
Abstract
Background
Results from recent clinical studies suggest that cerebrospinal fluid (CSF) biomarkers that are indicative of Alzheimer’s disease (AD) can be replicated in blood, e.g. amyloid-beta peptides (Aβ42 and Aβ40) and neurofilament light chain (NFL). Such data proposes that blood is a rich source of potential biomarkers reflecting central nervous system pathophysiology and should be fully explored for biomarkers that show promise in CSF. Recently, soluble fragments of the triggering receptor expressed on myeloid cells 2 (sTREM2) protein in CSF have been reported to be increased in prodromal AD and also in individuals with TREM2 rare genetic variants that increase the likelihood of developing dementia.
Methods
In this study, we measured the levels of plasma sTREM2 and plasma NFL using the MesoScale Discovery and single molecule array platforms, respectively, in 48 confirmed TREM2 rare variant carriers and 49 non-carriers.
Results
Our results indicate that there are no changes in plasma sTREM2 and NFL concentrations between TREM2 rare variant carriers and non-carriers. Furthermore, plasma sTREM2 is not different between healthy controls, mild cognitive impairment (MCI) or AD.
Conclusion
Concentrations of plasma sTREM2 do not mimic the recent changes found in CSF sTREM2.
Similar content being viewed by others

Introduction
There has been considerable progress in the search for blood-based biomarkers able to capture the clinical course and underlying pathophysiology of Alzheimer’s disease (AD), for review see [1]. Reduced plasma Aβ42/Aβ40 ratio [2,3,4] and increased neurofilament light chain (NFL) [5,6,7] are becoming consistently reported in AD, and encouragingly, these findings mimic the more established observations seen in cerebrospinal fluid (CSF) [8]. NFL is not a specific biomarker for AD [9]. Increases are observed in a number of neurodegenerative disorders [10,11,12], owing to its global reflection of axonal injury or degeneration. Yet, translating other co-pathology markers of neurodegeneration from CSF to blood has been less successful. For example, plasma total tau (T-tau) increases in AD [13] but it seems to have limited clinical utility, and phosphorylated form of tau (P-tau) has proven difficult to establish as a reliable measure in blood despite recent promise [14]. Likewise, TDP-43 [15], alpha-synuclein [16] and the post-synaptic dendritic biomarker, neurogranin [17], all have substantial and specific peripheral expression but levels appear to be unrelated to changes in the central nervous system.
The triggering receptor expressed on myeloid cells 2 (TREM2) is an innate immune receptor that guides essential functions of microglia. Rare variants in TREM2 strongly increase the likelihood of developing AD, frontotemporal dementia (FTD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) [18,19,20,21,22,23]. TREM2 is a type 1 transmembrane protein, and its ectodomain is shed at the plasma membrane by ADAM family proteases C-terminal at histidine 157 position [24,25,26,27]. The resulting soluble fragment (sTREM2) is released into the extracellular space and can be found in CSF and plasma [28, 29]. Recently, the concentrations of CSF sTREM2 have been shown to be increased in early symptomatic stages of sporadic [30,31,32,33,34] and autosomal dominant AD patients [35]. Interestingly, Aβ pathology and tau-related neurodegeneration may impact levels of CSF sTREM2 differently [33]. Moreover, it has been shown that the concentrations of CSF sTREM2 vary between different disease-associated TREM2 genetic variant carriers [32, 33].
Unlike CSF sTREM2, levels of sTREM2 in blood have been poorly investigated. In this study, we investigate plasma concentrations of sTREM2 in patients with AD and mild cognitive impairment (MCI) compared with aged-matched healthy controls. Furthermore, in a novel approach, we also report on blood concentrations of sTREM2 and NFL in TREM2 rare variant carriers.
