
Abstract
Background
Alzheimer's disease (AD) is frequently accompanied by sleep impairment, which can induce AD-related neurodegeneration. We herein investigated the sleep architecture, cognition, and cerebrospinal fluid (CSF) biomarkers (tau proteins and β-amyloid42) during AD progression from subjective cognitive impairment (SCI) to mild cognitive impairment (MCI) and eventually to AD dementia, and compared the results with cognitively normal (CN) subjects.
Methods
We included patients affected by SCI, MCI, mild AD, and moderate-to-severe AD in our study along with CN subjects as controls. All the subjects underwent nocturnal polysomnography to investigate sleep, neuropsychological testing to evaluate cognition, and lumbar puncture for CSF AD biomarkers assessment.
Results
Sleep (both rapid eye movement (REM) and non-REM sleep) and memory function are both progressively impaired during the course of AD from SCI to mild and subsequently to moderate AD. Further, sleep dysregulation appears earlier than cognitive deterioration, with a reduction of CSF β-amyloid42 level.
Conclusion
Sleep, memory, and CSF AD biomarkers are closely interrelated in AD progression from the earliest asymptomatic and preclinical stages of the disease related in AD since the earliest and preclinical stages of the disease.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive memory loss and a decline in global cognition [1]. A progressive alteration of cognition can be observed in the process of developing AD starting with subjective cognitive impairment (SCI) and progressing to mild cognitive impairment (MCI), which may likely represent the stage preceding AD dementia [2]. Neuropathologically, AD is characterized by progressive accumulation of both extracellular β-amyloid plaques and intracellular neurofibrillary tangles (NFT) of tau proteins, which result in a dramatic loss of neurons and synapses that affect the structural and physiological processes of the brain. Currently, β-amyloid42 (Aβ42), total tau (t-tau), and phosphorylated-tau (p-tau) proteins are the established cerebrospinal fluid (CSF) biomarkers that support the diagnosis of AD [3]. The CSF biomarkers of AD, Aβ42 in particular, begin to accumulate pathologically in the brain several years before the onset of cognitive deterioration wherein the subjects appear cognitively normal (CN) [4]. In this preclinical stage, however, behavioral modifications such as depression, anxiety, and sleep impairment can appear [5,6,7]. Accordingly, sleep dysregulation with a reduction of REM and slow-wave sleep may be observed in CN subjects that exhibit biomarkers of AD neurodegeneration [8,9,10]. Conversely, sleep disorders such as insomnia and obstructive sleep apnea may induce preclinical modifications of AD biomarkers and consequently trigger neurodegeneration by negatively modulating sleep and reducing its beneficial effects on brain health [11,12,13]. The reduction of sleep quality and an increase in nighttime wakefulness have been hypothesized to cause β-amyloid brain deposits from the early stages of AD [14,15,16]. Therefore, a bidirectional association seems to exist between sleep dysregulation and AD pathology with both contributing to the progressive alteration of cognitive processes and behavior.
The aim of this study was to investigate nocturnal sleep architecture changes, AD biomarkers in the CSF, and memory impairment across different levels of neurocognitive functioning and impairment ranging from the CN condition to SCI, MCI, and AD dementia.
Methods
Patients and study design
We included drug-naïve patients who were consecutively referred to the Neurological Clinic of the University Hospital of Rome “Tor Vergata” between January 2012 and July 2016. Inclusion criterion was patients with a diagnosis of SCI, MCI, or AD according to the recently proposed version of the diagnostic guidelines [1, 2, 17]. All patients underwent a diagnostic and experimental study protocol including history, physical and neurological examination, laboratory tests, standard neuropsychological evaluation, electroencephalogram (EEG), polysomnography (PSG), brain MRI, and lumbar puncture (LP) for CSF analysis. This protocol has been published in previous articles by our research group [11, 18,19,20].
Specifically, patients were considered affected by SCI if they reported subjective memory deficits reflecting a decline over the past 5 or 10 years, an absence of overt cognitive deficits, and cognitive performance in the normal range [2, 21,22,23]. The criteria used to define MCI due to AD included the following: cognitive concerns, objective evidence of cognitive impairment, normal functional activities, absence of dementia, and presence of AD biomarkers [17]. The diagnosis of AD was performed according to the recently proposed version of the diagnostic guidelines [1]. The biomarkers were considered as positive for AD when decreased CSF levels of Aβ42 were observed along with the following abnormalities: medial temporal lobe atrophy on MRI, cortical temporoparietal hypometabolism on 18F-fluorodeoxyglucose positron emission tomography ([18] FDG PET), and increased CSF levels of t-tau or p-tau [1]. Finally, we divided AD patients into the following two subgroups on the basis of their Mini-Mental State Examination (MMSE) profile: mild AD (mAD, MMSE ≥ 21) and moderate-severe AD (msAD, MMSE < 21) [18].
