
Abstract
Background
Gingivobuccal complex oral squamous cell carcinoma (GBC-OSCC) is an aggressive malignancy with high mortality often preceded by premalignant lesions, including leukoplakia. Previous studies have reported genomic drivers in OSCC, but much remains to be elucidated about DNA methylation patterns across different stages of oral carcinogenesis.
Results
There is a serious lack of biomarkers and clinical application of biomarkers for early detection and prognosis of gingivobuccal complex cancers. Hence, in search of novel biomarkers, we measured genome-wide DNA methylation in 22 normal oral tissues, 22 leukoplakia, and 74 GBC-OSCC tissue samples. Both leukoplakia and GBC-OSCC had distinct methylation profiles as compared to normal oral tissue samples. Aberrant DNA methylation increases during the different stages of oral carcinogenesis, from premalignant lesions to carcinoma. We identified 846 and 5111 differentially methylated promoters in leukoplakia and GBC-OSCC, respectively, with a sizable fraction shared between the two sets. Further, we identified potential biomarkers from integrative analysis in gingivobuccal complex cancers and validated them in an independent cohort. Integration of genome, epigenome, and transcriptome data revealed candidate genes with gene expression synergistically regulated by copy number and DNA methylation changes. Regularised Cox regression identified 32 genes associated with patient survival. In an independent set of samples, we validated eight genes (FAT1, GLDC, HOXB13, CST7, CYB5A, MLLT11, GHR, LY75) from the integrative analysis and 30 genes from previously published reports. Bisulfite pyrosequencing validated GLDC (P = 0.036), HOXB13 (P < 0.0001) promoter hypermethylation, and FAT1 (P < 0.0001) hypomethylation in GBC-OSCC compared to normal controls.
Conclusions
Our findings identified methylation signatures associated with leukoplakia and gingivobuccal complex cancers. The integrative analysis in GBC-OSCC identified putative biomarkers that enhance existing knowledge of oral carcinogenesis and may potentially help in risk stratification and prognosis of GBC-OSCC.
Graphical abstract

Similar content being viewed by others

Background
Oral squamous cell carcinoma (OSCC) arising in the gingivobuccal complex (GBC) is the most common malignancy in India attributable to tobacco abuse [1, 2]. Oral cancers have high morbidity and mortality as they are frequently diagnosed at an advanced stage, and managing them is challenging due to early invasion. Despite advances in treatment modalities, the prognosis of OSCC remains poor due to locoregional recurrence and nodal metastasis [3]. OSCCs are often preceded by oral premalignant lesions (OPLs), now termed as oral potentially malignant disorders (OPMDs), mainly leukoplakia, which has a transformation rate to OSCC between 0.13 and 34.0% [4]. Early diagnosis and primary prevention remain the best approaches for OSCC management. Histopathology is the gold standard for diagnosis, but it is not sufficient to predict transformation potential or disease progression. Human papillomavirus (HPV) infection has emerged as an important stratification factor associated with better prognosis and treatment de-escalation. In HPV-negative OSCC, several biomarkers have been proposed, but their effectiveness is yet to be proven in the clinic. These observations justify the need for robust biomarkers for early detection of high-risk OPMDs, treatment response, and prognosis to improve OSCC management [5].
Oral carcinogenesis is a complex multifactorial molecular process that involves the accumulation of genetic and epigenetic changes leading to aberrant copy number gains and upregulation of oncogenes as well as to copy number losses and downregulation of tumor suppressor genes (TSGs). Previously, we have reported genomic and transcriptomic profiles of leukoplakia and gingivobuccal complex cancers that suggest the possible involvement of epigenetic regulation [6, 7]. DNA methylation is the most commonly studied epigenetic mechanism that regulates gene expression, and a plethora of literature is available for DNA methylation-based biomarkers [8,9,10,11,12] with prognostic significance [13,14,15,16,17,18]. Hypomethylation of oncogenes and hypermethylation of TSGs are responsible for disease progression [19]. Genome-wide methylation patterns have been studied extensively in OSCC (Additional file 1: Table S1). However, these studies have small sample sizes, include samples from different anatomic sites, and mostly lack representation from OPMD samples. Few studies have integrated methylation data with gene expression (GE) or copy number alteration (CNA) data. Furthermore, validation and correlation of candidate biomarkers with clinical outcomes are lacking. To overcome these lacunae, we performed a comprehensive study and identified genome-wide DNA methylation signatures, gene expression, and copy number changes associated with different stages of oral carcinogenesis. We validated selected candidate genes from these signatures and evaluated their association with clinical outcomes in GBC-OSCC patients.
Results
Demographic and clinicopathological details of study samples
The study group consisted of a training set for discovery and an independent validation set. Clinicopathological and demographic details of the study participants are summarised in Table 1. Additional file 1: Tables S2 and S3 show the individual characteristics of leukoplakia and gingivobuccal complex oral cancer samples with follow-up data used in this study and previous studies [6, 7]. The validation set had similar characteristics to the training set. The grading of tumors is represented in Additional file 1: Table S3, with 63% of patients having advanced stage OSCC and 38% having lymph node positivity at presentation. The majority of the patients (69%) were solely tobacco consumers, while many (19%) consumed both tobacco and alcohol. The median follow-up of the validation set was 70 months. All samples were tested for the presence of HPV and were negative for HPV-DNA, which is consistent with our previous reports on gingivobuccal cancers [20].
Genome-wide DNA methylation analysis
We performed genome-wide DNA methylation in 22 normal buccal mucosa tissue samples from unrelated healthy subjects, 22 tissue samples from leukoplakia patients, and 74 tissue samples from gingivobuccal complex cancer patients using 850 K methylation arrays. Principal components analysis (PCA) of methylation sites in promoter regions identified two distinct clusters consisting of GBC-OSCCs and normals, respectively, while leukoplakia cases were spread across these two groups implying the absence of strong batch effects (Additional file 2: Figure S1).
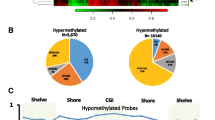
Differential methylation analyses performed across CpG sites, CpG islands, promoters, and genes is summarized in Additional file 1: Table S4. The list of the top 20 hypomethylated and hypermethylated targets for CpG sites and islands based on combined ranks are presented in Additional file 1: Tables S5–S8. Top 20 hypomethylated and hypermethylated protein-coding genes and promoters based on combined ranks are shown in Additional file 1: Tables S9–S12.. In the site-level analysis, 2296 and 17,076 differentially methylated CpG sites were reported in leukoplakia and GBC-OSCCs, respectively. Of these, 1885 (82.10%) were hypermethylated, and 411 (17.90%) were hypomethylated in leukoplakia. Consistent with these observations, GBC-OSCCs also showed more hypermethylation (72.92%) than hypomethylation (27.08%). By genomic location, the proportion of differential methylation was highest for chromosome 19 in leukoplakia (10.80%) and chromosome 1 in GBC-OSCCs (8.47%), whereas chromosome 21 showed the lowest differential methylation in both (0.78% and 0.67%, respectively; Additional file 2: Figure S2).
Promoter hypermethylation is one of the major mechanisms of gene regulation; hence, our analysis focused on methylation in promoter regions. Differentially methylated promoters (DMPs) are illustrated using volcano plots. Additional file 2: Figure S3 depicts the increase in aberrant methylation as the lesion progresses from OPMD to OSCC. We did not observe any significant associations of DMPs with clinicopathological parameters such as tumour stage, nodal status, and recurrence. Similar to CpG sites, the highest number of DMPs were located on chromosome 19 in leukoplakia (10.28%) and on chromosome 2 in GBC-OSCCs (7.53%), with chromosome 21 having the lowest DMPs in both conditions (Additional file 2: Figure S4). Further, we identified 846 DMPs (543 hypermethylated, 303 hypomethylated) in leukoplakia and 5111 DMPs (1984 hypermethylated, 3127 hypomethylated) in tumors.
The genes associated with the DMPs were enriched for their involvement in several biological processes. Both leukoplakia and GBC-OSCC shared hypermethylated promoters involved in the regulation of transcription, gene expression, biosynthetic and metabolic processes. The hypomethylated promoters were associated with the development, cell adhesion, and regulation of immune-mediated responses (Additional file 1: Tables S13–S14).
