
Abstract
Background
DNA methylation has started a recent revolution in genomics biology by identifying key biomarkers for multiple cancers, including oral squamous cell carcinoma (OSCC), the most common head and neck squamous cell carcinoma.
Methods
A multi-stage screening strategy was used to identify DNA-methylation-based signatures for OSCC prognosis. We used The Cancer Genome Atlas (TCGA) data as training set which were validated in two independent datasets from Gene Expression Omnibus (GEO). The correlation between DNA methylation and corresponding gene expression and the prognostic value of the gene expression were explored as well.
Results
The seven DNA methylation CpG sites were identified which were significantly associated with OSCC overall survival. Prognostic signature, a weighted linear combination of the seven CpG sites, successfully distinguished the overall survival of OSCC patients and had a moderate predictive ability for survival [training set: hazard ratio (HR) = 3.23, P = 5.52 × 10−10, area under the curve (AUC) = 0.76; validation set 1: HR = 2.79, P = 0.010, AUC = 0.67; validation set 2: HR = 3.69, P = 0.011, AUC = 0.66]. Stratification analysis by human papillomavirus status, clinical stage, age, gender, smoking status, and grade retained statistical significance. Expression of genes corresponding to candidate CpG sites (AJAP1, SHANK2, FOXA2, MT1A, ZNF570, HOXC4, and HOXB4) was also significantly associated with patient’s survival. Signature integrating of DNA methylation, gene expression, and clinical information showed a superior ability for prognostic prediction (AUC = 0.78).
Conclusion
Prognostic signature integrated of DNA methylation, gene expression, and clinical information provides a better prognostic prediction value for OSCC patients than that with clinical information only.
Similar content being viewed by others


Background
Oral squamous cell carcinoma (OSCC) is the most common head and neck squamous cell carcinoma (HNSCC), affecting approximately 48,000 individuals and causing 9500 deaths in the USA in 2016 [1]. The overall 5-year survival rate for OSCC is around 60% [2] and has only improved modestly over the past two decades despite considerable improvements in the treatment of OSCC [3, 4]. This can be attributed to limited understanding of OSCC carcinogenesis, development, progression, invasion, and metastasis [5], which sharply delays early diagnosis. Therefore, identification of molecular changes in significant oncogenes or tumor suppressor genes associated with OSCC will help improve survival prediction and early treatment [6, 7].
Epigenetic changes are inheritable and reversible, affecting the spatial conformation of DNA and its transcriptional activity [8]. DNA methylation changes may influence gene expression and interact with various positive and negative feedback mechanisms [9]. Therefore, aberrant methylation CpG sites have been considered potential prognostic factors not only in OSCC [10] but also in other cancers as well [11,12,13].
Previous studies have reported survival-related OSCC biomarkers at different omics levels, including somatic mutations [14], gene expression [15], miRNAs [16], and proteins [17]. Methylation markers have also been reported [18, 19]. However, these studies have relatively small sample sizes and are limited to a single epigenetic level. Therefore, more attention should be given to the relationship between methylation and gene expression [20].
In this study, we investigated the prognostic value of methylation biomarkers for OSCC overall survival. We generated a prognostic model using data from The Cancer Genome Atlas (TCGA), which are now continually hosted at the Genomics Data Commons (GDC), and validated our classifier using two independent external validation sets from Gene Expression Omnibus (GEO).
Methods
Study population
The training set including 313 OSCC cases were downloaded from the TCGA data portal accessed on March, 2016. Tumor sites of oral cavity, oral tongue, buccal mucosa, lip, alveolar ridge, hard palate, and floor of mouth were included. Patients were diagnosed during 1992–2013, and those with missing follow-up information were excluded. Of them, 32 OSCC patients had both tumor and adjacent non-tumor tissue samples, which was used as the discovery set to identify differential methylation CpG sites.
Clinical and DNA methylation data for validation set 1 and set 2 were obtained from GEO [accession number: GSE52793 [19] and GSE75537 [21]]. One sample in the validation set 2 were removed due to missing survival information. Clinical information was described in Table 1.