We enrolled a control group comprising CN subjects who underwent PSG, neuropsychological testing, brain MRI, and LP for diagnostic purposes (to rule out peripheral nerve diseases and/or chronic migraine).
Patients and controls were also required to fulfill the following entry criteria: no additional neurological or psychiatric disease; no intake of CNS active drugs; no use of caffeine, tobacco, and/or alcohol at the time of the sleep laboratory investigation. Contrarily, exclusion criteria for both patients and controls included the following: neoplastic or thyroid illness, diagnosis of conditions interfering with sleep quality such as symptomatic obstructive pulmonary disease, uncontrolled seizures, and abnormal cell count (> 4 cells/mcL) in the CSF sample.
Patients and controls provided their informed consent to the study, which was approved by the Independent Ethical Committee of the University Hospital of Rome “Tor Vergata.” Any anonymized data not published within the article will be shared on request from any qualified investigator.
Polysomnography
Patients and controls underwent two consecutive video-PSGs in order to evaluate nocturnal sleep (Somnomedics, Somnoscreen, SOMNOmedics GmbH-Randersacker, Germany). The signal was stored on a flash card using a common average reference and a time constant of 0.3 s. Electrodes were positioned according to the 10–20 International System. The montage consisted of two electrooculographic channels, three electromyographic channels (mentalis and anterior tibialis muscles), and eight EEG channels (F4, C4, O2, A2, F3, C3, O1, A1). Cardiorespiratory parameters were assessed by recording oronasal flow, thoracic and abdominal movements (plethysmography), pulse oximetry, and electrocardiogram. Patients and their caregivers were also instructed to maintain the usual sleep schedule and record it in a sleep diary during the week preceding the evaluation. The first-night sleep was considered as an adaptation period. Sleep analysis was performed according to the standard criteria on the second night of PSG monitoring [24]. The following standard parameters were computed: time in bed (TIB; time spent in bed between lights off and lights on), sleep onset latency (SL; the time-interval between the lights off and the first sleep epoch), total sleep time (TST; the actual sleep time without SL and awakenings), sleep efficiency (SE; the ratio between TST and TIB), REM sleep latency (LREM; the time-interval between the sleep onset and the first epoch of REM), stage 1 of non-REM sleep (N1), stage 2 of non-REM sleep (N2), stage 3 of non-REM sleep (N3), REM sleep (REM), and wakefulness after sleep onset (WASO). The percentages of the sleep stages were calculated over TST. Blinded researchers (CL, FP, FI) scored the PSG recordings on the basis of the international standard criteria of the American Academy of Sleep Medicine [24, 25]. PSG scorers also identified apnea/hypopnea events and scored leg movements based on the AASM international standard criteria [24]. Patients with Apnea-Hypopnea Index (AHI) > 15/h and/or periodic leg movement index (PLMI) > 15/h during the polysomnographic recording were ruled out.
CSF collection and analysis
All the CSF samples were obtained on the day after the second PSG recording by means of LP performed in the decubitus position using an atraumatic needle; the samples were collected between 8:00 AM and 9:30 AM (within 1–2 h after morning awakening) in polypropylene tubes using standard sterile techniques. The first 4 ml CSF sample was used for routine biochemistry analysis including total cell count and lactate levels. The second 4 ml CSF sample was centrifuged to eliminate cells and cellular debris and immediately frozen at − 80 °C to analyze t-tau, p-tau, and Aβ42 levels subsequently. CSF biomarkers’ levels were determined according to the previously published standard procedures using commercially available sandwich enzyme-linked immunosorbent assays (ELISA) (Innotest β-Amyloid 1–42, Innotest h-T-tau, Innotest Phospho-T-tau 181, Innogenetics, Ghent, Belgium) [26]. Aβ42, t-tau, and p-tau were classified based on previously established cutoff values: < 500 pg/mL for Aβ42, > 375 pg/mL for t-tau, and > 52 pg/mL for p-tau [27,28,29].
Neuropsychological assessments
The Mini-Mental State Examination (MMSE) was used to screen all participants within 48 h of PSG examination. Administration of the neuropsychological test battery required approximately 20 min, and scores were corrected for age and education level as described in a previous study [30]. Since memory is the most frequently altered domain in AD, we specifically evaluated short- and long-term memory using the Rey Auditory Verbal Learning Test (RAVLT). RAVLT consists of a list of 15 words that are read out to the subject five times. Measures include immediate recall (the sum of the words recalled in the five trials, RAVLT-I) and a 15-min delayed recall (the number of words recalled 15 min after the last word presentation, RAVLT-D).
