter lesions in Alzheimer’s disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol. 2017;134(3):459–73.
Bulk M, Abdelmoula WM, Nabuurs RJ, van der Graaf LM, Mulders CW, Mulder AA, et al. Postmortem MRI and histology demonstrate differential iron accumulation and cortical myelin organization in early-and late-onset Alzheimer’s disease. Neurobiol Aging. 2018;62:231–42.
Dawson HN, Cantillana V, Jansen M, Wang H, Vitek M, Wilcock D, et al. Loss of tau elicits axonal degeneration in a mouse model of Alzheimer’s disease. Neuroscience. 2010;169(1):516–31.
Desai MK, Guercio BJ, Narrow WC, Bowers WJ. An Alzheimer’s disease-relevant presenilin-1 mutation augments amyloid-beta-induced oligodendrocyte dysfunction. Glia. 2011;59(4):627–40.
Liao M-C, Ahmed M, Smith SO, Van Nostrand WE. Degradation of amyloid β protein by purified myelin basic protein. J Biol Chem. 2009;284(42):28917–25.
Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME, Bowers WJ. Triple-transgenic Alzheimer’s disease mice exhibit region‐specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia. 2009;57(1):54–65.
Hayden EY, Putman J, Nunez S, Shin WS, Oberoi M, Charreton M, et al. Ischemic axonal injury up-regulates MARK4 in cortical neurons and primes tau phosphorylation and aggregation. Acta Neuropathol Commun. 2019;7:1–12.
Walsh P, Sudre CH, Fiford CM, Ryan NS, Lashley T, Frost C, et al. CSF amyloid is a consistent predictor of white matter hyperintensities across the disease course from aging to Alzheimer’s disease. Neurobiol Aging. 2020;91:5–14.
Weaver NA, Doeven T, Barkhof F, Biesbroek JM, Groeneveld ON, Kuijf HJ, et al. Cerebral amyloid burden is associated with white matter hyperintensity location in specific posterior white matter regions. Neurobiol Aging. 2019;84:225–34.
Graff-Radford J, Arenaza-Urquijo EM, Knopman DS, Schwarz CG, Brown RD, Rabinstein AA, et al. White matter hyperintensities: relationship to amyloid and tau burden. Brain. 2019;142(8):2483–91.
Garnier-Crussard A, Bougacha S, Wirth M, Dautricourt S, Sherif S, Landeau B, et al. White matter hyperintensity topography in Alzheimer’s disease and links to cognition. Alzheimer’s Dement. 2022;18(3):422–33.
Laing KK, Simoes S, Baena-Caldas GP, Lao PJ, Kothiya M, Igwe KC, et al. Cerebrovascular disease promotes tau pathology in Alzheimer’s disease. Brain Commun. 2020;2(2):fcaa132.
Dadar M, Camicioli R, Duchesne S, Collins DL. Alzheimer’s Disease Neuroimaging I. The temporal relationships between white matter hyperintensities, neurodegeneration, amyloid beta, and cognition. Alzheimers Dement (Amst). 2020;12(1):e12091.
Moscoso A, Rey-Bretal D, Silva-Rodríguez J, Aldrey JM, Cortés J, Pías-Peleteiro J, et al. White matter hyperintensities are associated with subthreshold amyloid accumulation. NeuroImage. 2020;218:116944.
Soldan A, Pettigrew C, Zhu Y, Wang MC, Moghekar A, Gottesman RF, et al. White matter hyperintensities and CSF Alzheimer disease biomarkers in preclinical Alzheimer disease. Neurology. 2020;94(9):e950–e60.
Byun MS, Yi D, Lee JH, Choe YM, Sohn BK, Lee JY, et al. Korean brain aging study for the early diagnosis and prediction of Alzheimer’s Disease: methodology and baseline sample characteristics. Psychiatry Investig. 2017;14(6):851–63.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011;7(3):270–9.
Association AP. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR). Washington, DC: American Psychiatric Association; 2000.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–9.
Lee JH, Lee KU, Lee DY, Kim KW, Jhoo JH, Kim JH, et al. Development of the Korean Version of the Consortium to establish a Registry for Alzheimer’s Disease Assessment Packet (CERAD-K) clinical and neuropsychological assessment batteries. Journals Gerontol Ser B: Psychol Sci Social Sci. 2002;57(1):P47–P53.
DeCarli C, Mungas D, Harvey D, Reed B, Weiner M, Chui H, et al. Memory impairment, but not cerebrovascular disease, predicts progression of MCI to dementia. Neurology. 2004;63(2):220–7.
Lee DY, Lee KU, Lee JH, Kim KW, Jhoo JH, Kim SY, et al. A normative study of the CERAD neuropsychological assessment battery in the Korean elderly. J Int Neuropsychol Soc. 2004;10(1):72–81.
Wenham P, Price W, Blundell G. Apolipoprotein E genotyping by one-stage PCR. Lancet. 1991;337(8750):1158–9.
Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W et al. Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proceedings of the National Academy of Sciences. 2009;106(16):6820-5.
Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage. 2002;15(1):273–89.
Lowe VJ, Lundt ES, Senjem ML, Schwarz CG, Min H-K, Przybelski SA, et al. White matter reference region in PET studies of 11 C-Pittsburgh compound B uptake: effects of age and amyloid-β deposition. J Nucl Med. 2018;59(10):1583–9.
Schwarz CG, Senjem ML, Gunter JL, Tosakulwong N, Weigand SD, Kemp BJ, et al. Optimizing PiB-PET SUVR change-over-time measurement by a large-scale analysis of longitudinal reliability, plausibility, separability, and correlation with MMSE. NeuroImage. 2017;144:113–27.