Preprocessing of DNA methylation chip data
Genome-wide DNA methylation of the training set was profiled using Illumina Infinium HumanMethylation450 BeadChips Assay. Raw data (level 1 data from TCGA) were processed using R package minfi version 1.20.0 [22]. Background subtraction, quantile normalization, and quality control were performed subsequently. Low-quality probes were removed if they met the following criteria: (i) failed detection (P > 0.05) in ≥ 5% samples; (ii) coefficient of variance (CV) < 5%; (iii) methylated or unmethylated in all samples; (iv) single-nucleotide polymorphisms (SNPs) located in the assayed CpG dinucleotide [23]; and (v) did not map uniquely to the human reference genome (hg19) [24] or were on sex chromosomes [25]. Samples with > 5% undetectable probes also were excluded. BMIQ normalization was used for further type I and II probe correction [26].
DNA methylation data for validation sets were already normalized [19]. Quantile normalization was used to standardize all sample distributions.
Further, ComBat [27] was used to adjust batch effects among the three datasets using R package sva.
HPV status collection
Human papillomavirus (HPV) status of the training set was based on the molecular classification, with tumor samples having more than 1000 reads from RNA sequencing aligned to HPV sequences, or with evidence of genomic integrated HPV DNA, deemed HPV-positive [28]. HPV status of GSE75537 set was based on the evidence of HPV DNA. Due to the relative high missing rate, we used multivariate imputation by chained equations (MICE) to ensure the statistical power [29].
Preprocessing of gene expression data
Level 3 transcriptomic data of the training set were normalized by RSEM method [30]. All gene expression values were logarithmic transformed to approximate data to a normal distribution and then quantile normalized.
Sure independence screening method
The high-dimensional microarray data (> 450,000 probes) in contrast to the small number of cases (< 320) easily leads to overfitting [31]. Regularized penalized models such as LASSO can be used to identify important variables with non-zero coefficients [32]. In this study, sure independence screening (SIS) was used based on LASSO Cox penalized regression to identify candidate CpG sites and to construct a multi-CpG-based classifier for predicting overall survival [33]. This two-stage variable screening method is more stable and reliable and was performed with R package SIS.
Statistical analysis
Continuous variables were summarized as median value (range), and categorized variables were described by frequency (n) and proportion (%). Chi-square test was used for rate comparison. Volcano plot analysis was used to select CpG sites based on differential methylation value calculated as mean (βtumor) − mean (βnormal), combined with paired Student’s t test P values. We used Spearman’s rank correlation (r s ) to explore relationships between methylation and gene expression. Associations between characteristics and overall survival were evaluated by Cox proportional hazard models, while hazard ratio (HR) and 95% confidence interval (95% CI) were described as per 1% methylation increment.
Kaplan-Meier survival curves were drawn and compared among subgroups using log-rank tests. We predicted overall survival using the nearest neighbor method for receiver operating characteristic (ROC) curves of censored survival data [34]. Estimations of confidence intervals and P values of area under the curve (AUC) were based on bootstrap resampling.
VanderWeele’s mediation analysis was used to explore whether the prognostic effect of seven DNA methylation sites is mediated by affecting corresponding mRNA expression [35]. Total effect of methylation on survival (HRtotal) was decomposed to indirect effect (HRindirect) representing the effect of methylation mediated through affecting gene expression and direct effect (HRdirect) indicating the effect of methylation through mediators rather than regulating the expression.
Statistical analyses were performed using R version 3.3.0 (The R Foundation). P values were two-sided, and P < 0.05 was considered statistically significant.
Results
Candidate CpG sites
First, genome-wide differential methylation was identified from the discovery set of 32 OSCC patients which had both tumor and adjacent non-tumor tissues (Figs. 1 and 2a). The 1490 CpG sites with an absolute differential methylation of > 0.4 and paired t test P value of < 1 × 10−7 were identified (Fig. 2b).