Methods
Participants
Samples from a total of 97 participants were used for these analyses (Table 1). The majority of samples (n = 82) were from the longitudinal AD cohorts managed at King’s College London (KCL; AddNeuroMed [36], Kings Health Partners-Dementia Case Register (KHP-DCR) a UK clinic and population based study [37] and the MRC AD Biomarker study [38]). Further samples were obtained from DEGESCO (n = 11, Dementia Genetics Spanish Consortium [39, 40]) and EDAR (n = 4, beta amyloid oligomers in the early diagnosis of AD and as marker for treatment response [41]). Informed consent for all participants was obtained according to the Declaration of Helsinki (1991), and protocols and procedures were approved by the relevant local ethical committee at each site. The cohorts as mentioned above were genetically analysed to identify known or novel non-synonymous variants in exon 2 of the TREM2 gene, previously linked to pathogenic risk or predicted to be detrimental. Of the 48 participants identified with a TREM2 pathogenic variant (TREM2var, Table 1), 10 were controls, 10 had MCI and 28 had a dementia diagnosis (AD). Similar age-matched non-carrier control (n = 10), MCI (n = 8) and AD (n = 31) samples were also included.
Plasma measures of sTREM2 and NFL
Plasma sTREM2 was measured using an in-house electrochemoluminescent assay on the MesoScale Discovery SECTOR imager 6000 (MesoScale Discovery (MSD), Maryland, USA) using a method adapted from Kleinberger et al. [29]. The capture antibody was the biotinylated polyclonal goat anti-human TREM2 (0.25 μg/mL, R&D Systems, Minneapolis, USA), and the detector antibody was monoclonal mouse anti-human TREM2 (1 μg/mL, Santa Cruz Biotechnology, Texas, USA). A standard curve for calculations of unknowns was constructed using recombinant human TREM2 (4000–62.5 pg/mL, Sino Biological, Bejiing, China), and plasma samples were diluted 1:4 before being assayed. For a more comprehensive description of the method, please see [29]. For NFL, the commercially available NF-light assay on an HD-1 Simoa instrument (Quanterix, Lexington, MA, USA) was utilized. All biochemical analyses were performed at the Institute of Neurology at University College London (UCL).
Sample size and power calculation
In CSF, the effect size of sTREM2 between AD and control ranges between 1.077 and 1.539 (mean 1.272; source: Alzbiomarker, Alzforum). In applying a type error I (α) of 0.05, we reach a power (1-β) of 0.99 in our sample size of 97 participants (G*Power). However, the effect size of plasma sTREM2 is likely to be considerably lower than that of CSF sTREM2. Therefore, we examined the achieved power as a function of effect size (Cohen’s d), with a type error I (α) of 0.05 assuming 97 participants. In a medium effect size (Cohen’s d = 0.5), a power of 0.68 is achieved, whereas a large effect size (Cohen’s d = 0.8) computes a power of 0.97. Thus, we can reasonably state that there is a low probability of error type II in our results if the effect size was large.
Statistical analysis
Data normality was determined by the D’Agostino-Pearson test, and statistical evaluation was performed on log10-transformed data. After transformation, the data followed a normal distribution. All data analysis reported has been performed on log10-transformed sTREM2 and NFL, but the untransformed values are shown in descriptive tables and figures. To study the association of plasma measures with demographic data, a Pearson product-moment correlation was utilized for age and Mini-Mental State Examination (MMSE) whereas t test (sTREM2) or a one-way analysis of covariance (ANCOVA, NFL) for gender and APOE status. Only age was a significant predictor of plasma NFL; the subsequent analyses were therefore conducted including age as a confounder. A t test or ANCOVA were conducted to determine clinical group differences between blood biomarkers. ANCOVA analyses were followed by a Bonferroni-corrected post hoc pairwise comparison where appropriate. A partial correlation, adjusted by age, tested the association between plasma sTREM2 and plasma NFL. Statistical analysis was performed using IBM SPSS Statistics, version 25 (Armonk, NY, USA).