Statistical analysis
Demographic and clinical data of patients and controls were reported as frequency (N), percentage (%), mean, standard deviation (SD), and extreme values (minimum and maximum).
A one-way ANOVA was performed to compare sample characteristics according to pathologic or normal cognition profile/diagnosis. Moreover, if the ANOVA was significant, we carried out a post hoc analysis by applying Bonferroni’s correction to identify which groups were different from the others.
Correlation analysis (Pearson correlation coefficients) was conducted to study the strength of the relationship between variables. In particular, we used Cohen’s guidelines (r < |.10| negligible association, |.10| < r < |.30| weak, |.30| < r < |.50| moderate, r > |.50| strong) to interpret the effect size of correlations [31]. The significance level (p value) was also determined: any p value less than 0.05 would indicate that the result is not due to chance.
Data were submitted to principal component analysis (PCA), which was performed on variables chosen as a function of their proven or hypothetical relationship with cognitive profile and degeneration. PCA, a variant of factor analysis, is a data-driven analysis, and the output is a set of “components” with each of them explaining a part of the data variability. The ultimate purpose of PCA is to name and interpret components in physiological or clinical terms. The solution obtained by PCA has a simple format to describe the internal structure of the data set, which explained 62.5% of the total variance in our case [32, 33]. Estimation of the number of components accepted in the PCA solution was based on both Kaiser’s rule, which retains factors with eigenvalues > 1 and Cattell’s scree test, which retrieves the components corresponding to the last eigenvalue before they start to level off. Kaiser–Meyer–Olkin (KMO) Measure of Sampling Adequacy and Bartlett’s Test of Sphericity were used, before the extraction of the component, to assess the suitability of respondent data for PCA. The KMO index ranges from zero to one, with 0.50 considered suitable for factor analysis [34]. Bartlett’s Test of Sphericity should be significant (p < 0.05) for factor analysis to be relevant [35]. For the final solution, factors were constrained to remain uncorrelated (i.e., independent), and the solution structure was simplified by using orthogonal axis rotation with the varimax method. Measures were assigned to the component on which they showed the highest loading. We consider loadings > 0.6 to be very high, and loadings < 0.3 to be irrelevant. List-wise deletion was used to handle missing data.
All analyses were performed with Statistical Package for Social Sciences (SPSS) version, 2018. A p value less than 0.05 was considered statistically significant.
Results
Sample description and demographic data (one-way ANOVA analysis)
Descriptive information (mean, standard deviation, maximal, and minimal values) of the population, which was divided into groups (CN, SCI, MCI, mAD, and msAD) are summarized in Table 1. Altogether, 258 subjects were recruited for this study between January 2012 and July 2016. The whole population was distributed among five groups: two groups of AD patients (56 patients with mild AD and 48 patients with moderate-severe AD), one group of 59 MCI subjects, one group of 54 SCI subjects, and one group of 41 CN subjects.
One-way ANOVA analysis
Results from one-way ANOVA, which was performed to analyze and compare the included variables from the five groups of subjects, are presented in Table 1. With the exception of the SL variable (F = 1.07, p = 0.372), there were statistically significant differences between the groups, as determined by one-way ANOVA, for all the included variables.
To confirm whether differences occurred between groups, a post hoc analysis with Bonferroni’s correction was conducted, and the results are shown in Table 1.
CSF data (one-way ANOVA analysis)
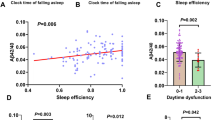
With reference to CSF biomarkers, we documented a significant reduction of CSF Aβ42 levels in MCI, mAD, and msAD patients compared to SCI and CN subjects (Fig. 1). Moreover, SCI patients presented significantly lower CSF Aβ42 levels than the CN subjects did (Table 1). CSF t-tau and p-tau levels did not differ between SCI and CN, but it was lower in these two groups than in MCI, mAD, and msAD groups (Table 1, Fig. 1).