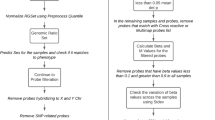
Flow chart indicating study design. We identified candidate CpG sites from 32 paired OSCC and adjacent non-tumor tissues by methylation 450k assay in the discovery set. Then, we excluded a large proportion of CpG sites that were unrelated to survival and developed prognostic scores by SIS. The seven-CpG-based classifier was validated in two independent datasets. Relationships between methylation and gene expression were also analyzed in the training dataset

Construction of the seven-CpG-based classifier. a Circos plot of epigenome-wide DNA methylation CpG sites. Results are presented as P values ordered by genomic position, including paired t test of the discovery set (green and red symbols) and univariable Cox regression analysis of the training set (orange and blue symbols). b Volcano plot comparing CpG methylation for OSCC tumor and non-tumor tissues. A total of 1490 CpG sites had an absolute value of differential methylation of > 0.4 and a paired t test P value of < 1 × 10−7 (blue dots). c Heatmap showing methylation of 15 CpG sites in tumor tissues and adjacent non-tumor tissues. d Coefficients of CpG sites calculated by univariate Cox regression and sure independence screening (SIS). After SIS selection, seven probes remained non-zero coefficients
Second, univariate Cox regression was used to evaluate their association with overall survival in the training set, which identified 15 CpG sites with P < 0.05. Further, SIS analysis was performed to further screen out a stable probe combination. Seven of the 15 candidate CpGs were identified, including cg13495205, cg07110405, cg03774514, cg09137696, cg19655456, cg03146625, and cg21546671 (Fig. 2c, Additional file 1: Table S1), mapped to AJAP1, SHANK2, FOXA2, MT1A, ZNF570, HOXC4, and HOXB4, respectively. Using coefficients generated from Cox model, we calculated a prognostic score for each patient based on individualized values of the seven genes (Fig. 2d): prognostic scoremethylation = 0.0054 × cg13495205 AJAP1 + 0.0318 × cg07110405 SHANK2 + 0.0256 × cg03774514 FOXA2 + 0.0063 × cg09137696 MT1A + 0.0013 × cg19655456 ZNF570 − 0.0297 × cg03146625 HOXC4 − 0.0157 × cg21546671 HOXB4 .
Prognostic signature for OSCC patients
We categorized patients into low-risk and high-risk groups using a cutoff prognostic score of 0.02, which was selected by the optimum cutoff value according to the highest χ 2 value defined by Kaplan-Meier survival analysis and log-rank test in the training set [36]. As a weighted linear combination model of seven CpG sites, higher prognostic score was significantly associated with shorter survival in the training set (HR = 3.23; 95% CI 2.18–4.77; P = 5.52 × 10−10; Fig. 3a). A significant different proportion of patients in the low-risk group (23.3%) and high-risk group (54.4%) were followed until death (χ 2 = 28.48; P = 9.45 × 10−8). Results remained significant after adjustment for HPV status, age, gender, clinical stage, smoking status, and tumor grade (HRadjust = 3.14; 95% CI 1.89–5.22; P = 9.57 × 10−6; Additional file 1: Table S2).

Prognostic signature and OSCC patient survival. Left panels show Kaplan-Meier survival analyses of patients, which are categorized into low-risk and high-risk groups using a cutoff value of 0.02, for the a training set, b validation set 1, and c validation set 2. P values were calculated using log-rank test, and HR indicates hazard ratio. Right panels show time-dependent ROC curves of different months used to evaluate patient survival, with risk score using the nearest neighbor method
The prognostic signature with the same classifier cutoff (0.02) were successfully validated in the two validation sets, respectively. In validation set 1, there was a 2.79-fold higher risk of death for the high-risk group compared to the low-risk group (HR = 2.79; 95% CI 1.23–6.33; P = 0.010; Fig. 3b). In validation set 2, there was a 3.69-fold higher risk of death for the high-risk group compared to the low-risk group (HR = 3.69; 95% CI 1.25–10.85; P = 0.011; Fig. 3c). After controlling for HPV status, age, gender, clinical stage, smoking status, and grade in validation set 2, the results retained statistical significance (HRadjust = 2.96; 95% CI 0.53–7.26; P = 0.031; Additional file 1: Table S2).
Further, prediction ability of the prognostic signature was evaluated for 5-year overall survival. Time-dependent AUCs were 0.76 in the training set (95% CI 0.67–0.82; P < 0.001), 0.67 in validation set 1 (95% CI 0.54–0.78; P = 0.005), and 0.66 in validation set 2 (95% CI 0.50–0.79; P = 0.030) (Fig. 3a–c, right panels).