Results
Forty-eight confirmed TREM2 rare variant carriers and 49 non-carriers were included in the study. The TREM2 rare variant carrier group comprised of 10 different variants: p.Q33X, p.D39N, p.R47H, p.P59L, p.R62H, p.D87N, p.T96K, p.Q691H, p.H703Y and p.L868R. The demographical characteristics of the cohort are described in Table 1. There were no differences in age between TREM2 rare variant carriers (M = 75.2, SD = 7.3) and non-carriers (M = 76.1, SD = 6.7). The inclusion of gender was very similar across groups (carriers, 26/48 female [54.2%]; non-carriers, 27/49 female [55.1%]). Finally, there was no statistically significant difference in MMSE (carriers, M = 23.1, SD = 6.4; non-carriers, M = 23.3, SD = 5.0) or clinical diagnosis between the two groups (Table 1).
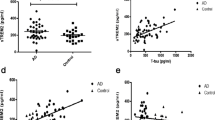
In the whole cohort, plasma sTREM2 was not associated with age (r = 0.060; P = 0.562), gender (P = 0.083), APOE ε4 status (P = 0.237) or MMSE (r = − 0.018; P = 0.858). There were no differences in the levels of plasma sTREM2 between TREM2 rare variant carriers (M = 7346 ng/L, SD = 5526 ng/L) and non-carriers (M = 8750 ng/L, SD = 5265 ng/L; t(95) = 1.696, P = 0.093; Fig. 1a). There were no significant differences in plasma sTREM2 between carriers of different TREM2 rare variants (F(3, 84) = 1.68, P = 0.177, Fig. 1b). Note, we only included TREM2 rare variants with > 2 individuals per group in this analysis. Adding clinical diagnosis as covariate did not change the result (P = 0.171). Next, we tested whether plasma sTREM2 levels differ between clinical diagnoses, regardless of the TREM2 rare variant status, between AD (M = 8859 ng/L, SD = 5951 ng/L), MCI (M = 6204 ng/L, SD = 2572 ng/L) and controls (M = 7352 ng/L, SD = 5318 ng/L). Plasma sTREM2 levels were not different between these groups (F(2, 94) = 1.84, P = 0.164, Fig. 1c). Adjusting for the effect of age and gender did not change the result of sTREM2 in plasma.

The concentrations of plasma sTREM2. No change in the levels of sTREM2 between TREM2 rare variant carriers and non-carriers (a, b). A non-significant increase in sTREM2 was observed in AD patients compared to MCI and aged-matched controls (c)
As expected, plasma NFL concentrations were significantly associated with age (r = 0.202, P = 0.047). After accounting for the effect of age, plasma NFL levels were found to be associated with MMSE (r = − 0.353, P = 0.0004) and tended to be higher in females (M = 25.6, SD = 13.1 ng/L) compared to males (M = 25.1, SD = 22.8 ng/L; P = 0.079). Levels were not affected by APOE ε4 status (P = 0.899). There were no differences in plasma NFL between TREM2 rare variant carriers (M = 24.6 ng/L, SD = 19.1 ng/L) and non-carriers (M = 26.1 ng/L, SD = 17.1 ng/L) (F(1, 94) = 0.505, P = 0.479, Fig. 2a), after adjusting for the effect of age. Similar to plasma sTREM2, there were no differences in plasma NFL when comparing the different TREM2 rare variants (F(3, 83) = 0.113, P = 0.952, Fig. 2b). This remained true when correcting for the effect of the clinical diagnosis (P = 0.633). As expected, differences in plasma NFL were observed between the three clinical groups (F(2, 93) = 4.89, P = 0.010). The Bonferroni-corrected post hoc pairwise comparison demonstrated that the AD group (M = 29.0 ng/L, SD = 21.6 ng/L) had significantly higher levels of NFL compared to controls (M = 18.8 ng/L, SD = 7.2 ng/L; P = 0.025) but not to MCI (M = 20.5 ng/L, SD = 8.1 ng/L; P = 0.096, Fig. 2c). A further adjustment for the effect of gender did not change these NFL findings.