Graphical representation of ANOVA among SCI, MCI, mAD, msAD, and control groups. Box and whisker plots show the median (lines), 25th to 75th percentiles (boxes), and full spread (whiskers) of all the variables. a CSF Aβ42 levels. b CSF t-tau levels. c p-tau levels
PSG data (one-way ANOVA analysis)
Analysis of all the PSG variables included in this study was also performed (Table 1). Regarding TIB, we documented a significantly higher TIB in mAD, SCI, and MCI patients compared to msAD. Considering each PSG variable, we observed a significant reduction of both REM sleep and SE in SCI patients compared to CN subjects without any other significant difference in the remaining sleep macrostructural parameters (Fig. 2). TST was lower in CN, SCI, and MCI compared to mAD patients who in turn showed a lower TST than msAD patients. SE was higher in CN subjects than in SCI patients and then became progressively lower in MCI, mAD, and msAD patients. The mAD patients showed the highest LREM compared to SCI and CN subjects. N1 was higher in mAD and msAD patients than in MCI patients who in turn showed a higher N1 compared to SCI and CN subjects (Fig. 2). N3 was lower in msAD compared to mAD patients who themselves showed lower N3 than the MCI and SCI patients, and CN subjects (Fig. 2). REM sleep was significantly reduced in both mAD and msAD compared to MCI patients; moreover, MCI patients showed a significantly lower REM sleep compared to SCI patients who themselves showed reduced REM sleep compared to CN subjects. Finally, WASO was higher in all the pathological groups (SCI, MCI, mAD, and msAD) compared to the CN group.

Graphical representation of ANOVA among SCI, MCI, mAD, msAD, and control groups. Box and whisker plots show the median (lines), 25th to 75th percentiles (boxes), and full spread (whiskers) of all the variables. a REM sleep. b Stage 1 of non-REM sleep (N1). c Stage 3 of non-REM sleep (N3)
Neuropsychological data (one-way ANOVA analysis)
As expected, a significant reduction of MMSE scores was observed in MCI, mAD, and msAD patients compared to both SCI and CN subjects; moreover, MCI patients presented significantly higher MMSE scores than mAD and msAD patients, and the mAD group presented significantly higher MMSE scores compared to the msAD group (Table 1).
Concerning the tests investigating memory, a significant progressive reduction of RAVLT-I scores was observed in MCI, mAD, and msAD patients compared to SCI and CN subjects (Fig. 3). Considering RAVLT-D scores, the comparison between mAD and msAD groups did not show significant differences, but RAVLT-D scores of mAD and msAD patients were lower than MCI patients, who in turn showed lower scores than SCI and CN subjects (Fig. 3). Notably, SCI patients did not show pathological scores in RAVLT-D and RAVLT-I tests (Table 1).

Graphical representation of ANOVA among SCI, MCI, mAD, msAD, and control groups. Box and whisker plots show the median (lines), 25th to 75th percentiles (boxes), and full spread (whiskers) of all the variables. a Rey Auditory Verbal Learning Test (RAVLT) – Immediate Recall (RAVLT-I). b RAVLT – Delayed Recall (RAVLT-D)
Correlation analysis
The correlation matrix shown in Table 2 contains the Pearson correlation coefficients (and the significance level) between the variables, which denotes the strength of the relationship between the variables. Overall, moderate to strong correlations (r ≥ 0.50) were found between CSF biomarkers and both neuropsychological and polysomnography data (Table 2).
Principal component analysis
PCA was performed on the following: age, scores of MMSE, RAVLT-I, RAVLT-D, CSF Aβ42 level, t-tau and p-tau levels, and a standard set of polysomnography parameters including SL, TST, LREM, N1, N3, REM, and WASO. Some variables were not considered including gender, which was not significantly associated with any variable, N2 because it presented split loadings on all factors, and SE and TIB as they were computed from other polysomnographic parameters. Was excluded as it presented split loadings on all factors, and SE and TIB were excluded as they were computed from other polysomnographic parameters. A three-component solution that explained 62.5% of the total variance was identified by performing PCA with varimax rotation. The KMO measure of sampling adequacy met the “meritorious” criterion (KMO = 0.86) to perform the principal component analysis. Moreover, for these data, Bartlett’s test of Sphericity was highly significant (p < 0.001).
The contribution of the three factors to sample variance was 53.5%, 18.1%, and 11.9%.
Factor solution after varimax rotation is presented in Table 3. Overall, with the exception of a few cross-loading variables, each factor defines a distinct cluster of interrelated variables.
Contribution to component 1 mainly derived from RAVLT-I (0.78), N1 (− 0.76), MMSE (0.74), REM (0.73), N3 (0.73), RAVLT-D (0.71), Aβ42 (0.67), and TST (0.60). Notably, N1 load on the first component was of the opposite sign in comparison to other variables thereby suggesting an inverse correlation among them. Component 2 explains the association between CSF p-tau (0.82), t-tau (0.82), and age (0.55). Contribution to component 3 mainly derived from REML (0.66), WASO (0.64), and SL (0.59).