Sensitivity analysis for the seven-CpG-based signature
Due to the small sample size of HPV-positive cases, the training set and validation set 2 were merged to explore the relationship between the prognostic score and HPV status. Using multiple linear regression adjusted for age, gender, stage, smoking status, and grade, the score was not associated with HPV status (β = − 0.01; 95% CI − 0.23–0.20; P = 0.898). In the subgroup analysis stratified by HPV status, the seven-CpG-based signature was significant both within HPV-positive cases (HR = 3.33; 95% CI 1.07–10.37; P = 0.027; Fig. 4a) and HPV-negative cases (HR = 2.65; 95% CI 1.73–4.04; P = 5.80 × 10−6; Fig. 4b).

Subgroup and stratification analysis of the seven-CpG-based signature. Subgroup analysis for HPV+ cases (a) and HPV− cases (b) in the imputed combined dataset. c Kaplan-Meier curves plotting overall survival of the combined three datasets for respective prognostic score categories. d Subgroup analysis with clinical stage of the combined training set and validation set 2
Using the data merged of training and validation sets, as shown in Fig. 4c, prognostic score showed a stronger association with overall survival (log-rank P = 2.74 × 10−10). Stratified analyses by clinical characteristics (clinical stage, age, gender, smoking status, and grade) retained statistical significance (Fig. 4d and Additional file 1: Figure S2).
Relationship of CpG methylation, gene expression, and prognosis
Methylation and expression quantitative trait loci (meQTL) relationship for the seven CpG sites was performed in the training set. Expression and methylation data were both available in 308 cases of training set (Table 2). Methylation level of CpG sites at the promoter region and 1st exon region was moderately correlated with the corresponding gene expression for AJAP1 (r s = − 0.15; P = 0.009), HOXB4 (r s = − 0.41; P = 6.23 × 10−14), MT1A (r s = − 0.31; P = 2.39 × 10−8), ZNF570 (r s = − 0.64; P < 2.20 × 10−16), and SHANK2 (r s = 0.31; P = 2.93 × 10−8). Methylation of the other two CpG sites located in the gene body of HOXC4 (r s = − 0.01; P = 0.806) and FOXA2 (r s = 0.05; P = 0.340) was not observed any correlation with the gene expression (Fig. 5a–g, left panels). These genes’ expression was also significantly associated with patient’s overall survival (Fig. 5a–g, right panels).

Association between gene expression and methylation. Left panels show correlation of a AJAP1, b SHANK2, c FOXA2, d MT1A, e ZNF570, f HOXC4, and g HOXB4 expression (X-axis) with methylation (Y-axis). Right panels show Kaplan-Meier survival plots of gene expression from the TCGA cohort. HR indicates hazard ratio. Correlation coefficients and hypothesis tests were based on Spearman rank correlation tests. Patients were categorized into high-risk and low-risk groups by an optimum cutoff point according to the highest χ 2 value. h ROC curves for expression of the seven genes (left) and combinations of different types of data (right), including clinical characteristics (Clin), gene expression (Exp), and methylation (Methy)
Prognostic score using the expression of seven genes was also calculated (scoreexpression = − 0.115 × AJAP1 + 0.089 × SHANK2 + 0.147 × FOXA2 + 0.111 × MT1A − 0.173 × ZNF570 + 0.030 × HOXC4 + 0.789 × HOXB4), which was significantly associated with the prognosis (dichotomized by median, HR = 2.20; 95% CI 1.47–3.29; P = 1.22 × 10−4). After adjustment for HPV status, age, gender, clinical stage, smoking status, and grade, the result was still significant (HR = 3.41; 95% CI 1.98–5.89; P = 1.07 × 10−5) (Additional file 1: Figure S3). In addition, it effectively predicted 5-year survival (AUC = 0.65; 95% CI 0.54–0.72; P = 0.001) (Fig. 5h, left panel).
Combination of clinical information, expression, and methylation data (AUC = 0.78) showed a superior prediction ability in comparison to the model using clinical data only (AUC = 0.53) or clinical and expression data (AUC = 0.67) (Fig. 5h, right panel).
Furthermore, VanderWeele’s mediation analysis was used to explore the underlying mediation pathway of methylation, mRNA expression, and overall survival (Fig. 6a). Scoreexpression, the linear combination of seven genes’ mRNA expression, was treated as mediator in the overall mediation model. The prognostic effect of methylation signature was significantly mediated through affecting their mRNA expression (HRindirect = 1.08; 95% CI 1.02–1.15; P = 0.008; proportion mediated, 11.27%). Sensitivity analysis by excluding each gene expression from scoreexpression retained statistical significance (Fig. 6b).