Van Baarsen K, Kleinnijenhuis M, Jbabdi S, Sotiropoulos SN, van Grotenhuis J. Walsum AvC. A probabilistic atlas of the cerebellar white matter. NeuroImage. 2016;124:724–32.
Jack CR Jr, Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimer’s Dement. 2017;13(3):205–16.
Park J-C, Han S-H, Yi D, Byun MS, Lee JH, Jang S, et al. Plasma tau/amyloid-β1–42 ratio predicts brain tau deposition and neurodegeneration in Alzheimer’s disease. Brain. 2019;142(3):771–86.
Tsai JZ, Peng SJ, Chen YW, Wang KW, Li CH, Wang JY, et al. Automated segmentation and quantification of white matter hyperintensities in acute ischemic stroke patients with cerebral infarction. PLoS ONE. 2014;9(8):e104011.
Kantarci K, Murray ME, Schwarz CG, Reid RI, Przybelski SA, Lesnick T, et al. White-Matter integrity on DTI and the pathologic staging of Alzheimer’s disease. Neurobiol Aging. 2017;56:172–9.
Strain JF, Smith RX, Beaumont H, Roe CM, Gordon BA, Mishra S, et al. Loss of white matter integrity reflects tau accumulation in Alzheimer disease defined regions. Neurology. 2018;91(4):e313–e8.
Strain JF, Barthelemy N, Horie K, Gordon BA, Kilgore C, Aschenbrenner A, et al. CSF tau phosphorylation at Thr205 is associated with loss of white matter integrity in autosomal dominant Alzheimer disease. Neurobiol Dis. 2022;168:105714.
Sachdev PS, Zhuang L, Braidy N, Wen W. Is Alzheimer’s a disease of the white matter? Curr Opin Psychiatry. 2013;26(3):244–51.
Maezawa I, Zimin PI, Wulff H, Jin LW. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem. 2011;286(5):3693–706.
di Penta A, Moreno B, Reix S, Fernandez-Diez B, Villanueva M, Errea O, et al. Oxidative stress and proinflammatory cytokines contribute to demyelination and axonal damage in a cerebellar culture model of neuroinflammation. PLoS ONE. 2013;8(2):e54722.
Pietroboni AM, Colombi A, Carandini T, Scarpini E, Galimberti D, Bozzali M. The role of amyloid-beta in White Matter damage: possible common pathogenetic mechanisms in neurodegenerative and demyelinating diseases. J Alzheimers Dis. 2020;78(1):13–22.
Steiner M, Dunn E, Born L. Hormones and mood: from menarche to menopause and beyond. J Affect Disord. 2003;74(1):67–83.
Colafella KMM, Denton KM. Sex-specific differences in hypertension and associated cardiovascular disease. Nat Rev Nephrol. 2018;14(3):185–201.
Pike CJ, Carroll JC, Rosario ER, Barron AM. Protective actions of sex steroid hormones in Alzheimer’s disease. Front Neuroendocrinol. 2009;30(2):239–58.
Keuss SE, Coath W, Nicholas JM, Poole T, Barnes J, Cash DM, et al. Associations of beta-amyloid and vascular Burden with Rates of Neurodegeneration in cognitively normal members of the 1946 British birth cohort. Neurology. 2022;99(2):e129–e41.
Liu C, Li Y, Nwosu A, Ang TFA, Liu Y, Devine S, et al. Sex-specific biomarkers in Alzheimer’s disease progression: Framingham Heart Study. Alzheimers Dement (Amst). 2022;14(1):e12369.
Lohner V, Pehlivan G, Sanroma G, Miloschewski A, Schirmer MD, Stocker T, et al. Relation between sex, menopause, and White Matter hyperintensities: the Rhineland Study. Neurology. 2022;99(9):e935–e43.
Acknowledgements
We thank all the members of the KBASE Research Group for their contribution. The investigators within the KBASE Research Group contributed to the design and implementation of KBASE and/or provided data but did not participate in analysis or writing of this report. The list of members of the KBASE Research Group can be found at http://kbase.kr/ and are provided as supplementary material. We thank AVID Radiopharmaceuticals for providing the precursor of [18F] AV-1451.
Funding
This study was supported by grants from the Ministry of Science, ICT, and Future Planning, Republic of Korea (Grant No: NRF-2014M3C7A1046042 & NRF-2020R1G1A1099652) and grants of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant No: HI18C0630 & HI19C0149), and a grant from the National Institute of Aging, United States of America (U01AG072177).
Author information
Authors and Affiliations
Contributions
WJC and DYL designed and conceptualized the study.WJC, DY, HA, MSB, YYC, JMC, KK, HC, GJ, KMK, CHS, YSL, YKK, and DYL acquired, analyzed and interpreted the data.WJC, DY, and DYL drafted and revised the manuscript for intellectual content.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study protocol was approved by the institutional review boards of the Seoul National University Hospital (C-1401-027-547) and the Seoul Metropolitan Government-Seoul National University (SMG-SNU) Boramae Medical Center (26-2015-60), in Seoul, South Korea; and we conducted it in accordance with the recommendations of the current version of the Declaration of Helsinki. Written informed consent was obtained from all participants involved in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Consortia
For the KBASE Research Group.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cha, WJ., Yi, D., Ahn, H. et al. Association between brain amyloid deposition and longitudinal changes of white matter hyperintensities. Alz Res Therapy 16, 50 (2024). https://doi.org/10.1186/s13195-024-01417-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-024-01417-8