DNA methylation changes in leukoplakia and gingivobuccal complex cancers
The Venn diagram in Additional file 2: Figure S5 (A) depicts DMPs identified by comparison between (1) OPL vs normal, (2) tumor vs normal, and (3) tumor vs OPL. The heatmap of the top 20 hyper and hypomethylated promoters in leukoplakia and OSCC revealed a methylation signature different from normal, including promoters of well-established cancer genes such as PPP1R1C, EID3, DLEC1, HOXA7, CDKN1B, MIR9-1, TSHZ3, FAM84A and CDH4 in leukoplakia. Similarly, hypermethylation of SHISA3, KCNA3, NPY, and hypomethylation of NUMB, BST2, MAPK13 promoters, which have previously been implicated in other cancers, robustly separate normal and GBC-OSCC samples (Fig. 1). Selecting the top 200 DMPs and retaining only protein-coding genes in common between leukoplakia and GBC-OSCC revealed 45 hypermethylated promoters (Additional file 1: Table S15). Hypermethylation of TSGs, such as CDKN1B, NR1H2, ZFP82, and SHISA3, and other novel gene promoters might play important roles during oral carcinogenesis. These shared 45 promoters were also differentially methylated in a separate comparison between oral potentially malignant disorders and early-stage GBC-OSCC suggesting that these are early methylation events and may facilitate early detection of potentially cancerous lesions (Additional file 2: Figure S5(B)). We did not detect any significant DMPs between early- and late-stage GBC-OSCC. Reassuringly, we found substantial overlap between our study and previously published studies (Additional file 1: Table S1) in which established promoter methylation markers such as FGF3, SOX17, RUNX1, WT1 were found to be associated with clinicopathological features.

Heatmap of top 20 hypo- and hypermethylated promoters in leukoplakia and OSCC. A Differentially methylated promoters in OPL (grey) when compared to normal (light grey). B Differentially methylated promoters in early-stage OSCC (grey) and advanced-stage OSCC (dark grey) when compared to normal (light grey). The hypo- and hypermethylated promoters are represented in blue and red, respectively. All methylation values were log-transformed and scaled
Integrative analysis
To find prognostic markers of GBC-OSCC, we performed an integrative analysis of DNA methylation with GE and CNA [6, 7] data using the R-package CNAmet. CNAmet facilitates the identification of putative driver genes, which are synergistically regulated by methylation and CNA. The integrative analysis by CNAmet provided gene lists in four categories summarized in Additional file 1: Table S16. We found 17 genes with hypomethylation or copy number gain and increased expression, including cancer-associated genes such as AGO1, GHR and FAT1, which might be potential oncogenes. Similarly, we identified 57 potential TSGs, of which at least 13 have been implicated in cancer, including HOXB13, ZNF350, ZNF331, CNTNAP2, NEFH, and ABR with reduced expression due to hypermethylation or loss. In addition, we found 21 genes with hypermethylation or gain and three genes with hypomethylation or loss, which are less common and are usually indicative of a methylation change farther from the promoter. These 21 genes with hypermethylation or gain included oncogenes UCHL1, MLLT11, SLC35F2, SEMA6D, and ARHGEF39 and tumor suppressors SOD2, RASSF2, and TRIM13. The hypomethylation or loss category included three genes, none of which are known drivers in cancer.
Survival analysis
We applied regularised Cox regression with L1 penalty (LASSO) on the methylation and CNA data of the genes selected by CNAmet to identify potential survival-associated markers. We found 32 candidate prognostic markers (Table 2). The top predictors based on LASSO estimates included FAT1, JAK3, ARHGEF39, CD1C, HOXB13, GLDC, and TRIM13. For most genes in Table 2, exactly one of the methylation changes or the CNA is associated with a change in survival; the three genes for which both changes are associated with survival are FEZ2, MLLT11, and ZNF350. We validated 32 candidate prognostic markers from integrative analysis using the TCGA_HNSC set from the MethCNA database (Additional file 1: Table S17). We found 87.5% of genes were in agreement with methylation status from our data and TCGA cohort, except for FAT1, GHR, LY75, and MED7. Overall, we observed high concordance between our analysis of our data and the TCGA data strengthening our results.
Validation of candidate marker genes
To validate the results of the integrative analysis, we performed bisulfite pyrosequencing for promoter methylation and TaqMan real-time PCR for CNAs. Three genes were selected based on their role in other cancers, namely HOXB13, GLDC, and FAT1. Pyrosequencing of their gene promoters was performed in independent samples of 127 GBC-OSCCs and 20 healthy controls. The CpG site of interest covered by each sequencing primer is listed in Additional file 1: Table S18. Figure 2A shows the average percentage methylation values for each of the promoters compared to normals. Significant promoter hypermethylation was observed in HOXB13 (P < 0.0001), GLDC (P = 0.036), and hypomethylation was observed in FAT1 (P < 0.0001). Thus, the pyrosequencing data confirmed the findings based on the methylation array data. There was no significant difference in methylation levels between early and late-stage OSCC except for FAT1 (Additional file 2: Figure S6).

Validation of promoter methylation and copy number targets. A Pyrosequencing analysis showing differential promoter methylation between normals and OSCC for HOXB13, GLDC, and FAT1. B, C panel demonstrates the frequency of copy number targets selected from integrative analysis and previously published reports6,7
For validation of CNAs, we performed real-time PCR for five candidate genes from the integrative analysis along with additional genes from previously reported chromosomal locus associated with poor survival [6, 7]. The detailed CNA frequency of each target compared to control DNA and CNAs of the TCGA-HNSCC dataset from the Genome Data Commons (GDC) Data portal are represented in Additional file 1: Table S19 and Fig. 2B, C. The majority of the samples did not show a CNA for the targets from the integrative analysis. The frequencies of copy number gains were 3.1–11% of samples for CST7, GHR, and MLLT11; in addition, 5.5% of samples showed copy number losses for CYB5A. Interestingly, LY75 showed copy number gains (17.3%) instead of the expected losses. The gain/amplification was highest for DVL1 (95.9%) and ANO1 (100%) from the 1p36.32 and 11q13.3 loci, respectively. The genes on the 11q22 amplicon, BIRC2, BIRC3, and YAP1, also showed high copy number gains and amplification in some samples.
We analysed the association between validation targets and clinicopathological parameters Additional file 1: Table S20. We found that NADK gain (BH P-value = 0.044) was associated with the pathological stage. Further, we studied the correlation of biomarkers among themselves Additional file 1: Table S21. We found ISG15 and NADK (R = 0.665) on band 1p36.33 showed moderate correlation. A strong correlation was observed between BIRC2 and BIRC3 (R = 0.778) as well as BIRC2 and YAP1 (R = 0.884) present in the 11q22 locus.
To evaluate the association of biomarkers with clinical outcomes, Kaplan–Meier (KM) survival analysis was performed using the log-rank test. KM plots are provided in Fig. 3A–E. We did not observe a strong correlation between methylation and CNA targets selected from the integrative analysis to clinical outcomes. However, we observed a trend toward poor survival. Univariate Cox regression analysis (Additional file 1: Table S22) performed on validation targets indicated that ISG15 may be associated with better clinical outcomes. A gain in ISG15 was nominally significantly associated with better recurrence-free survival (RFS) [HR (95% CI):0.40 (0.2–0.80), P = 0.010], disease-specific survival (DSS) [HR:0.30 (0.13–0.71), P = 0.006] and overall survival (OS) [HR:0.31 (0.14–0.69), P = 0.004], but not after correction for multiple testing. In addition, CYB5A [HR:3.067 (1.21–7.76), P = 0.018] and CASP4 [HR:3.35(1.32–8.55), P = 0.011] were nominally associated with poor RFS. Multivariable analysis with nominal P-values adjusted for the confounding factors revealed ISG15 was associated with RFS [HR:0.47 (0.23–0.99), P = 0.049], DSS [HR:0.32 (0.13–0.77), P = 0.011] and OS [HR:0.33 (0.15–0.75), P = 0.008], Table 3. The predictive value of ISG15 was determined by the receiver operator characteristic (ROC), which showed an area under the curve (AUC) of 0.711 (95% CI 0.57–0.85, P = 0.007), 0.712 (95% CI:0.57–0.85, P = 0.008) and 0.715 (95% CI 0.58–0.85, P = 0.006) for RFS, DSS, and OS, respectively. The optimal cutoff value determined using Youden’s index for DSS was 60.67 with a sensitivity of 65.4% and a specificity of 80%, Fig. 3F. Further, we stratified the validation data according to node and stage to assess the robustness of prognostic markers. Stratification according to nodal status and stage revealed new prognostic markers Additional file 1: Table S23–S25. Univariate Cox regression in N0 samples showed that BIRC2, BIRC3, along with other markers, were associated with poor RFS, DSS, and OS, suggesting different molecular signatures of N0 and N + tumors.