Mediation analysis for methylation prognostic signature through mRNA expression. a Diagram of mediation model. b Methylation signature from the seven CpG sites was treated as “exposure”; mediator was the linear combination of the corresponding seven genes’ expression level (scoreexpression) (Overall model). Total prognostic effect in hazard ratio (HR) were described as direct effect (HRdirect), indirect effect (HRindirect), corresponding 95% confidence interval (95% CI), and the proportion of effect mediated (M%). Further, sensitivity analyses were performed by excluding each gene from scoreexpression, respectively, which retained statistical significance for mediation effect
Discussion
Cancer involves a complex regulatory network, integrating multiple biomarkers into an aggregated model could improve prognostic value compared with single biomarker [37]. The biomarkers discovery for OSCC have been reported in several studies [38, 39], but few of them used more than two datasets or explore the biomarkers across different omics. In this study, we developed an OSCC prognostic classifier model that includes seven CpG sites and validated the model using two independent external datasets. Results show that the prognostic signature was significantly associated with OSCC patient overall survival and had certain prediction abilities in the three datasets tested. OSCC patients with higher prognostic scores tended to have poorer clinical outcomes. Further, the gene expression of the corresponding CpG sites were also associated with overall survival. The integrated model of methylation and expression could add prognostic predictive value based on the clinical information (e.g., HPV status, age, clinical stage, and grade).
We used a three-step selection method to screen out significant biomarkers from more than 320,000 CpG sites after quality control. Differential methylation analysis using paired tissue data as the first step excluded 99.5% of probes. To exclude probes unrelated to survival, we evaluated their prognostic values by univariable Cox regression as the second step. However, the Cox model is not suitable for accurate modeling due to the low sample size/variable ratio and unstable variable combination [40]. To overcome the problem, SIS, a method based on a LASSO penalized model, was used to select a more stable and reliable set of CpG sites for further modeling. It first screened all included variables and discarded the irrelevant features with weak correlation to overall survival, then applied LASSO to estimate sensitivity from the selected genomic instability data [41].
The Cancer Genetics Web [42] suggests that research on OSCC biomarkers is still not comprehensive enough. Our study provides seven significant prognostic genes at the epigenetic and transcriptomic levels. Among the seven genes corresponding to candidate CpG sites, six have been reported as cancer-related genes. AJAP1, a novel tumor suppressor gene, is associated with survival in esophageal squamous cell carcinoma [43], hepatocellular carcinoma [44], and glioma [45]. Demethylation of hypermethylated AJAP1 reactivates its mRNA expression [43]. SHANK2 might cooperate with EMS1 to encode cytoskeleton-associated proteins implicated in tumor cell motility and invasiveness in OSCC [46]. It is also hypermethylated in prostate cancer tissues compared with paired non-tumor tissues [47]. FOXA2 is implicated in increased relapses and risk prognostic value in triple-negative/basal-like breast tumors [48]. Conversely, FOXA2 also is downregulated in lung cancer through epigenetic silencing of hypermethylation [49]. Further experiments are needed to verify the function of FOXA2 in OSCC. MT1A, which regulates cell growth and differentiation, has been described as a hypermethylated CpG biomarker for OSCC [50]. MT1A overexpression is also associated with HNSCC [51]. HOXC4 and HOXB4 are hypermethylated and downregulated in high-risk groups, and hypermethylation of HOXB4 is inversely correlated with decreased expression as an epigenetic biomarker for OSCC [52]. In addition, HOXC4 triggers similar molecular alterations as HOXB4 [53] and also is involved in some cancers [54, 55]. ZNF570 belongs to the large zinc finger gene family, which has been reported that is useful in the detection of HNSCC [56]. Although this gene’s function is still not known well, we measured a strong negative correlation of ZNF570 methylation and expression, both of which were significant in patient prognosis. Therefore, additional experiments are required; ZNF570 may represent a novel OSCC biomarker.
In addition to DNA methylation, mRNA expression levels of seven genes also affect prognosis significantly. Around 11% of methylation prognostic effect is mediated through affecting corresponding gene expression. Interestingly, most of the methylation’s effect may act, beyond affecting expression, but gene function [57], which warrants further functional experiments.