Discussion
Different lines of evidence suggested that AD neurodegeneration starts several years before the appearance of clinical symptoms, which are not exclusively cognitive but also behavioral (depression, anxiety, sleep fragmentation) [36]. Conversely, insomnia and sleep-disordered breathing (SDB) that frequently affect the elderly have recently been associated with an increased risk of developing AD. This association between sleep disorders and AD has been supposed based on the evidence that sleep dysregulation can induce pathological changes in β-amyloid and tau metabolism in the brain [11,12,13,14,15,16]. Moreover, in healthy subjects, it has been demonstrated that not just chronic but even a single night of sleep deprivation can alter CSF levels of Aβ42 in addition to reducing cognitive performances [37, 38]. Therefore, it is currently under debate if sleep alteration represents a symptom or a possible cause of AD.
In this comprehensive study, we demonstrated that sleep impairment and dysregulation is present before the clinical appearance of objective cognitive deterioration and dementia by analyzing PSG recordings, assessing CSF biomarkers, and performing cognitive tests in subjects ranging from CN to AD dementia. In particular, subjects complaining of SCI already showed pathological modification of sleep architecture (SE, REM, and WASO), which was significantly different from that of CN controls. Moreover, we documented that REM sleep is altered in the preclinical stage of AD and that it is linked to β-amyloid pathology and memory loss.
To better interpret the findings of this study, we applied an exploratory PCA to analyze simultaneously the relationship between sleep and the variables putatively related to cognitive profile and neurodegeneration. The three components obtained by PCA were relatively “pure” with respect to the variables that loaded in each of them, and each variable tended to load heavily in only one component thus making the attribution of the physiological value to the factorial solution relatively easy.
An understanding of the temporal sequence between alterations in sleep architecture and dementia onset remains inadequate in the scientific literature. Accordingly, sleep impairment has been considered as either an early marker of AD pathology or a risk factor for AD. Following the first hypothesis, cross-sectional studies demonstrate that sleep architecture not only progressively deteriorates in patients with dementia but can also be dysregulated from the earlier stages of cognitive impairment [18, 39]. In accordance with the second supposition, REM sleep dysregulation with an increase in latency and a reduction in quantity has recently been associated with the incipient risk of dementia in CN subjects [10]. Further, sleep disturbances such as SDB and insomnia have been associated with the incipient risk of cognitive impairment and AD [8, 11, 14]. Following this line of evidence, orexinergic system dysregulation, sleep-wake cycle impairment, behavioral disturbances, and impaired generation of slow-wave sleep oscillations have been associated with cortical β-amyloid pathology and AD [12, 13, 18,19,20, 40,41,42]. Moreover, sleep influences the generation and clearance of β-amyloid by the aggregation of isoform 42 into oligomers and the deposition of brain plaques [43,44,45]. All these effects seem to be mediated by the functioning of the glymphatic system, which ensures the clearance of extracellular beta-amyloid and other toxic substrates during sleep [12].
Progressive cognitive deterioration and memory loss are the main clinical features of AD pathology. However, several studies suggest that sleep impairment is also a frequent and highly disruptive neuropsychiatric symptom associated with AD [6]. Epidemiological studies have documented that sleep disturbances occur in several patients affected by AD, and increase in frequency with the progression of the disease [6, 46]. Additionally, sleep disturbances can precipitate dementia symptoms with a negative impact on the cognitive and behavioral domains [6, 18]. After a thorough literature purview on the mutual interference of sleep, CSF AD biomarkers, and memory functioning, we examined not only whether sleep architecture is associated with the progression of AD in subjects affected by different stages of AD pathology, but also whether the modifications of sleep architecture correlated with cognitive performances and CSF AD biomarkers.
For this purpose, we performed PCA and documented the mutual interplay among a combination of variables related to sleep architecture (REM, N1, N3, and TST), CSF Aβ42 levels, and both global cognitive functioning and memory impairment (MMSE, RAVLT-I, and RAVLT-D) in Component 1. Our finding suggests a strong association between these parameters, but it does not give any indication on the possible causal link among them. Therefore, component 1 linking sleep to CSF β-amyloid concentrations and cognition during the progressive stages of the AD process along with the correlations supplemented the previously hypothesized association between AD pathological biomarkers and sleep dysregulation. Furthermore, it confirms the evidence that sleep fragmentation with reduced REM and N3 sleep is associated not only with β-amyloid pathology but also with tau neurodegeneration [10, 42]. Beyond the already stated association between sleep and cognition, the present findings additionally link β-amyloid and tau neurodegeneration to sleep dysregulation and cognitive impairment in the AD process. Notably, brain regions and networks involved in the control of the sleep-wake rhythm can be affected by AD pathology, which is often clinically complicated by both circadian rhythm disruption and sleep disturbances including night-time awakenings and non-REM/REM sleep dysregulation [3, 47, 48]. Component 2 of the PCA reflected a combination of variables related to CSF levels of the biomarkers attributed to both neurodegeneration (t-tau and p-tau) and the patient’s age. Age represents a risk factor for AD and cognitive deterioration [43]. Low Aβ42 and high t-tau and p-tau concentrations in the CSF are biomarkers of AD, reflecting brain deposition of amyloid plaques and NFT. Since the concentration of CSF biomarkers of AD is associated with age, following the second component of PCA, we confirmed prior evidence that suggested that the association between aging and tau pathology also influences memory performance [43].