Kaplan–Meier (KM) plots and receiver operating characteristic (ROC) curve. KM plots for A CYB5A and B CASP4 losses were associated with poor recurrence free survival (RFS). ISG15 gain showed better C RFS, D Disease specific survival (DSS) and E Overall survival (OS). F illustrates the prognostic potential of ISG15 as analysed by a ROC curve
Discussion
We have presented a comprehensive study that provides genome-wide methylation profiles associated with oral premalignant lesions and gingivobuccal complex cancers. We report a methylation signature of leukoplakia and GBC-OSCC, which can be used for identifying high-risk precancerous lesions having the potential of malignant transformation. Another feature of the study is that we integrated methylation data with genomic copy number data and transcriptomic data to identify 32 genes with prognostic significance synergistically regulated by copy number and methylation. We report previously unrecognized differential methylation of FAT1, HOXB13, and GLDC in GBC-OSCC tissues. Further, validation experiments revealed that losses of CYB5A and CASP4 were associated with poor RFS, while gain of ISG15 was identified as a potential prognostic marker for better RFS, DSS, and OS.
We performed genome-wide DNA methylation analysis that reported 846 (303 hypomethylated, 543 hypermethylated) and 5111 (3127 hypomethylated, 1984 hypermethylated) DMPs in leukoplakia and GBC-OSCCs, respectively. The methylation profile of leukoplakia and tumors are different from normals, and methylation aberrations increase as the lesion progresses. The top 200 DMPs common between leukoplakia and tumor and retaining only protein-coding genes revealed 45 hypermethylated promoters. In our literature search, we found consistent previous reports regarding 21 of these 45 genes (Additional file 1: Table S15). Among the genes adjacent to these promoters, CDKN1B, ZFP82, SHISA3, GPX7, and IRF8 are known TSGs [21,22,23,24,25]. The hypermethylation of KCNA3, IRF8, and ZNF529 was also reported by Foy et al. [26] in leukoplakia transforming into OSCC, implying important early events which might play a role in disease progression. Hence, these markers can be helpful in stratifying potentially malignant lesions and need to be further investigated. Nevertheless, there is a significant overlap between our findings and previously published reports on methylation in OSCC (Additional file 1: Table S1); many of these differentially methylated genes were reported in an extensive review by Flausino et al. [27].
The transformation of leukoplakia to malignant carcinoma involves the accumulation of genomic and epigenomic alterations [28,29,30,31,32]. The recurrent CNA (gains and losses) and aberrant methylation (hypo- and hypermethylation) have been linked to apoptosis evasion, metastasis, and therapy resistance [33, 34]. Hence, to search for prognostic biomarkers, we integrated DNA methylation, GE, and CNA and identified 32 candidate marker genes with potential prognostic value in gingivobuccal complex cancers. Hypomethylation and amplification-dependent gene upregulation were observed for seven potential oncogenes associated with poor survival. Of these seven genes, hypomethylation of FAT1 and GHR was also observed in leukoplakia, indicating early oncogenic events. FAT1 regulates cell–cell contact and acts as a tumor suppressor or oncogene in a context-dependent manner [35]. Multiple studies reported mutation, deletion, and hypermethylation of FAT1 in OSCC [36,37,38,39]. Conversely, upregulation has been linked with cell migration and invasion through β-catenin localisation in oral cancers [40]. A recent study in OSCC reported that FAT1 may act as an oncogene, and its overexpression was associated with poor prognosis affecting proliferation, migration, and apoptosis [41]. Here, for the first time, we report that FAT1 is regulated by promoter hypomethylation that could lead to its overexpression, supporting its oncogenic role in OSCC. The growth hormone receptor (GHR) is overexpressed in many solid tumors [42,43,44] and promotes tumor proliferation, progression, and metastasis in breast cancer via the BRAF/MEK/ERK pathway [44]. We observed a GHR copy number gain in 10% of OSCC tissues. It has been suggested that the knockdown of FAT1 and GHR enhances the sensitivity toward drugs, making them therapeutic drug targets [45,46,47]. Other genes from this category are CST7, CD1C, AGO1, CLDN12, and FEZ2. CST7 and CD1C are involved in immune modulation [48, 49]. Hypomethylation or gain of CST7 has not been reported in OSCC but has been proposed as a prognostic biomarker for other carcinomas [50]. Previously, upregulation of AGO1 and CLDN12 has been linked to progression and metastasis [51, 52]. Both hypomethylation and gain of FEZ2 were associated with poor survival, consistent with a report by Yang et al. [53] in pancreatic ductal adenocarcinoma (PDAC). Overall, the CNA and methylation status of these genes can serve as a guide for the identification and design of therapeutic drug targets.
In the hyperloss category, we identified 13 potential TSGs linked with poor clinical outcome. We found 4 genes (LY75, ZNF83, ZNF160, ZNF331) common in leukoplakia and OSCC, while 9 genes appeared only in OSCC, suggesting their role in disease advancement. Among these, HOXB13 (involved in skin development) and GLDC (a component of the glycine cleavage system) are both known TSGs that are epigenetically silenced in many cancers [54,55,56] and promote apoptosis and autophagy [57, 58], respectively. However, no literature evidence is available for the methylation status of these genes in the context of oral cancer. We found HOXB13 and GLDC to be novel TSGs in OSCC, inactivated through promoter hypermethylation. Further, we validated CNAs of CYB5A and LY75. CYB5A (18q22.3) is a membrane-bound mitochondrial hemoprotein involved in regulating cellular redox balance [59]. The significant downregulation of CYB5A has been reported in cancers [60, 61]; however, the mechanism underlying the antitumor effect of CYB5A has not been explored much. CYB5Ais reported to be deleted, induces autophagy-mediated cell death in PDAC [62]. Consistent with this report in PDAC, we found loss of CYB5A to be associated with shortened patient survival. LY75 encodes a mannose receptor involved in the immune and inflammatory response [63]. Recently, Mehdi et al. [64] showed hypomethylation and overexpression of LY75 regulate EMT phenotype and metastatic potential in ovarian cancer. Interestingly, we observed copy number gains in LY75 rather than losses. Another significant gene in this category, JAK3, is involved in the JAK-STAT signalling pathway, reported to be methylated in bladder cancer [65]. Lastly, we also reported a cluster of zinc finger proteins (ZNF350, ZNF331, ZNF160, ZNF83) located on 19q13.41, which belong to a Krüppel-type family of transcriptional repressors, and have previously been shown to be epigenetically silenced and downregulated in multiple cancers [66, 67] including head and neck cancers [68]. The remaining genes in the hyperloss category are ECHDC3, KRTCAP3, LURAP1, and MAMDC4, none of which are known drivers in cancer.
In the hypergain category, we identified seven genes. Interesting candidates include MLLT11 and TRIM13, whose CNAs or differential methylation status have not been reported in OSCC. MLLT11 (1q21.3) is an oncogene dysregulated in some acute myeloid leukemia patients with t(1;11)(q21;q23) translocation and also facilitates the progression of solid tumors [69, 70]. Recurrent gains of the 1q21.3 region and upregulation of MLLT11 were reported in the Wilms tumor [71]. We report MLLT11 CNA in 11% of OSCC samples. TRIM13 is a member of the tripartite motif (TRIM) family downregulated in multiple neoplasms [72,73,74] and acts as TSG by inducing apoptosis [73]. MLLT11 has been shown to play a dual function in malignancy, including both promotion and inhibition of cancer progression, and therefore may be relevant in OSCC prognosis and management. While MLLT11 acts as an oncogene, TRIM13 acts as a TSG; hence genes from the hypergain category are enigmatic and can be interpreted only with the aid of additional functional studies. Lastly, in the hypoloss category, we did not find any literature evidence for MED7 and TAAR5.
Numerous studies have identified prognostic factors such as tumor size, cancer stage, surgical margins, nodal involvement, grade, perineural invasion, lymphovascular invasion, and extranodal extension associated with disease-free survival and overall survival [75,76,77,78,79]. Previously, recurrent focal alterations in OSCC were found to be associated with poor survival; however, the prognostic significance of CNAs at the single-gene level remains to be elucidated [6, 7]. Hence, in search of novel biomarkers, we validated important targets from previous studies and observed that CNA status differs for each gene even though the nearby genes are present on the same amplicon. In this study, copy number loss in CASP4, and gain in ISG15 were associated with survival. Amplification of the 1p36.33 locus was found in progressive leukoplakia [30] as well as in OSCC [7]. Particularly, Interferon Stimulated Gene 15 (ISG15) present on 1p36.33 locus encodes a ubiquitin-like protein associated with antiviral response and regulation of key cellular processes [80] and has prognostic significance in multiple cancers [81,82,83]. The ubiquitin domains in ISG15 make it possible for ISG15 to be conjugated to some other proteins via lysine residues. The conjugated ISG15 (ISGylation) has pro-tumorigenic activity, while the extracellular free form has anti-tumorigenic activity in breast cancer [84] and is being considered as a tumor-associated antigen for cancer immunotherapy [85]. In OSCC, ISG15 was reported to be upregulated and to affect migration and lymph node metastasis [86]. Interestingly, for the first time, we report that a gain of ISG15 is associated with better relapse-free, disease-specific, and overall survival, making it a potential prognostic marker of OSCC suitable for further validation. In our study, loss of CASP4 and the tumor suppressor ATM on 11q22.3 was associated with lymph node involvement. CASP4 is a component of non-canonical inflammasome that induces pyroptosis [87]. Previous studies have demonstrated that higher expression of CASP4 can serve as a prognostic marker in glioma and renal cell carcinoma [88, 89]. Alternatively, few studies have reported the loss and downregulation of CASP4 was associated with poor prognosis in head and neck squamous cell carcinoma [90,91,92]. Similar to these findings, we also observed the loss of CASP4 was associated with poor RFS.