However, our study has some limitations. First, baseline information for GEO validation set 1 is unavailable, so multivariable analysis could not be made in the validation phase for this dataset. Second, due to the small sample size of some groups in the stratification analysis, like the HPV-positive cases, the results should be taken with caution since the sample size is insufficient. Third, further studies are needed to verify the biological function of some genes.
Conclusions
This study suggests that the developed seven-CpG-based signature coupled with gene expression is a useful and practical tool to improve prognostic value and survival prediction of OSCC, indicating it may have new applications for appropriate clinical adjuvant trials. Future studies including these molecular methylation and/or gene expression biomarkers, HPV status, age, other clinical characteristics, and different therapy effects will be useful for developing future personalized treatments.
Abbreviations
- 95% CI:
-
95% confidence interval
- AUC:
-
Area under the curve
- CV:
-
Coefficient of variance
- GDC:
-
Genomic Data Commons
- GEO:
-
Gene Expression Omnibus
- HNSCC:
-
Head and neck squamous cell carcinoma
- HPV:
-
Human papillomavirus
- HR:
-
Hazard ratio
- MICE:
-
Multivariate imputation by chained equations
- OSCC:
-
Oral squamous cell carcinoma
- ROC:
-
Receiver operating
- SIS:
-
Sure independence screening
- SNP:
-
Single-nucleotide polymorphism
- TCGA:
-
The Cancer Genome Atlas
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008;371(9625):1695–709.
Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45(4–5):301–8.
Scully C, Bagan JV. Recent advances in oral oncology 2007: imaging, treatment and treatment outcomes. Oral Oncol. 2008;44(3):211–5.
Lo WY, Tsai MH, Tsai Y, Hua CH, Tsai FJ, Huang SY, et al. Identification of over-expressed proteins in oral squamous cell carcinoma (OSCC) patients by clinical proteomic analysis. Clin Chim Acta. 2007;376(1–2):101–7.
Pitiyage G, Tilakaratne WM, Tavassoli M, Warnakulasuriya S. Molecular markers in oral epithelial dysplasia: review. J Oral Pathol Med. 2009;38(10):737–52.
Choi S, Myers JN. Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dent Res. 2008;87(1):14–32.
Arantes LM, de Carvalho AC, Melendez ME, Carvalho AL, Golonibertollo EM. Methylation as a biomarker for head and neck cancer. Oral Oncol. 2014;50(6):587–92.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–68.
Hasegawa M, Nelson HH, Peters E, Ringstrom E, Posner M, Kelsey KT. Patterns of gene promoter methylation in squamous cell cancer of the head and neck. Oncogene. 2002;21(27):4231–6.
Gu J, Berman D, Lu C, Wistuba II, Roth JA, Frazier M, et al. Aberrant promoter methylation profile and association with survival in patients with non-small cell lung cancer. Clin Cancer Res. 2006;12(24):7329–38.
Fujiya K, Ashida T, Maemoto A, Orii F, Fujiki T, Fujiya M, et al. High detection rate of aberrant methylation of p16 gene in the serum/plasma in patients with colorectal cancer. Gastroenterology. 2000;118(4):A1387.
Hoque MO, Feng Q, Toure P, Dem A, Critchlow CW, Hawes SE, et al. Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J Clin Oncol. 2006;24(26):4262–9.
Mroz EA, Tward AD, Pickering CR, Myers JN, Ferris RL, Rocco JW. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer. 2013;119(16):3034–42.
Lohavanichbutr P, Méndez E, Holsinger FC, Rue TC, Zhang Y, Houck J, et al. A 13-gene signature prognostic of HPV-negative OSCC: discovery and external validation. Clin Cancer Res. 2013;19(5):1197–203.
Gombos K, Szele E, Gocze K, Somlai K, Pajkos G, Ember I, et al. miRNA expression profiles of oral squamous cell carcinomas. Anticancer Res. 2013;33(4):1511–7.
Chauhan SS, Kaur J, Kumar M, Matta A, Srivastava G, Alyass A, et al. Prediction of recurrence-free survival using a protein expression-based risk classifier for head and neck cancer. Oncogene. 2015;4(4):e147.
Foy JP, Pickering CR, Papadimitrakopoulou VA, Jelinek J, Lin SH Jr, WW, et al. New DNA methylation markers and global DNA hypomethylation are associated with oral cancer development. Cancer Prev Res. 2015;8(11):1027.