The concentrations of plasma NFL. No change in the concentrations of NFL between TREM2 rare variants and non-carriers (a, b). A significant increase in NFL was observed in AD patients compared to MCI and aged-matched controls which was not influenced by TREM2 mutation status (c)
In the whole cohort, when correcting for age, a significant positive correlation between plasma sTREM2 and plasma NFL was observed (r = 0.245, P = 0.016). However, this correlation is driven by the symptomatic individuals (MCI and AD) as no significant association of plasma sTREM2 and plasma NFL was observed in the control group (r = 0.250, P = 0.302) but a tendency in the symptomatic group (r = 0.223, P = 0.053).
Discussion
This is the first study to have comprehensively investigated if the levels of plasma sTREM2 and a neurodegenerative marker, plasma NFL, differ between TREM2 rare variant carriers and non-carriers. Our main finding demonstrates that there are no significant differences in plasma sTREM2 and NFL between these groups. Furthermore, we also show that plasma sTREM2 does not differ between controls, MCI and AD.
While sTREM2 has been extensively studied in CSF, only a few studies have reported sTREM2 in blood. Piccio and co-workers [28] demonstrated that serum sTREM2 levels did not differ between multiple sclerosis, other inflammatory neurologic diseases and non-inflammatory controls. Kleinberger et al. [29] found no difference in plasma sTREM2 between healthy controls, AD and FTD. The lack of separation between AD and controls was then independently replicated [32]. In a different approach, Ohara et al. showed that increased serum sTREM2 is associated with increased risk to develop dementia in Japanese population [42]. Herein, we also demonstrate that plasma sTREM2 is not different between TREM2 rare variant carriers and non-carriers, nor with respect to clinical diagnosis. Therefore, although readily detectable, sTREM2 in blood is not useful to discriminate those people with a clinical diagnostic group or those with a TREM2 variant associated with AD. It should be noted that sTREM2 in blood most likely has a peripheral rather than a central nervous system (CNS) origin from microglia. This is in agreement with the ubiquitous expression pattern of TREM2 at both mRNA and protein levels in Human Protein Atlas (https://www.proteinatlas.org/ENSG00000095970-TREM2/tissue). TREM2 is highly expressed in cells from a myeloid lineage, such as monocytes, macrophages, Kupffer cells or osteoclasts [43,44,45,46].
Previously, it was shown that both plasma and CSF sTREM2 were useful to detect those with homozygous TREM2 mutations (e.g. p.T66 M, p.W198X, p.Q33X and p.Y38C) that lead to Nasu-Hakola disease or an FTD-like syndrome [47, 48]. In these diseases, sTREM2 in blood or CSF is almost absent, which is in line with the impaired cell surface transport and shedding that occur with these mutations [29, 49]. In contrast, heterozygous TREM2 rare variant carriers have a less obvious and inconsistent pattern of CSF sTREM2. While the TREM2 p.R47H rare variant is associated with increased CSF sTREM2, other TREM2 variants (e.g. p.L211P; T96K/L211P/W191X) have been reported to be associated with decreased or unchanged levels of CSF sTREM2 (e.g. R62H) [32, 33] (Deming et al. Sci Trans Med 2019 in press). Our findings could be explained by how these rare variants differently affect the processing and shedding of TREM2. For example, individual variants are believed to impact TREM2 production (Q33X), expression or turnover at the cell surface (R47H) [29, 50]; α-secretase cleavage of the extracellular soluble ectodomain (H157Y) [27, 51]; and/or ligand binding (R47H, R62H, T96K, H157Y) [52,53,54]. Surprisingly, we did not find such differences in plasma, suggesting a different regulation of TREM2 in the periphery and the CNS. We also investigated plasma NFL, but here, we also found no differences between TREM2 rare variant carriers and non-carriers, even when introducing clinical diagnosis as a co-variable, suggesting that the neuronal injury is no different in AD irrespective of whether someone has a TREM2 rare variant. These findings are agreement with clinical data that find AD cases with a TREM2 p.R47H rare variant are clinically indistinguishable from other AD, albeit those with a TREM2 rare variant generally have an earlier age of onset [55]. Finally, the finding that sTREM2 is associated with NFL in AD is consistent with the association of CSF sTREM2 with T-tau, another marker of neurodegeneration, and suggests that inflammatory response might be coupled to neurodegeneration [56].