Although the data presented here indicate the interplay between CNA, GE, and methylation, there are some limitations. The CNAmet software may have missed genes that are not regulated synergistically, co-expressed, or co-methylated. This study underlines the need for functional validation of such datasets in an independent cohort. The clinical outcome of leukoplakia patients was not available; hence they were not included for further validation. Furthermore, while performing genome-wide methylation analysis, it is important to note that the differentially methylated sites and regions reported are sensitive to the choice of |∆β| threshold (Additional file 1: Table S26). Given the reduced spread of \(\Delta \beta\) in DMRs in our study, we use a more relaxed threshold to be more inclusive for the downstream integrative analysis. However, exploring a less permissive threshold and shifting |∆β| threshold from 0.2 to 0.25 for genome-wide CpG sites, we drop two-thirds of the hypermethylated sites. Similarly, moving the |∆β| threshold from 0.1 to 0.25, we retain only 2 and 62 DMPs in leukoplakia and GBC-OSCCs, respectively. While we recover several top established DMPs, including, KCNA3, NPY, SHISA3, ZNF529 from the literature, and ZNF160 from the integrative analysis, a stricter threshold increases the chance of dropping several well-studied and prospective markers. This is exemplified by further moving the |∆β| to 0.5, which resulted in only 9 differentially methylated sites in GBC-OSCCs and no significant regions. Thus, there is a trade-off between sensitivity and specificity. A quantitative study exploring this trade-off could help improve the discovery of prospective markers.
Despite these concerns, the strength of the study relies on the integration of three data types from the same patients, enabling a more comprehensive understanding of the underlying molecular mechanisms in oral carcinogenesis [93]. A key strength of our study design is that we have utilized normal samples from healthy individuals rather than noncancerous tissue adjacent to cancerous tissue from affected individuals to avoid the effects of field cancerization [94]. Another strength is that we used only tumors from GBC sites to avoid heterogeneity since different anatomic sites have different methylation profiles [95]. Lastly, in contrast to many previous studies, our study also validated selected candidates in an independent set of samples, further bolstering our findings.
Conclusion
Overall, our study identified potential candidate genes in oral carcinogenesis, which needs functional validation. The global methylation signature common between leukoplakia and gingivobuccal complex cancers may help in identifying potential malignant lesions. Differential promoter methylation analysis revealed FAT1, GLDC, and HOXB13 to be associated with oral carcinogenesis. Copy number changes in CYB5A, CASP4, and ISG15 were found to be potential predictors of survival. The data provides novel insight into OSCC carcinogenesis and prognosis. This study highlights the importance of integration of data across the genomic, epigenomic, and transcriptomic levels to identify novel genes with prognostic and therapeutic potential in gingivobuccal complex cancer.
Materials and methods
Sample collection and micro-dissection
The institutional ethics committee of Tata Memorial Hospital approved the study (Project No-218 of 2016). Written informed consent was obtained from all study participants. Frozen tissues of treatment-naïve, pathologically diagnosed, and surgically resected gingivobuccal tumor and leukoplakia tissues were obtained from Tata Memorial Hospital. The non-inflamed gingivobuccal tissues obtained from healthy individuals with no previous history of cancer were obtained from Nair Dental College. Cryosectioning, DNA-RNA extraction and HPV detection were performed as described in the Additional file 3: Additional informations. The training set included 74 OSCCs, 22 leukoplakia, and 22 normals, and the validation set consisted of 127 GBC-OSCCs and 20 normals.
DNA methylation profiling and analysis
Genome-wide DNA methylation profiling was performed on 74 GBC-OSCC samples, 22 leukoplakia samples, and 22 normal tissues from healthy individuals using Infinium Methylation EPIC (850 K) BeadChips (Illumina, San Diego, CA, USA). The bisulfite conversion of gDNA and hybridization details are provided in the Additional file 3: Additional informations. The raw files have been submitted to the Gene Expression Omnibus (GEO) with accession number GSE204943.
DNA methylation data (.IDAT files) were processed and analysed using the R Bioconductor packages RnBeads (v 1.12.1) and RnBeads.hg19 (v 1.13.1). The RnBeads pipeline included quality control assessment, preprocessing, normalization, exploratory, and differential methylation analyses. Through quality control, preprocessing, and normalization steps, after filtering SNP-enriched probes, probes with unreliable measurements (Greedycut algorithm), context-specific probes, and other filtering procedures, 820,193 probes were retained across 118 samples. Differential methylation analyses were performed across CpG sites, CpG islands, promoters, and genes. The standard methylation β values are defined as the ratio of methylated to overall probe intensities and summarize the methylation status of each probe. RnBeads also combines data for multiple probes in a CpG island, in a promoter, or in a gene so that each of these types of genomic units gets a single averaged β value in each sample. Combined ranks were calculated based on differences in mean methylation, log ratio in mean methylation, and P-value from limma or t-test. Genome-wide CpG sites, promoters, CpG islands, and genes with adjusted false discovery rate (FDR) P-values < 0.05 and |∆β|> {0.2, 0.1, 0.1, 0.1}, respectively, were reported as significantly differentially methylated. The RnBeads pipeline also performed gene set enrichment analyses.
Integrative analysis
We used the R-package CNAmet [96] to integrate methylation data with previously published CNA (accession numbers-GSE85514, GSE23831) and GE data (accession numbers-GSE85195, GSE23558) [6, 7]. CNA calls were previously done with GISTIC [97]. Since CNAmet requires continuous gene expression data and binary copy number gain, copy number loss, hyper-, and hypomethylation data, all of the equal dimensions, the data were processed as follows. We focused on significant DMPs of protein-coding genes in OSCC and subsequently identified 449 common protein-coding genes with expression, methylation, and CNA data shared across 58 tumor samples. We prepared the four binarized matrices- hypomethylation, hypermethylation, loss, and gain as follows. The β values of the promoters in GBC-OSCC samples were first standardized with respect to the mean and standard deviation of normal samples (z-score). A promoter in a sample was considered to be hypomethylated and assigned a value of 1 if the z-score was less than − 2.576, and 0 otherwise. Similarly, a promoter in a sample was considered to be hypermethylated and assigned a value of 1 if the z-score was greater than 2.576, and 0 otherwise. The chosen cut-offs ensured the inclusion of the most differentially methylated promoters. For the CNA data, a gene in a sample was considered deleted and assigned a value of 1 if the GISTIC value was in {− 1, − 2}, and 0 otherwise. Similarly, a gene in a sample was considered amplified and assigned a value of 1 if the GISTIC value was in {1, 2}, and 0 otherwise. For GE, we used previously published data [6, 7]. Subsequently, we evaluated the effects of all four combinations of methylation and CNA data (hypermethylation with loss, hypomethylation with loss, hypermethylation with gain, and hypomethylation with gain) on gene expression using CNAmet with default parameters. For each combination of methylation and CNA data, CNAmet prioritizes the genes (CNAmet hits) according to a synergistic score, and a corresponding FDR corrected P-value. Literature-supported evidence was obtained by querying CNAmet results on CancerMine [98].
Survival analysis
We retained the top significant genes (FDR corrected P-value < 0.1) that were in agreement with the methylation status from our differential methylation analysis and performed survival analysis on these hits. We used disease-specific survival times recorded in months for 17 dead of disease (DOD events) samples and 53 right-censored tumor samples. For each combination of CNAmet hits, we created a design matrix with stage, sex, age, ꞵ values of the hits, and GISTIC values of the hits. We applied regularised Cox regression with L1 penalty (LASSO) to select prognostic markers associated with survival. We compared our data with the TCGA data for HNSC as represented in the MethCNA database [99]. MethCNA is a publicly available database that integrates genomic and epigenomic data from exactly the same DNA specimen. We particularly compared 32 candidate prognostic hits from integrative analysis to the promoters in head and neck squamous cell carcinoma (TCGA_HNSC). The TCGA_HNSC data includes cancer samples from all the anatomic sites of the head and neck region including the oral cavity, larynx, alveolar ridge, oropharynx, floor of mouth, hypopharynx, oral tongue, and tonsil.