Finally, component 3 of the PCA combined three sleep measures (REML, WASO, and SL) related to sleep quality/fragmentation; this finding further supports our hypothetical model on the role of sleep fragmentation and night-time wakefulness, which can reduce the beneficial effects of sleep against the neurodegenerative processes. Consistently, sleep fragmentation and related nocturnal arousals are accompanied by a significant increase in N1 that is associated with a decrease in N3 and REM. Further, more detrimental effects on sleep are induced by sleep fragmentation than partial sleep deprivation thus supporting the evidence that the brain tolerates sleep deprivation better than sleep fragmentation [44, 49]. Accordingly, sleep fragmentation is associated with cognitive decline and the risk of subsequent AD [50].
Our results consistently demonstrate that REM sleep dysregulation is associated with a more marked damage in the sleep architecture (featured by a more consistent sleep fragmentation) that worsens during the progression of AD. These findings coupled with those of the previous investigation that showed that REM sleep deterioration is associated with the increase in dementia risk further suggests that future research should take into account the role of REM sleep in the pathology of AD [10]. Nevertheless, the mechanisms linking REM sleep to the AD process remain to be well understood. The loss of cholinergic function may underpin REM sleep impairment during the AD process since cholinergic neurons are important determinants of REM sleep [51]. Loss of cholinergic function, degeneration of cholinergic projections in the basal forebrain, and changes in acetylcholine release are established fingerprints of AD neurodegeneration [52]. Moreover, the orexinergic system is found to be dysregulated in AD in both animal models and human studies [18, 53]. According to this evidence, orexinergic signaling malfunction can be related to REM sleep dysregulation thus promoting sleep architecture damage and β-amyloid pathology [18, 53]. Therefore, damage in the cholinergic pathway may cause a dysregulation of the orexinergic system, and both systems may negatively influence sleep, cognition, and AD neurodegenerative processes.
The novelties of this study include the large sample size of patients experiencing different stages of AD and the comparison to a control group comprising CN subjects. All the subjects underwent a very comprehensive protocol evaluating sleep, global cognition, memory functioning, and CSF AD biomarkers. Our study was the first to include subjects affected by SCI, which can represent a very early preclinical stage of AD pathology in humans and to compare them not only to MCI and AD patients but also to CN subjects. Pertinently, SCI was recently defined as a clinical condition indicating an increased risk of AD progression in patients especially when biomarkers consistent with AD are present [2]. Notably, our study documented both non-REM and REM sleep dysregulation during the AD process from the earliest stages of the disease onwards. Finally, the PCA gives further clarity to this data since it allows us to globally analyze all the parameters and interpret them as reciprocally related. In particular, sleep, cognition, and CSF AD biomarkers’ levels appear to be mutually related throughout the different stages of AD; this indicates that sleep is a potential therapeutic target for disease-modifying strategies. Conversely, the main limitation of this study is the non-longitudinal evaluation of the sample subjects.
Conclusion
Through exploring the interplay between different parameters in our model including not only patients affected by AD at different stages but also CN subjects, we identified three different and independent components manifesting a strong relationship among all the analyzed parameters. These include the following: component 1 linking sleep architecture, neurocognitive and memory functioning, and Aβ42 and tau proteins levels, component 2 linking aging to neurodegeneration, and component 3 linking sleep fragmentation to REM sleep dysregulation. Hence, this study suggests that sleep dysregulation is not just a risk factor but may also serve as an early marker of AD. To confirm this evidence, a long-term, longitudinal study on patients affected by SCI is necessary in order to track and understand the modifications of sleep, cognition, and neurodegenerative biomarkers.
Availability of data and materials
Any anonymized data not published within the article will be shared by request from any qualified investigator.
Change history
08 May 2020
An amendment to this paper has been published and can be accessed via the original article.
References
McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263–9.