This study has some limitations. Despite our study recruiting the highest number of TREM2 rare variant carriers to date, some individual variants were represented by only very small numbers, which precluded a comparison between them. Additionally, these samples did not have the core AD CSF biomarkers to confirm diagnosis, disease stage or link analyses individually to tau or amyloid burden, and hence, we were limited to using clinical diagnosis. The main strengths are the fact that we used very reliable and well-established assays for both sTREM2 and NFL. The study was designed in such a way that an important total number of TREM2 rare variants were carefully matched to a control group in terms of age, gender and clinical diagnosis.
Conclusion
This study, for the first time, demonstrates that the levels of sTREM2 and NFL in plasma do not differ between TREM2 rare variant carriers and non-carriers. Furthermore, we confirm previous reports that sTREM2 is not changed in AD or MCI compared with aged-matched controls. Therefore, we conclude that although plasma sTREM2 may be useful to detect TREM2 homozygous mutations, plasma sTREM2 is not a reliable biomarker to detect TREM2 rare variant status nor suspected AD.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- ANCOVA:
-
Analysis of covariance
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- FTD:
-
Frontotemporal dementia
- MCI:
-
Mild cognitive impairment
- MMSE:
-
Mini-Mental State Examination
- MSD:
-
MesoScale Discovery
- NFL:
-
Neurofilament light chain
- PD:
-
Parkinson’s disease
- P-tau:
-
Phosphorylated tau
- Simoa:
-
Single molecule array
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
- T-tau:
-
Total tau
References
Hampel H, O'Bryant SE, Molinuevo JL, Zetterberg H, Masters CL, Lista S, et al. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. 2018;14(11):639–52.
Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Dore V, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature. 2018;554(7691):249–54.
Janelidze S, Stomrud E, Palmqvist S, Zetterberg H, van Westen D, Jeromin A, et al. Plasma beta-amyloid in Alzheimer’s disease and vascular disease. Sci Rep. 2016;6:26801.
Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, Schneider T, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13(8):841–9.
Mattsson N, Andreasson U, Zetterberg H, Blennow K, Alzheimer’s Disease Neuroimaging I. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2017;74(5):557–566.
Lewczuk P, Ermann N, Andreasson U, Schultheis C, Podhorna J, Spitzer P, et al. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimers Res Ther. 2018;10(1):71.
Ashton NJ, Leuzy A, Lim YM, Troakes C, Hortobagyi T, Hoglund K, et al. Increased plasma neurofilament light chain concentration correlates with severity of post-mortem neurofibrillary tangle pathology and neurodegeneration. Acta Neuropathol Commun. 2019;7(1):5.
Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15(7):673–84.
Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90(8):870-81.
Hansson O, Janelidze S, Hall S, Magdalinou N, Lees AJ, Andreasson U, et al. Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology. 2017;88(10):930–7.
Rohrer JD, Woollacott IO, Dick KM, Brotherhood E, Gordon E, Fellows A, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87(13):1329–36.
Byrne LM, Rodrigues FB, Blennow K, Durr A, Leavitt BR, Roos RAC, et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. Lancet Neurol. 2017;16(8):601–9.
Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, et al. Plasma tau in Alzheimer disease. Neurology. 2016;87(17):1827–35.
Mielke MM, Hagen CE, Xu J, Chai X, Vemuri P, Lowe VJ, et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement. 2018;14(8):989–97.
Foulds P, McAuley E, Gibbons L, Davidson Y, Pickering-Brown SM, Neary D, et al. TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degeneration. Acta Neuropathol. 2008;116(2):141–6.
Barbour R, Kling K, Anderson JP, Banducci K, Cole T, Diep L, et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener Dis. 2008;5(2):55–9.