Bisulfite pyrosequencing
The promoter methylations of HOXB13, GLDC, and FAT1 were validated by bisulfite pyrosequencing in an independent 127 GBC-OSCCs and 20 normal samples. In order to cover more CpG sites, we designed two primer sets for each gene targeting CpG locus represented by the 850 K BeadChip. The details of PCR amplification and bisulfite pyrosequencing are given in Additional file 3: Additional informations and Additional file 1: Table S18. The methylation percentages for each CpG locus in controls and GBC-OSCC cases were calculated by the PyroMark Q96ID Software (Qiagen).
Copy number alteration (CNA) analysis for validation
The copy numbers of candidate genes were validated by TaqMan qPCR. The detailed protocol and selected copy number assays are provided in Additional file 3: Additional informations and Additional file 1: Table S27. We chose two copy number assays targeting different regions of the same gene to strengthen the results for targets from the integrative analysis. The copy number was calculated using CopyCaller software v2.1 (Applied Biosystems). The CopyCaller-predicted copy number was used for the analysis, and more than two copies were considered a gain. One copy was considered a partial loss, and zero copies was considered a complete loss.
Statistical and survival analyses for validation
The statistical analysis of the validation set was performed using IBM SPSS v20 software. Mann–Whitney two-tailed tests were applied to compare methylation between tumor and normal. The categorical data are presented as frequency and percentage. We categorise the methylation data as hypermethylated (z-score ≥ 2.576) and hypomethylated (z-score ≤ − 2.576). The possibility of an association between different biomarkers and clinicopathological parameters was tested using the χ2 test and Fisher’s exact test wherever applicable. Correlation between pairs of biomarkers was computed using Spearman rank correlation. Survival was quantified as the number of days between surgery and disease-specific death (DSS), disease recurrence (RFS), or death due to any other cause (OS) and analysed by Kaplan–Meier curves and log-rank tests. The predictive value of ISG15 gain associated with survival was determined by the area under the receiver operating characteristic (ROC) curve, and an optimal cutoff point was determined using the Youden index [100]. The univariate Cox proportional hazard models were fitted for all available variables separately, and a multivariable Cox model was then applied to adjust for the potential confounders age, sex, stage, and nodal status. All tests were performed two-tailed, and a P-value < 0.05 was considered nominally significant. All tests for association between targets and clinical outcomes, as well as for correlations among targets (Additional file 1: Tables S20–S24) were corrected for multiple testing using the Benjamini-Hochberg (BH) method to control the FDR at 0.05.
Availability of data and materials
The dataset supporting the conclusions of this article is available in Gene Expression Omnibus (GEO); https:// www.ncbi.nlm.nih.gov/geo with accession number GSE204943. Token Code to access the GEO submission GSE204943-mnwxqokgdbkzhez.
Abbreviations
- AUC:
-
Area under the curve
- BH:
-
Binyamini–Hochberg (method to correct for multiple testing)
- CNA:
-
Copy number alteration
- DMP:
-
Differentially methylated promoters
- DSS:
-
Disease-specific survival
- FDR:
-
False discovery rate
- GBC:
-
Gingivobuccal complex
- GE:
-
Gene expression
- GEO:
-
Gene expression omnibus
- HPV:
-
Humana papillomavirus
- HR:
-
Hazard ratio
- LASSO:
-
Least absolute shrinkage and selection operator
- OPL:
-
Oral premalignant lesions
- OS:
-
Overall survival
- OSCC:
-
Oral squamous cell carcinoma
- RFS:
-
Recurrence-free survival
- ROC:
-
Receiver operating characteristic
- TSG:
-
Tumor suppressor genes
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Pathak KA, Gupta S, Talole S, Khanna V, Chaturvedi P, Deshpande MS, Pai PS, Chaukar DA, D’Cruz AK. Advanced squamous cell carcinoma of lower gingivobuccal complex: patterns of spread and failure. Head Neck. 2005;27(7):597–602.
Chi AC, Day TA, Neville BW. Oral cavity and oropharyngeal squamous cell carcinoma—an update. CA Cancer J Clin. 2015;65(5):401–21.
Warnakulasuriya S, Ariyawardana A. Malignant transformation of oral leukoplakia: a systematic review of observational studies. J Oral Pathol Med. 2016;45(3):155–66.
Coletta RD, Yeudall WA, Salo T. Grand challenges in oral cancers. Front Oral Health. 2020;1:3.
Ambatipudi S, Gerstung M, Pandey M, Samant T, Patil A, Kane S, Desai RS, Schaffer AA, Beerenwinkel N, Mahimkar MB. Genome-wide expression and copy number analysis identifies driver genes in gingivobuccal cancers. Genes Chromosomes Cancer. 2012;51(2):161–73.
Bhosale PG, Cristea S, Ambatipudi S, Desai RS, Kumar R, Patil A, Kane S, Borges AM, Schaffer AA, Beerenwinkel N, Mahimkar MB. Chromosomal alterations and gene expression changes associated with the progression of leukoplakia to advanced gingivobuccal cancer. Transl Oncol. 2017;10(3):396–409.
Locke WJ, Guanzon D, Ma C, Liew YJ, Duesing KR, Fung KYC, Ross JP. DNA methylation cancer biomarkers: translation to the clinic. Front Genet. 2019;10:1150.
Arantes LM, de Carvalho AC, Melendez ME, Carvalho AL, Goloni-Bertollo EM. Methylation as a biomarker for head and neck cancer. Oral Oncol. 2014;50(6):587–92.
Romanowska K, Sobecka A, Rawluszko-Wieczorek AA, Suchorska WM, Golusinski W. Head and neck squamous cell carcinoma: epigenetic landscape. Diagnostics (Basel). 2020;11:1.
Gazdzicka J, Golabek K, Strzelczyk JK, Ostrowska Z. Epigenetic modifications in head and neck cancer. Biochem Genet. 2020;58(2):213–44.
Zhou C, Shen Z, Ye D, Li Q, Deng H, Liu H, Li J. The association and clinical significance of CDKN2A promoter methylation in head and neck squamous cell carcinoma: a meta-analysis. Cell Physiol Biochem. 2018;50(3):868–82.
Gissi DB, Fabbri VP, Gabusi A, Lenzi J, Morandi L, Melotti S, Asioli S, Tarsitano A, Balbi T, Marchetti C, Montebugnoli L. Pre-operative evaluation of DNA methylation profile in oral squamous cell carcinoma can predict tumor aggressive potential. Int J Mol Sci. 2020;21:18.
Hier J, Vachon O, Bernstein A, Ibrahim I, Mlynarek A, Hier M, Alaoui-Jamali MA, Maschietto M, da Silva SD. Portrait of DNA methylated genes predictive of poor prognosis in head and neck cancer and the implication for targeted therapy. Sci Rep. 2021;11(1):10012.
Kurokawa T, Nakagawa T, Matsusaka K, Fukuyo M, Mima M, Misawa K, Rahmutulla B, Ikeda JI, Hanazawa T, Okamoto Y, Kaneda A. Establishment of epigenetic markers to predict irradiation efficacy against oropharyngeal cancer. Cancer Sci. 2020;111(4):1407–16.
Shen S, Wang G, Shi Q, Zhang R, Zhao Y, Wei Y, Chen F, Christiani DC. Seven-CpG-based prognostic signature coupled with gene expression predicts survival of oral squamous cell carcinoma. Clin Epigenet. 2017;9:88.
Virani S, Bellile E, Bradford CR, Carey TE, Chepeha DB, Colacino JA, Helman JI, McHugh JB, Peterson LA, Sartor MA, Taylor JM, Walline HM, Wolf GT, Rozek LS. NDN and CD1A are novel prognostic methylation markers in patients with head and neck squamous carcinomas. BMC Cancer. 2015;15:825.
Choudhury JH, Ghosh SK. Promoter hypermethylation profiling identifies subtypes of head and neck cancer with distinct viral, environmental, genetic and survival characteristics. PLoS ONE. 2015;10(6): e0129808.
Camuzi D, Simao TA, Dias F, Ribeiro Pinto LF, Soares-Lima SC. Head and neck cancers are not alike when tarred with the same brush: an epigenetic perspective from the cancerization field to prognosis. Cancers (Basel). 2021;13:22.
Bhosale PG, Pandey M, Desai RS, Patil A, Kane S, Prabhash K, Mahimkar MB. Low prevalence of transcriptionally active human papilloma virus in Indian patients with HNSCC and leukoplakia. Oral Surg Oral Med Oral Pathol Oral Radiol. 2016;122(5):609–18.
Bencivenga D, Stampone E, Aulitto A, Tramontano A, Barone C, Negri A, Roberti D, Perrotta S, Della Ragione F, Borriello A. A cancer-associated CDKN1B mutation induces p27 phosphorylation on a novel residue: a new mechanism for tumor suppressor loss-of-function. Mol Oncol. 2021;15(4):915–41.