Molinuevo JL, Rabin LA, Amariglio R, et al. Implementation of subjective cognitive decline criteria in research studies. Alzheimers Dement. 2017;13(3):296–311.
Van Harten AC, Visser PJ, Pijnenburg YA, et al. Cerebrospinal fluid Aβ42 is the best predictor of clinical progression in patients with subjective complaints. Alzheimers Dement. 2013;9(5):481–7.
Colijn MA, Grossberg GT. Amyloid and tau biomarkers in subjective cognitive impairment. J Alzheimers Dis. 2015;47:1–8.
Scaricamazza E, Colonna I, Sancesario GM, et al. Neuropsychiatric symptoms differently affect mild cognitive impairment and Alzheimer's disease patients: a retrospective observational study. Neurol Sci. 2019;40(7):1377–82.
Bliwise DL. Sleep disorders in Alzheimer's disease and other dementias. Clin Cornerstone. 2004;6(Suppl 1A):S16–28.
Sheikh F, Ismail Z, Mortby ME, et al. Prevalence of mild behavioral impairment in mild cognitive impairment and subjective cognitive decline, and its association with caregiver burden. Int Psychogeriatr. 2018;30(2):233–44.
Lauriola M, Esposito R, Delli Pizzi S, et al. Sleep changes without medial temporal lobe or brain cortical changes in community-dwelling individuals with subjective cognitive decline. Alzheimers Dement. 2017;13(7):783–91.
Lutsey PL, Misialek JR, Mosley TH, et al. Sleep characteristics and risk of dementia and Alzheimer's disease: the atherosclerosis risk in communities study. Alzheimers Dement. 2018;14(2):157–66.
Pase MP, Himali JJ, Grima NA, et al. Sleep architecture and the risk of incident dementia in the community. Neurology. 2017;89(12):1244–50.
Liguori C, Mercuri NB, Izzi F, et al. Obstructive sleep apnea is associated with early but possibly modifiable Alzheimer’s disease biomarkers changes. Sleep. 2017;40(5). https://doi.org/10.1093/sleep/zsx011
Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–7.
Roh JH, Jiang H, Finn MB, et al. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J Exp Med. 2014;211(13):2487–96.
Ju YE, McLeland JS, Toedebusch CD, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70(5):587–93.
Ooms S, Overeem S, Besse K, et al. Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 2014;71(8):971–7.
Ju Y, Ooms S, Sutphen C, et al. Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain. 2017;140(8):2104–11.
Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270–9.
Liguori C, Romigi A, Nuccetelli M, et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014;71(12):1498–505.
Liguori C, Mercuri NB, Nuccetelli M, Izzi F, Bernardini S, Placidi F. Cerebrospinal fluid orexin levels and nocturnal sleep disruption in Alzheimer's disease patients showing neuropsychiatric symptoms. J Alzheimers Dis. 2018;66(3):993–9.
Liguori C, Nuccetelli M, Izzi F, et al. Rapid eye movement sleep disruption and sleep fragmentation are associated with increased orexin-a cerebrospinal-fluid levels in mild cognitive impairment due to Alzheimer's disease. Neurobiol Aging. 2016;40:120–6.
Reisberg B, Shulman MB, Torossian C, Leng L, Zhu W. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6(1):11–24.
Jessen F, Wiese B, Bachmann C, et al. Prediction of dementia by subjective memory impairment: effects of severity and temporal association with cognitive impairment. Arch Gen Psychiatry. 2010;67(4):414–22.
Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. 2014;13(6):614–29.
Iber C, Ancoli-Israel S, Chesson A, Quan S. The AASM Manual for the scoring of sleep and associated events: rules, terminology and technical specifications. 1st ed. Westchester: American Academy of Sleep Medicine; 2007. IL
Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM manual for the scoring of sleep and associated events. Deliberations of the sleep apnea definitions task force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8(5):597–619.
Duits FH, Teunissen CE, Bouwman FH, et al. The cerebrospinal fluid “Alzheimer profile”: easily said, but what does it mean? Alzheimers Dement. 2014;10(6):713–723.e2.
Mulder C, Verwey NA, van der Flier WM, et al. Amyloid-beta(1-42), total tau, and phosphorylated tau as cerebrospinal fluid biomarkers for the diagnosis of Alzheimer disease. Clin Chem. 2010;56(2):248–53.
Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59(3):512–9.
Liguori C, Chiaravalloti A, Nuccetelli M, et al. Hypothalamic dysfunction is related to sleep impairment and CSF biomarkers in Alzheimer’s disease. J Neurol. 2017;264(11):2215–23.
Torelli F, Moscufo N, Garreffa G, et al. Cognitive profile and brain morphological changes in obstructive sleep apnea. Neuroimage. 2011;54(2):787–93.