Kvartsberg H, Portelius E, Andreasson U, Brinkmalm G, Hellwig K, Lelental N, et al. Characterization of the postsynaptic protein neurogranin in paired cerebrospinal fluid and plasma samples from Alzheimer’s disease patients and healthy controls. Alzheimers Res Ther. 2015;7(1):40.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–27.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–16.
Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, et al. Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol Aging. 2014;35(3):726 e11–9.
Borroni B, Ferrari F, Galimberti D, Nacmias B, Barone C, Bagnoli S, et al. Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging. 2014;35(4):934 e7–10.
Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener. 2013;8:19.
Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014;71(4):449–53.
Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J Biol Chem. 2013;288(46):33027–36.
Feuerbach D, Schindler P, Barske C, Joller S, Beng-Louka E, Worringer KA, et al. ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci Lett. 2017;660:109–14.
Schlepckow K, Kleinberger G, Fukumori A, Feederle R, Lichtenthaler SF, Steiner H, et al. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med. 2017;9(10):1356–65.
Thornton P, Sevalle J, Deery MJ, Fraser G, Zhou Y, Stahl S, et al. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol Med. 2017;9(10):1366–78.
Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008;131(Pt 11):3081–91.
Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6(243):243ra86.
Suarez-Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med. 2016;8(5):466–76.
Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol Neurodegener. 2016;11:3.
Piccio L, Deming Y, Del-Aguila JL, Ghezzi L, Holtzman DM, Fagan AM, et al. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016;131(6):925–33.
Suarez-Calvet M, Morenas-Rodriguez E, Kleinberger G, Schlepckow K, Araque Caballero MA, Franzmeier N, et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-beta pathology. Mol Neurodegener. 2019;14(1):1.
Rauchmann BS, Schneider-Axmann T, Alexopoulos P, Perneczky R, Alzheimer’s Disease Neuroimaging I. CSF soluble TREM2 as a measure of immune response along the Alzheimer’s disease continuum. Neurobiol Aging 2019;74:182–190.
Suarez-Calvet M, Araque Caballero MA, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci Transl Med. 2016;8(369):369ra178.
Lovestone S, Francis P, Kloszewska I, Mecocci P, Simmons A, Soininen H, et al. AddNeuroMed--the European collaboration for the discovery of novel biomarkers for Alzheimer’s disease. Ann N Y Acad Sci. 2009;1180:36–46.
Hye A, Riddoch-Contreras J, Baird AL, Ashton NJ, Bazenet C, Leung R, et al. Plasma proteins predict conversion to dementia from prodromal disease. Alzheimers Dement. 2014;10(6):799–807 e2.
Hye A, Lynham S, Thambisetty M, Causevic M, Campbell J, Byers HL, et al. Proteome-based plasma biomarkers for Alzheimer’s disease. Brain. 2006;129(Pt 11):3042–50.
Ruiz A, Dols-Icardo O, Bullido MJ, Pastor P, Rodriguez-Rodriguez E, Lopez de Munain A, et al. Assessing the role of the TREM2 p.R47H variant as a risk factor for Alzheimer’s disease and frontotemporal dementia. Neurobiol Aging. 2014;35(2):444 e1–4.
Benitez BA, Cooper B, Pastor P, Jin SC, Lorenzo E, Cervantes S, et al. TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol Aging. 2013;34(6):1711 e15–7.
Reijs BLR, Ramakers I, Elias-Sonnenschein L, Teunissen CE, Koel-Simmelink M, Tsolaki M, et al. Relation of odor identification with Alzheimer’s disease markers in cerebrospinal fluid and cognition. J Alzheimers Dis. 2017;60(3):1025–34.
Ohara T, Hata J, Tanaka M, Honda T, Yamakage H, Yoshida D, et al. Serum soluble triggering receptor expressed on myeloid cells 2 as a biomarker for incident dementia: the Hisayama study. Ann Neurol. 2019;85(1):47–58.
Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001;194(8):1111–22.
Schmid CD, Sautkulis LN, Danielson PE, Cooper J, Hasel KW, Hilbush BS, et al. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem. 2002;83(6):1309–20.