Ye L, Xiang T, Fan Y, Zhang D, Li L, Zhang C, He X, Xiang Q, Tao Q, Ren G. The 19q13 KRAB Zinc-finger protein ZFP82 suppresses the growth and invasion of esophageal carcinoma cells through inhibiting NF-kappaB transcription and inducing apoptosis. Epigenomics. 2019;11(1):65–80.
Shahzad N, Munir T, Javed M, Tasneem F, Aslam B, Ali M, Mutahir Z, Akhtar Ali M, Umer M, Ahmad M, Farooq K, Hassan U, Mustafa T, Anjum RS, Shakoori AR. SHISA3, an antagonist of the Wnt/beta-catenin signaling, is epigenetically silenced and its ectopic expression suppresses growth in breast cancer. PLoS ONE. 2020;15(7): e0236192.
Chen Z, Hu T, Zhu S, Mukaisho K, El-Rifai W, Peng DF. Glutathione peroxidase 7 suppresses cancer cell growth and is hypermethylated in gastric cancer. Oncotarget. 2017;8(33):54345–56.
Liang J, Lu F, Li B, Liu L, Zeng G, Zhou Q, Chen L. IRF8 induces senescence of lung cancer cells to exert its tumor suppressive function. Cell Cycle. 2019;18(23):3300–12.
Foy JP, Pickering CR, Papadimitrakopoulou VA, Jelinek J, Lin SH, William WN Jr, Frederick MJ, Wang J, Lang W, Feng L, Zhang L, Kim ES, Fan YH, Hong WK, El-Naggar AK, Lee JJ, Myers JN, Issa JP, Lippman SM, Mao L, Saintigny P. New DNA methylation markers and global DNA hypomethylation are associated with oral cancer development. Cancer Prev Res (Phila). 2015;8(11):1027–35.
Flausino CS, Daniel FI, Modolo F. DNA methylation in oral squamous cell carcinoma: from its role in carcinogenesis to potential inhibitor drugs. Crit Rev Oncol Hematol. 2021;164: 103399.
Abe M, Yamashita S, Mori Y, Abe T, Saijo H, Hoshi K, Ushijima T, Takato T. High-risk oral leukoplakia is associated with aberrant promoter methylation of multiple genes. BMC Cancer. 2016;16:350.
Asokan GS, Jeelani S, Gnanasundaram N. Promoter hypermethylation profile of tumour suppressor genes in oral leukoplakia and oral squamous cell carcinoma. J Clin Diagn Res. 2014;8(10):ZC09-12.
Cervigne NK, Machado J, Goswami RS, Sadikovic B, Bradley G, Perez-Ordonez B, Galloni NN, Gilbert R, Gullane P, Irish JC, Jurisica I, Reis PP, Kamel-Reid S. Recurrent genomic alterations in sequential progressive leukoplakia and oral cancer: drivers of oral tumorigenesis? Hum Mol Genet. 2014;23(10):2618–28.
Ghosh A, Das C, Ghose S, Maitra A, Roy B, Majumder PP, Biswas NK. Integrative analysis of genomic and transcriptomic data of normal, tumour, and co-occurring leukoplakia tissue triads drawn from patients with gingivobuccal oral cancer identifies signatures of tumour initiation and progression. J Pathol. 2022;257(5):593–606.
Das D, Maitra A, Panda CK, Ghose S, Roy B, Sarin R, Majumder PP. Genes and pathways monotonically dysregulated during progression from normal through leukoplakia to gingivo-buccal oral cancer. NPJ Genom Med. 2021;6(1):32.
Castilho RM, Squarize CH, Almeida LO. Epigenetic modifications and head and neck cancer: implications for tumor progression and resistance to therapy. Int J Mol Sci. 2017;18:7.
Kou F, Wu L, Ren X, Yang L. Chromosome abnormalities: new insights into their clinical significance in cancer. Mol Ther Oncolytics. 2020;17:562–70.
Chen ZG, Saba NF, Teng Y. The diverse functions of FAT1 in cancer progression: good, bad, or ugly? J Exp Clin Cancer Res. 2022;41(1):248.
India Project Team of the International Cancer Genome C. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat Commun. 2013;4:2873.
Nakaya K, Yamagata HD, Arita N, Nakashiro KI, Nose M, Miki T, Hamakawa H. Identification of homozygous deletions of tumor suppressor gene FAT in oral cancer using CGH-array. Oncogene. 2007;26(36):5300–8.
Faraji F, Ramirez SI, Anguiano Quiroz PY, Mendez-Molina AN, Gutkind JS. Genomic hippo pathway alterations and persistent YAP/TAZ activation: new hallmarks in head and neck cancer. Cells. 2022;11:8.
Lin LH, Chou CH, Cheng HW, Chang KW, Liu CJ. Precise identification of recurrent somatic mutations in oral cancer through whole-exome sequencing using multiple mutation calling pipelines. Front Oncol. 2021;11: 741626.
Nishikawa Y, Miyazaki T, Nakashiro K, Yamagata H, Isokane M, Goda H, Tanaka H, Oka R, Hamakawa H. Human FAT1 cadherin controls cell migration and invasion of oral squamous cell carcinoma through the localization of beta-catenin. Oncol Rep. 2011;26(3):587–92.
Lan T, Ge Q, Zheng K, Huang L, Yan Y, Zheng L, Lu Y, Zheng D. FAT1 upregulates in oral squamous cell carcinoma and promotes cell proliferation via cell cycle and DNA repair. Front Oncol. 2022;12: 870055.
Yan HZ, Wang HF, Yin Y, Zou J, Xiao F, Yi LN, He Y, He BS. GHR is involved in gastric cell growth and apoptosis via PI3K/AKT signalling. J Cell Mol Med. 2021;25(5):2450–8.
Verreault M, Segoviano Vilchis I, Rosenberg S, Lemaire N, Schmitt C, Guehennec J, Royer-Perron L, Thomas JL, Lam TT, Dingli F, Loew D, Ducray F, Paris S, Carpentier C, Marie Y, Laigle-Donadey F, Rousseau A, Pigat N, Boutillon F, Bielle F, Mokhtari K, Frank SJ, de Reynies A, Hoang-Xuan K, Sanson M, Goffin V, Idbaih A. Identification of growth hormone receptor as a relevant target for precision medicine in low-EGFR expressing glioblastoma. Clin Transl Med. 2022;12(7): e939.
Zhu X, Li Y, Xu G, Fu C. Growth hormone receptor promotes breast cancer progression via the BRAF/MEK/ERK signaling pathway. FEBS Open Biol. 2020;10(6):1013–20.
Hsu TN, Huang CM, Huang CS, Huang MS, Yeh CT, Chao TY, Bamodu OA. Targeting FAT1 inhibits carcinogenesis, induces oxidative stress and enhances cisplatin sensitivity through deregulation of LRP5/WNT2/GSS signaling axis in oral squamous cell carcinoma. Cancers (Basel). 2019;11:12.
Gao S, Ni Q, Wu X, Cao T. GHR knockdown enhances the sensitivity of HCC cells to sorafenib. Aging (Albany NY). 2020;12(18):18127–36.
Meng Y, Zhou B, Pei Z, Chen Y, Chang D. The nuclear-localized GHR is involved in the cell proliferation of gastric cancer, and pegvisomant may be an important potential drug to inhibit the proliferation of gastric cancer cells. Biochem Cell Biol. 2022;24:1–11.
Senjor E, Perisic Nanut M, Breznik B, Mitrovic A, Mlakar J, Rotter A, Porcnik A, Lah Turnsek T, Kos J. Cystatin F acts as a mediator of immune suppression in glioblastoma. Cell Oncol (Dordr). 2021;44(5):1051–63.
Kwiecien I, Rutkowska E, Raniszewska A, Rzepecki P, Domagala-Kulawik J. Modulation of the immune response by heterogeneous monocytes and dendritic cells in lung cancer. World J Clin Oncol. 2021;12(11):966–82.
Yang C, Yu T, Liu Z, Ye X, Liao X, Wang X, Han C, Zhu G, Qin W, Peng T. Cystatin F as a key family 2 cystatin subunit and prognostic biomarker for earlystage pancreatic ductal adenocarcinoma. Oncol Rep. 2019;42(1):79–90.
Wang M, Zhang L, Liu Z, Zhou J, Pan Q, Fan J, Zang R, Wang L. AGO1 may influence the prognosis of hepatocellular carcinoma through TGF-beta pathway. Cell Death Dis. 2018;9(3):324.