Langevin SM, Butler RA, Eliot M, Pawlita M, Maccani JZ, Mcclean MD, et al. Novel DNA methylation targets in oral rinse samples predict survival of patients with oral squamous cell carcinoma. Oral Oncol. 2014;50(11):1072–80.
Shaw RJ, Hall GL, Lowe D, Liloglou T, Field JK, Sloan P, et al. The role of pyrosequencing in head and neck cancer epigenetics: correlation of quantitative methylation data with gene expression. Arch Otolaryngol Head Neck Surg. 2008;134(3):251–6.
Krishnan NM, Dhas K, Nair J, Palve V, Bagwan J, Siddappa G, et al. A minimal DNA methylation signature in oral tongue squamous cell carcinoma links altered methylation with tumor attributes. Mol Cancer Res. 2016;14:805.
Aryee MJ. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–9.
Sandoval J, Mendezgonzalez J, Nadal E, Chen G, Carmona FJ, Sayols S, et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J Clin Oncol. 2013;31(32):4140–7.
Price EM, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, et al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6(1):1–15.
Chen Y, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–9.
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–96.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–27.
Network CGA. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–82.
White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med. 2011;30(4):377–99.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323.
Pineda S, Real FX, Kogevinas M, Carrato A, Chanock SJ, Malats N, et al. Integration analysis of three omics data using penalized regression methods: an application to bladder cancer. Hum Hered. 2015;11(12):100.
Tibshirani R. Regression shrinkage and selection via the lasso: a retrospective. J R Stat Soc. 2011;73(3):273–82.
Fan J, Lv J. Sure independence screening for ultrahigh dimensional feature space. J R Stat Soc. 2008;70(5):883–911.
Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56(2):337–44.
Naimi AI, VanderWeele TJ. Explanation in causal inference: methods for mediation and interaction. Eur J Epidemiol. 2016;31:1065–6.
Camp RL, Dolledfilhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10(21):7252–9.
Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433–7.
Sailer V, Holmes EE, Gevensleben H, Goltz D, Dröge F, Franzen A, et al. PITX3 DNA methylation is an independent predictor of overall survival in patients with head and neck squamous cell carcinoma. Clin Epigenetics. 2017;9(1):12.
Yang CM, Wang TH, Chen HC, Li SC, Lee MC, Liou HH, et al. Aberrant DNA hypermethylation-silenced SOX21-AS1 gene expression and its clinical importance in oral cancer. Clin Epigenetics. 2016;8(1):129.
Simon R, Altman DG. Statistical aspects of prognostic factor studies in oncology. Br J Cancer. 1994;69(6):979–85.
Tibshirani R. Regression shrinkage selection via the LASSO. J R Stat Soc. 2011;73(3):273–82.
SJ C. Home Page, Cancer Genetics Web: http://www.cancer-genetics.org/index.htm. Accessed 27 Feb 2017.
Tanaka H, Kanda M, Koike M, Iwata N, Shimizu D, Ezaka K, et al. Adherens junctions associated protein 1 serves as a predictor of recurrence of squamous cell carcinoma of the esophagus. Int J Oncol. 2015;47(5):31–65.
Ezaka K, Kanda M, Sugimoto H, Shimizu D, Oya H, Nomoto S, et al. Reduced expression of adherens junctions associated protein 1 predicts recurrence of hepatocellular carcinoma after curative hepatectomy. Ann Surg Oncol. 2015;22(3):1499–507.
Zeng L, Fee BE, Rivas MV, Lin J, Adamson DC. Adherens junctional associated protein-1: a novel 1p36 tumor suppressor candidate in gliomas (review). Int J Oncol. 2014;45(1):13–7.
Freier K, Sticht C, Hofele C, Flechtenmacher C, Stange D, Puccio L, et al. Recurrent coamplification of cytoskeleton-associated genesEMS1 andSHANK2 withCCND1 in oral squamous cell carcinoma. Genes Chromosomes Cancer. 2006;45(2):118–25.
Devaney JM, Wang S, Furbertharris P, Apprey V, Ittmann M, Wang BD, et al. Genome-wide differentially methylated genes in prostate cancer tissues from African-American and Caucasian men. Epigenetics. 2015;10(4):1–10.