Paloneva J, Mandelin J, Kiialainen A, Bohling T, Prudlo J, Hakola P, et al. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med. 2003;198(4):669–75.
Kiialainen A, Hovanes K, Paloneva J, Kopra O, Peltonen L. Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiol Dis. 2005;18(2):314–22.
Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71(3):656–62.
Guerreiro R, Bilgic B, Guven G, Bras J, Rohrer J, Lohmann E, et al. Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol Aging. 2013;34(12):2890 e1–5.
Kleinberger G, Brendel M, Mracsko E, Wefers B, Groeneweg L, Xiang X, et al. The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J. 2017;36(13):1837–53.
Park JS, Ji IJ, An HJ, Kang MJ, Kang SW, Kim DH, et al. Disease-associated mutations of TREM2 alter the processing of N-linked oligosaccharides in the golgi apparatus. Traffic. 2015;16(5):510–8.
Jiang T, Tan L, Chen Q, Tan MS, Zhou JS, Zhu XC, et al. A rare coding variant in TREM2 increases risk for Alzheimer’s disease in Han Chinese. Neurobiol Aging. 2016;42:217 e1–3.
Kober DL, Alexander-Brett JM, Karch CM, Cruchaga C, Colonna M, Holtzman MJ, et al. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife. 2016;5:e20391.
Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK, et al. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 2017;13(4):381–7.
Sudom A, Talreja S, Danao J, Bragg E, Kegel R, Min X, et al. Molecular basis for the loss-of-function effects of the Alzheimer’s disease-associated R47H variant of the immune receptor TREM2. J Biol Chem. 2018;293(32):12634–46.
Slattery CF, Beck JA, Harper L, Adamson G, Abdi Z, Uphill J, et al. R47H TREM2 variant increases risk of typical early-onset Alzheimer’s disease but not of prion or frontotemporal dementia. Alzheimers Dement. 2014;10(6):602–8 e4.
Gispert JD, Suarez-Calvet M, Monte GC, Tucholka A, Falcon C, Rojas S, et al. Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer’s disease. Alzheimers Dement. 2016;12(12):1259–72.
Acknowledgements
NJA is funded by the Wallenburg Centre for Molecular and Translational. MSC is funded by the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie action grant agreement no. 752310. RSV acknowledges funding from grant n1 PI160235 to RSV (ISCIII, Spain, and FEDER, EU). KB is supported by the Torsten Söderberg Foundation, Stockholm, Sweden. HZ is a Wallenberg Academy Fellow supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), the Swedish State Support for Clinical Research (#ALFGBG-720931) and the UK Dementia Research Institute at UCL.
Funding
The study has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement no. 115976. This Joint Undertaking receives support from the European Union’s Horizon 2020 Research and Innovation Programme and EFPIA (AKH and HZ), the UK Dementia Research Institute (AKH and HZ) and the Olav Thon Foundation. This study is an independent research partly funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. NFL measurements were performed on equipment purchased through a Wellcome Trust Multi-User Equipment Grant. The plasma samples were collected with funding through the EU FP6 programme (AddNeuroMed).
Author information
Authors and Affiliations
Contributions
NJA, AKH and HZ provided the initial concept and study design. AH performed all the data acquisition. NJA and MSA performed the data analysis, interpreted the data and co-wrote the manuscript. All authors contributed to the content of the publication, and critically reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
KB has served as a consultant or at advisory boards for Alector, Alzheon, CogRx, Biogen, Lilly, Novartis and Roche Diagnostics, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg, all unrelated to the work presented in this paper. HZ has served at scientific advisory boards for Roche Diagnostics, Wave, Samumed and CogRx; has given lectures in symposia sponsored by Alzecure and Biogen; and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg. The other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ashton, N.J., Suárez-Calvet, M., Heslegrave, A. et al. Plasma levels of soluble TREM2 and neurofilament light chain in TREM2 rare variant carriers. Alz Res Therapy 11, 94 (2019). https://doi.org/10.1186/s13195-019-0545-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-019-0545-5