Kolchakova D, Moten D, Batsalova T, Dzhambazov B. Tight junction protein claudin-12 is involved in cell migration during metastasis. Biomolecules. 2021;11:5.
Yang C, Wang X, Qiu C, Zheng Z, Lin K, Tu M, Zhang K, Jiang K, Gao W. Identification of FEZ2 as a potential oncogene in pancreatic ductal adenocarcinoma. PeerJ. 2022;9: e12736.
Min HL, Kim J, Kim WH, Jang BG, Kim MA. Epigenetic silencing of the putative tumor suppressor gene GLDC (glycine dehydrogenase) in gastric carcinoma. Anticancer Res. 2016;36(1):179–87.
Geng H, Liu G, Hu J, Li J, Wang D, Zou S, Xu X. HOXB13 suppresses proliferation, migration and invasion, and promotes apoptosis of gastric cancer cells through transcriptional activation of VGLL4 to inhibit the involvement of TEAD4 in the Hippo signaling pathway. Mol Med Rep. 2021;24:4.
Sui BQ, Zhang CD, Liu JC, Wang L, Dai DQ. HOXB13 expression and promoter methylation as a candidate biomarker in gastric cancer. Oncol Lett. 2018;15(6):8833–40.
Zhuang H, Wu F, Wei W, Dang Y, Yang B, Ma X, Han F, Li Y. Glycine decarboxylase induces autophagy and is downregulated by miRNA-30d-5p in hepatocellular carcinoma. Cell Death Dis. 2019;10(3):192.
Okuda H, Toyota M, Ishida W, Furihata M, Tsuchiya M, Kamada M, Tokino T, Shuin T. Epigenetic inactivation of the candidate tumor suppressor gene HOXB13 in human renal cell carcinoma. Oncogene. 2006;25(12):1733–42.
Schenkman JB, Jansson I. The many roles of cytochrome b5. Pharmacol Ther. 2003;97(2):139–52.
Fujisawa Y, Sakaguchi K, Ono H, Yamaguchi R, Kato F, Kagami M, Fukami M, Ogata T. Combined steroidogenic characters of fetal adrenal and Leydig cells in childhood adrenocortical carcinoma. J Steroid Biochem Mol Biol. 2016;159:86–93.
Guo H, Liang S, Wang Y, Zhou S, Yin D, Zhang S, Wang J, Wu D, Ma K, Liu Y, Sun L, Ji C, Li X, Zhou H, Yang G, Guo X, Cui T, Li Z, Liu Y, Wang J, Liu L. Cytochrome B5 type A alleviates HCC metastasis via regulating STOML2 related autophagy and promoting sensitivity to ruxolitinib. Cell Death Dis. 2022;13(7):623.
Giovannetti E, Wang Q, Avan A, Funel N, Lagerweij T, Lee JH, Caretti V, van der Velde A, Boggi U, Wang Y, Vasile E, Peters GJ, Wurdinger T, Giaccone G. Role of CYB5A in pancreatic cancer prognosis and autophagy modulation. J Natl Cancer Inst. 2014;106(1):346.
Martinek J, Lin J, Kim KI, Wang VG, Wu TC, Chiorazzi M, Boruchov H, Gulati A, Seeniraj S, Sun L, Marches F, Robson P, Rongvaux A, Flavell RA, George J, Chuang JH, Banchereau J, Palucka K. Transcriptional profiling of macrophages in situ in metastatic melanoma reveals localization-dependent phenotypes and function. Cell Rep Med. 2022;3(5): 100621.
Mehdi S, Bachvarova M, Scott-Boyer MP, Droit A, Bachvarov D. LY75 ablation mediates mesenchymal-epithelial transition (MET) in epithelial ovarian cancer (EOC) cells associated with DNA methylation alterations and suppression of the Wnt/beta-catenin pathway. Int J Mol Sci. 2020;21:5.
Lopez JI, Angulo JC, Martin A, Sanchez-Chapado M, Gonzalez-Corpas A, Colas B, Ropero S. A DNA hypermethylation profile reveals new potential biomarkers for the evaluation of prognosis in urothelial bladder cancer. APMIS. 2017;125(9):787–96.
Tanaka H, Kuwano Y, Nishikawa T, Rokutan K, Nishida K. ZNF350 promoter methylation accelerates colon cancer cell migration. Oncotarget. 2018;9(95):36750–69.
Nie C, Han X, Wei R, Leonteva A, Hong J, Du X, Wang J, Zhu L, Zhao Y, Xue Y, Zhou H, Tian W. Association of ZNF331 and WIF1 methylation in peripheral blood leukocytes with the risk and prognosis of gastric cancer. BMC Cancer. 2021;21(1):551.
Gaykalova DA, Vatapalli R, Wei Y, Tsai HL, Wang H, Zhang C, Hennessey PT, Guo T, Tan M, Li R, Ahn J, Khan Z, Westra WH, Bishop JA, Zaboli D, Koch WM, Khan T, Ochs MF, Califano JA. Outlier analysis defines zinc finger gene family DNA methylation in tumors and saliva of head and neck cancer patients. PLoS ONE. 2015;10(11): e0142148.
Hu J, Li G, Liu L, Wang Y, Li X, Gong J. AF1q mediates tumor progression in colorectal cancer by regulating AKT signaling. Int J Mol Sci. 2017;18:5.
Tiberio P, Lozneanu L, Angeloni V, Cavadini E, Pinciroli P, Callari M, Carcangiu ML, Lorusso D, Raspagliesi F, Pala V, Daidone MG, Appierto V. Involvement of AF1q/MLLT11 in the progression of ovarian cancer. Oncotarget. 2017;8(14):23246–64.
Krepischi ACV, Maschietto M, Ferreira EN, Silva AG, Costa SS, da Cunha IW, Barros BDF, Grundy PE, Rosenberg C, Carraro DM. Genomic imbalances pinpoint potential oncogenes and tumor suppressors in Wilms tumors. Mol Cytogenet. 2016;9:20.
Li H, Qu L, Zhou R, Wu Y, Zhou S, Zhang Y, Cheng B, Ni J, Huang H, Hou J. TRIM13 inhibits cell migration and invasion in clear-cell renal cell carcinoma. Nutr Cancer. 2020;72(7):1115–24.
Xu L, Wu Q, Zhou X, Wu Q, Fang M. TRIM13 inhibited cell proliferation and induced cell apoptosis by regulating NF-kappaB pathway in non-small-cell lung carcinoma cells. Gene. 2019;715: 144015.
Chen WX, Cheng L, Xu LY, Qian Q, Zhu YL. Bioinformatics analysis of prognostic value of TRIM13 gene in breast cancer. Biosci Rep. 2019;39:3.
Boonpoapichart S, Punyavong P, Jenwitheesuk K, Surakunprapha P, Winaikosol K. Significant prognostic factors influencing the survival difference of oral tongue squamous cell carcinoma. Plast Reconstr Surg Glob Open. 2021;9(10): e3889.
Dolens EDS, de Morais EF, Paranaiba LMR, Rangel A, Almangush A, Salo T, Brennan PA, Coletta RD. Prognostic significance of the neural invasion in oral squamous cell carcinoma. J Oral Pathol Med. 2023.
Huang S, Zhu Y, Cai H, Zhang Y, Hou J. Impact of lymphovascular invasion in oral squamous cell carcinoma: a meta-analysis. Oral Surg Oral Med Oral Pathol Oral Radiol. 2021;131(3):319–28.
Suresh GM, Koppad R, Prakash BV, Sabitha KS, Dhara PS. Prognostic indicators of oral squamous cell carcinoma. Ann Maxillofac Surg. 2019;9(2):364–70.
Thomas B, Stedman M, Davies L. Grade as a prognostic factor in oral squamous cell carcinoma: a population-based analysis of the data. Laryngoscope. 2014;124(3):688–94.
Sandy Z, da Costa IC, Schmidt CK. More than meets the ISG15: emerging roles in the DNA damage response and beyond. Biomolecules. 2020;10:11.
Chen RH, Du Y, Han P, Wang HB, Liang FY, Feng GK, Zhou AJ, Cai MY, Zhong Q, Zeng MS, Huang XM. ISG15 predicts poor prognosis and promotes cancer stem cell phenotype in nasopharyngeal carcinoma. Oncotarget. 2016;7(13):16910–22.
Qu T, Zhang W, Qi L, Cao L, Liu C, Huang Q, Li G, Li L, Wang Y, Guo Q, Guo Y, Ren D, Gao Y, Wang J, Meng B, Zhang B, Cao W. ISG15 induces ESRP1 to inhibit lung adenocarcinoma progression. Cell Death Dis. 2020;11(7):511.
Kariri YA, Alsaleem M, Joseph C, Alsaeed S, Aljohani A, Shiino S, Mohammed OJ, Toss MS, Green AR, Rakha EA. The prognostic significance of interferon-stimulated gene 15 (ISG15) in invasive breast cancer. Breast Cancer Res Treat. 2021;185(2):293–305.