Cohen J. Statistical power analysis for the behavioural sciences. 2nd ed. New York: Lawrence Erlbaum; 1988.
Bastuji H, García-Larrea L. Sleep/wake abnormalities in patients with periodic leg movements during sleep: factor analysis on data from 24-h ambulatory polygraphy. J Sleep Res. 1999;8(3):217–23.
Yong AG, Pearce S. A beginner’s guide to factor analysis: focusing on exploratory factor analysis. Tutorials Quantitative Methods Psychol. 2013;9:79–94.
Kaiser HF. A second generation little jiffy. Psychometrika. 1970;35:401–16.
Bartlett MS. Test of significance in factor analysis. Br J Psychol. 1950;3:77–85.
Aisen PS, Cummings J, Jack CR Jr, Morris JC, Sperling R, Frölich L. On the path to 2025: understanding the Alzheimer’s disease continuum. Alzheimers Res Ther. 2017;9(1):60.
Shokri-Kojori E, Wang GJ, Wiers CE, et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A. 2018;115(17):4483–8.
Lucey BP, Hicks TJ, McLeland JS, et al. Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann Neurol. 2018;83(1):197–204.
Prinz PN, Vitaliano PP, Vitiello MV, et al. Sleep, EEG and mental function changes in senile dementia of the Alzheimer’s type. Neurobiol Aging. 1982;3(4):361–70.
Liguori C, Mercuri NB, Nuccetelli M, et al. Obstructive sleep apnea may induce orexinergic system and cerebral β-amyloid metabolism dysregulation: is it a further proof for Alzheimer's disease risk? Sleep Med. 2019;56:171–6.
Mander BA, Marks SM, Vogel JW, et al. β-Amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. 2015;18(7):1051–7.
Slats D, Claassen JA, Lammers GJ, et al. Association between hypocretin-1 and amyloid-β42 cerebrospinal fluid levels in Alzheimer's disease and healthy controls. Curr Alzheimer Res. 2012;9(10):1119–25.
Paternicò D, Galluzzi S, Drago V, et al. Cerebrospinal fluid markers for Alzheimer's disease in a cognitively healthy cohort of young and old adults. Alzheimers Dement. 2012;8(6):520–7.
Stepanski E, Lamphere J, Roehrs T, Zorick F, Roth T. Experimental sleep fragmentation in normal subjects. Int J Neurosci. 1987;33(3–4):207–14.
Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–12.
Guarnieri B, Adorni F, Musicco M, et al. Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross-sectional study on 431 patients. Dement Geriatr Cogn Disord. 2012;33(1):50–8.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–59.
Ju YE, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology--a bidirectional relationship. Nat Rev Neurol. 2014;10(2):115–9.
Martin SE, Wraith PK, Deary IJ, Douglas NJ. The effect of nonvisible sleep fragmentation on daytime function. Am J Respir Crit Care Med. 1997;155(5):1596–601.
Lim AS, Kowgier M, Yu L, Buchman AS, Bennett DA. Sleep fragmentation and the risk of incident Alzheimer's disease and cognitive decline in older persons. Sleep. 2013;36(7):1027–32.
Vazquez J, Baghdoyan HA. Basal forebrain acetylcholine release during REM sleep is significantly greater than during waking. Am J Physiol Regul Integr Comp Physiol. 2001;280(2):R598–601.
Mesulam M, Shaw P, Mash D, Weintraub S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol. 2004;55(6):815–28.
Kang JE, Lim MM, Bateman RJ, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326(5955):1005–7.
Acknowledgements
We thank all the Staff of the Sleep Laboratory.
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
CL is responsible for the study concept, acquisition of data, interpretation of data, and drafting the manuscript. NBM is responsible for the study supervision and critical revision of the manuscript for important intellectual content. FI is responsible for the critical revision of the manuscript for important intellectual content. MS is responsible for the acquisition of data. FP is responsible for the study supervision and critical revision of the manuscript for important intellectual content. ADP is responsible for the analysis of data and drafting the manuscript. All authors read and approved the final manuscript.
Authors’ information
CL and ADP had full access to all data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Patients and controls provided their informed consent to the study, which was approved by the Independent Ethical Committee of the University Hospital of Rome “Tor Vergata.”
Consent for publication
All authors received the consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liguori, C., Placidi, F., Izzi, F. et al. Sleep dysregulation, memory impairment, and CSF biomarkers during different levels of neurocognitive functioning in Alzheimer’s disease course. Alz Res Therapy 12, 5 (2020). https://doi.org/10.1186/s13195-019-0571-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-019-0571-3