Perez-Balaguer A, Ortiz-Martínez F, García-Martínez A, Pomares-Navarro C, Lerma E, Peiró G. FOXA2 mRNA expression is associated with relapse in patients with triple-negative/basal-like breast carcinoma. Breast Cancer Res Treat. 2015;153(2):465–74.
Basseres DS, D’Alò F, Yeap BY, Löwenberg EC, Gonzalez DA, Yasuda H, et al. Frequent downregulation of the transcription factor Foxa2 in lung cancer through epigenetic silencing. Lung Cancer. 2012;77(1):31–7.
Li YF, Hsiao YH, Lai YH, Chen YC, Chen YJ, Chou JL, et al. DNA methylation profiles and biomarkers of oral squamous cell carcinoma. Epigenetics. 2015;10(3):229–36.
Raudenska M, Sztalmachova M, Gumulec J, Fojtu M, Polanska H, Balvan J, et al. Prognostic significance of the tumour-adjacent tissue in head and neck cancers. Tumor Biol. 2015;36(12):9929–39.
Xavier FC, Destro MF, Duarte CM, Nunes FD. Epigenetic repression of HOXB cluster in oral cancer cell lines. Arch Oral Biol. 2014;59(8):783–9.
Céline Auvray AD, Pflumio F, Haddad R, Amsellem S, Miri-Nezhad A, Broix L, Yacia A, Bulle F, Fichelson S, Vigon I. HOXC4 homeoprotein efficiently expands human hematopoietic stem cells and triggers similar molecular alterations as HOXB4. Haematologica. 2012;97(2):168–78.
Risk MC, Knudsen BS, Coleman I, Dumpit RF, Kristal AR, Lemeur N, et al. Differential gene expression in benign prostate epithelium of men with and without prostate cancer: evidence for a prostate cancer field effect. J Urol. 2008;179(4):460.
Shinawi T, Hill VK, Krex D, Schackert G, Gentle D, Morris MR, et al. DNA methylation profiles of long- and short-term glioblastoma survivors. Epigenetics. 2013;8(2):149–56.
Gaykalova DA, Vatapalli R, Wei Y, Tsai H-L, Wang H, Zhang C, et al. Outlier analysis defines zinc finger gene family DNA methylation in tumors and saliva of head and neck cancer patients. PLoS One. 2015;10(11):e0142148.
Schübeler D. Function and information content of DNA methylation. Nature. 2015;517(7534):321–6.
Acknowledgements
We thank the patients and investigators who participated in TCGA and GEO for providing data.
Funding
This study was supported by the National Natural Science Foundation of China (81,473,070 and 81,530,088 to F.C., 81,402,764 to Y.W., 81,373,102 to Y.Z., and 81,402,763 to R.Z.) and the Natural Science Foundation of Jiangsu, China (No. BK20140907 to Y.W.). It is also supported by Nanjing Medical University international exchange and cooperation project (C018 to S.S.)
Availability of data and materials
TCGA: https://tcga-data.nci.nih.gov, now hosted at GDC: https://portal.gdc.cancer.gov/
GSE52793: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52793
GSE75537: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75537
Author information
Authors and Affiliations
Contributions
SS, GW, DCC, and FC contributed to the study design. RZ and YW contributed to data collection. SS and YW performed statistical analysis and interpretation. SS drafted the manuscript. All authors contributed to critical revision of the final manuscript and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Consent for publication
All participants gave written informed consent. All authors have reviewed the manuscript and consented for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1: Table S1.
Annotation for seven CpG sites selected by SIS. Table S2 Cox regression analysis of clinical characteristics and risk scores. Figure S1. Boxplot depicting beta-values of seven CpG sites after ComBat processing in training and validation datasets. Figure S2. Kaplan-Meier survival analyses of patients subgrouped by (A) age divided by median value (60 years), (B) gender, (C) smoking status, or (D) grade. Figure S3. Kaplan-Meier survival analyses of the gene expression prognostic score. Low-risk and high-risk patients were divided by the median value. (DOCX 832 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shen, S., Wang, G., Shi, Q. et al. Seven-CpG-based prognostic signature coupled with gene expression predicts survival of oral squamous cell carcinoma. Clin Epigenet 9, 88 (2017). https://doi.org/10.1186/s13148-017-0392-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-017-0392-9