Tecalco-Cruz AC, Ramirez-Jarquin JO, Cruz-Ramos E. Regulation and action of interferon-stimulated gene 15 in breast cancer cells. Hum Cell. 2020;33(4):954–62.
Wood LM, Pan ZK, Seavey MM, Muthukumaran G, Paterson Y. The ubiquitin-like protein, ISG15, is a novel tumor-associated antigen for cancer immunotherapy. Cancer Immunol Immunother. 2012;61(5):689–700.
Chen YL, Wu WL, Jang CW, Yen YC, Wang SH, Tsai FY, Shen YY, Chen YW. Interferon-stimulated gene 15 modulates cell migration by interacting with Rac1 and contributes to lymph node metastasis of oral squamous cell carcinoma cells. Oncogene. 2019;38(23):4480–95.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–5.
Tian G, Li Q, Niu L, Luo Y, Wang H, Kang W, Fang X, Bai S, Yuan G, Pan Y. CASP4 can be a diagnostic biomarker and correlated with immune infiltrates in gliomas. Front Oncol. 2022;12:1025065.
Li T, Liu N, Zhang G, Chen M. CASP4 and CASP8 as newly defined autophagy-pyroptosis-related genes associated with clinical and prognostic features of renal cell carcinoma. J Cancer Res Ther. 2022;18(7):1952–60.
Shibamoto M, Hirata H, Eguchi H, Sawada G, Sakai N, Kajiyama Y, Mimori K. The loss of CASP4 expression is associated with poor prognosis in esophageal squamous cell carcinoma. Oncol Lett. 2017;13(3):1761–6.
Li H, Wang J, Zeng Z, Fu X, Zhang W. Expression and correlation of apoptosis-related gene c-IAP2 and caspase-4 in head and cervical undifferentiation squamous cell carcinoma. Lin Chuang Er Bi Yan Hou Ke Za Zhi. 2003;17(12):739–41.
Li H, Zeng ZH, Wang LH, Wang JQ. Expression and correlation of apoptosis-related gene c-IAP2 and caspase-4 in sinonasal squamous carcinoma. Zhonghua Er Bi Yan Hou Ke Za Zhi. 2004;39(6):324–7.
Serafini MS, Lopez-Perez L, Fico G, Licitra L, De Cecco L, Resteghini C. Transcriptomics and epigenomics in head and neck cancer: available repositories and molecular signatures. Cancers Head Neck. 2020;5:2.
Sorroche BP, Talukdar FR, Lima SCS, Melendez ME, de Carvalho AC, de Almeida GC, De Marchi P, Lopes M, Ribeiro Pinto LF, Carvalho AL, Herceg Z, Arantes L. DNA methylation markers from negative surgical margins can predict recurrence of oral squamous cell carcinoma. Cancers (Basel). 2021;13:12.
Soares-Lima SC, Mehanna H, Camuzi D, de Souza-Santos PT, Simao TA, Nicolau-Neto P, Almeida Lopes MS, Cuenin C, Talukdar FR, Batis N, Costa I, Dias F, Degli Esposti D, Boroni M, Herceg Z, Ribeiro Pinto LF. Upper aerodigestive tract squamous cell carcinomas show distinct overall DNA methylation profiles and different molecular mechanisms behind WNT signaling disruption. Cancers (Basel). 2021;13:12.
Louhimo R, Hautaniemi S. CNAmet: an R package for integrating copy number, methylation and expression data. Bioinformatics. 2011;27(6):887–8.
Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41.
Lever J, Zhao EY, Grewal J, Jones MR, Jones SJM. CancerMine: a literature-mined resource for drivers, oncogenes and tumor suppressors in cancer. Nat Methods. 2019;16(6):505–7.
Deng G, Yang J, Zhang Q, Xiao ZX, Cai H. MethCNA: a database for integrating genomic and epigenomic data in human cancer. BMC Genom. 2018;19(1):138.
Youden WJ. Index for rating diagnostic tests. Cancer. 1950;3(1):32–5.
Acknowledgements
The authors thank all participants of the study. ICMR National Tumour Tissue Repository, Tata Memorial Centre; ACTREC Biorepository and Department of Pathology, Tata Memorial Centre is acknowledged for providing tumor tissues for analysis. The authors thank Dr. Rajiv S. Desai (Department of Oral Pathology & Microbiology, Nair Hospital Dental College, Mumbai) for providing control tissue samples. We are grateful to Dr. Simona Cristea for her help in identifying the targets. We thank Dr. Nafisa H. Balasinor and Mamata Datar Alavani, ICMR-National Institute for Research in Reproductive and Child Health (NIRRCH), for providing the pyrosequencing facility and help in the experiments. We thank Sadhana Kannan, Jitendra Gawde, and Subham Kumar Mohanta for their help in statistical analysis.
Funding
This work was supported by a grant from the Terry Fox International Foundation and intramural support from Tata Memorial Centre. This research was supported in part by the Intramural Research Program of the National Institutes of Health, NLM, and NCI. Part of this study was supported by SystemsX.ch, the Swiss Initiative in Systems Biology, under Grant No. RTD 2013/152, evaluated by the Swiss National Science Foundation.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: MI, MBM. Performed the experiments: MI, SA, PGB. Analyzed the data: MI, SS, AAS, NB, MBM. Contributed reagents/materials/analysis tools: MBM, NB, AAS. Wrote the paper: MI, SS, AAS, NB, MBM. Assessment of clinical annotation, histopathological evaluation, and IHC grading: AP. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Institute Ethics committee (IEC) of Tata Memorial Centre, Project number 218 of 2016. Written informed consent was obtained from all the study participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table S1
: Literature of genome wide DNA methylation analysis in OSCC. Table-S2: Detailed demographic and clinicopathological characteristics of the leukoplakia samples. Table S3: Detailed demographic clinicopathological and characteristics of the OSCC samples. Table S4: Summary of genomewide differentially methylated regions. Table S5: Top 20 differentially methylated CpG sites identified across the leukoplakia samples. Table S6: Top 20 differentially methylated CpG sites identified across the OSCC samples. Table S7: Top 20 differentially methylated CpG islands identified across the leukoplakia samples. Table S8: Top 20 differentially methylated CpG islands identified across the OSCC samples. Table S9: Top 20 differentially hypermethylated and hypomethylated promoters identified across the leukoplakia samples. Table S10: Top 20 differentially hypermethylated and hypomethylated promoters identified across the OSCC samples. Table S11: Top 20 differentially hypermethylated and hypomethylated genes identified across the leukoplakia samples. Table S12: Top 20 differentially hypermethylated and hypomethylated genes identified across the OSCC samples. Table S13: Gene enrichment analysis of top 100 differentially methylated promoters in leukoplakia based on combined rank. Table S14: Gene enrichment analysis of top 100 differentially methylated promoters in OSCC based on combined rank. Table S15: List of differentially methylated promoters common between leukoplakia and OSCC. Table S16: Candidate target gene list obtained from integrative analysis of copy number, gene expression, and DNA methylation data using CNAmet. Table S17: Comparison of our data with TCGA_HNSC cohort. Table S18: Details for pyrosequencing primers. Table S19: Copy number alteration distribution among candidate genes. Table S20: Association between biomarkers and clinicopathological parameters. Table S21: Correlation among different targets. Table S22: Univariate Cox analysis of the association between markers and clinical outcome. Table S23: Univariate Cox analysis of the association between markers and clinical outcome in N0 and N+ group. Table S24: Univariate Cox analysis of the association between markers and clinical outcome in early-stage and advanced-stage groups. Table S25: Prognostic significance of biomarkers based on nodal status and stage of the disease. Table S26: Summary of genomewide differentially methylated regions as a function of deltathreshold. Table S27: Details for TaqMan qPCR CNV assays.
Additional file 2. Figure S1
: Principal component analysisof methylation sites in the promoter region. Figure S2: Distribution of differential methylation at CpG sites in leukoplakia and OSCC. Figure S3: Volcano plots of −log10against the average methylation differenceshowing differentially hypo and hyper methylated promoters in OPL vs normal, tumor vs normal and tumor vs OPL. Figure S4: Distribution of differentially methylated promoters. Figure S5: Venn diagram. Figure S6: Boxplots showing differential promoter methylation between early and advanced-stage OSCC.
Additional file 3
. Additional information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Inchanalkar, M., Srivatsa, S., Ambatipudi, S. et al. Genome-wide DNA methylation profiling of HPV-negative leukoplakia and gingivobuccal complex cancers. Clin Epigenet 15, 93 (2023). https://doi.org/10.1186/s13148-023-01510-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-023-01510-z