
Abstract
Background
Fetal overgrowth “programs” an elevated risk of type 2 diabetes in adulthood. Epigenetic alterations may be a mechanism in programming the vulnerability. We sought to characterize genome-wide alterations in placental gene methylations in fetal overgrowth and the associations with metabolic health biomarkers including leptin, adiponectin and fetal growth factors.
Results
Comparing genome-wide placental gene DNA methylations in large-for-gestational-age (LGA, an indicator of fetal overgrowth, n = 30) versus optimal-for-gestational-age (OGA, control, n = 30) infants using the Illumina Infinium Human Methylation-EPIC BeadChip, we identified 543 differential methylation positions (DMPs; 397 hypermethylated, 146 hypomethylated) at false discovery rate < 5% and absolute methylation difference > 0.05 after adjusting for placental cell-type heterogeneity, maternal age, pre-pregnancy BMI and HbA1c levels during pregnancy. Twenty-five DMPs annotated to 20 genes (QSOX1, FCHSD2, LOC101928162, ADGRB3, GCNT1, TAP1, MYO16, NAV1, ATP8A2, LBXCOR1, EN2, INCA1, CAMTA2, SORCS2, SLC4A4, RPA3, UMAD1,USP53, OR2L13 and NR3C2) could explain 80% of the birth weight variations. Pathway analyses did not detect any statistically significant pathways after correcting for multiple tests. We validated a newly discovered differentially (hyper-)methylated gene-visual system homeobox 1 (VSX1) in an independent pyrosequencing study sample (LGA 47, OGA 47). Our data confirmed a hypermethylated gene—cadherin 13 (CDH13) reported in a previous epigenome-wide association study. Adiponectin in cord blood was correlated with its gene methylation in the placenta, while leptin and fetal growth factors (insulin, IGF-1, IGF-2) were not.
Conclusions
Fetal overgrowth may be associated with a large number of altered placental gene methylations. Placental VSX1 and CDH13 genes are hypermethylated in fetal overgrowth. Placental ADIPOQ gene methylations and fetal circulating adiponectin levels were correlated, suggesting the contribution of placenta-originated adiponectin to cord blood adiponectin.
Similar content being viewed by others

Background
Fetal overgrowth, as indicated by high birth weight or large-for-gestational-age (LGA), is associated with elevated risks of metabolic syndrome and type 2 diabetes in adulthood [1,2,3]. The fetus may adapt to adverse environmental cues during gestation that may have long-lasting impact on the vulnerability to a number of chronic diseases [4, 5]—a phenomenon known as developmental “programming.” Such programming in fetal growth restriction may be partly attributable to reduced β-cell mass [6], whereas less is understood about the programming mechanisms in excessive fetal growth.
Epigenetic changes especially in DNA methylation regulating gene expression play a central role in fetal development [7] and may be a mechanism in developmentally programming the vulnerability to metabolic syndrome related disorders. During fetal development, the placenta, through the production of various enzymes and hormones, plays an important role in regulating fetal growth and development [8]. Emerging studies in humans have associated birth weight with placental DNA methylation, but most of these studies have been focused on gene-specific methylation changes in the placentas in small-for-gestational-age (SGA) newborns. We are aware of only two small studies (n < 20) [9, 10] on differentially methylated genes in LGA in the placenta. No studies have assessed the associations between LGA-associated placental differential gene methylations and fetal circulating levels of metabolic health biomarkers. Therefore, we sought to evaluate placental gene DNA methylation alterations in LGA, and explore the associations with fetal circulating (cord blood) metabolic health biomarkers including leptin, adiponectin and fetal growth factors [insulin, insulin-like growth factor I (IGF-I) and IGF II].
Results
Characteristics of study subjects
Table 1 presents maternal and neonatal characteristics of the 30 pairs of study subjects. Comparing LGA versus birth weight optimal-for-gestational-age (OGA) control subjects, there were no significant differences in maternal age, ethnicity, parity, education, smoking, gestational hypertension, family history of hypertension, family history of diabetes, maternal blood glycated hemoglobin (HbA1c) levels during the 2nd and 3rd trimesters of pregnancy, and gestational age at delivery. Women bearing a LGA fetus had higher pre-pregnancy body mass index (BMI mean: 24.0 vs. 22.0 kg/m2) and were more likely to have a cesarean section delivery (73.3% vs. 26.7%). As expected, average birth weight and birth length were substantially higher in LGA vs. OGA newborns.
LGA newborns had significantly higher cord blood IGF-I concentrations (mean: 88.8 vs.68.6 ng/mL, P = 0.006), and lower HMW adiponectin concentrations (14.6 vs. 20.5 μg/mL, P = 0.014) (Table 1). There were no significant differences in cord blood insulin, C-peptide, proinsulin, leptin, IGF-II and total adiponectin concentrations.
Differentially methylated positions (DMPs)
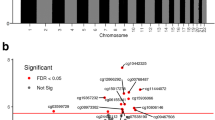
Adjusting for maternal age, pre-pregnancy BMI, whole blood HbA1c levels at the second and third trimesters of pregnancy and the identified four principal components (from principal component analysis) representing placental cell types (other co-variables were excluded since they were similar and did not affect the comparisons), a total of 543 CpG sites were differentially methylated positions (DMPs) comparing LGA and OGA groups accounting for multiple tests with false discovery rate (FDR) < 5% and absolute methylation difference (delta beta) > 0.05, including 397 hypermethylated and 146 hypomethylated DMPs (Fig. 1, Additional file 2: Table S1). These loci were distributed over 316 genes (232 hypermethylated genes, 84 hypomethylated genes).

Volcano plot of differentially methylated positions (DMPs) in placental gene DNAs comparing large-for-gestational-age (LGA, birth weight > 90th percentile) versus optimal-for-gestational-age (OGA, 25–75th percentiles, control) newborns. DMPs in the upper left and right quadrants (colored) are differentially methylated at false discovery rate (FDR) < 5%
The top 50 DMPs (25 hypermethylated, 25 hypomethylated CpG sites) are presented in Tables 2 and 3. The hypermethylated loci were annotated to 14 genes (EN2, LOC283999, CADM2, ADGB, KRTAP13-4, CRMP1, GFRA1, NRXN1, VSX1, PPFIA2, PLXNC1, DNAJB5, DAOA and ZPLD1). The hypomethylated sites were annotated to 13 genes (FAM155A, C21orf34, WNT5B, DTNA, OPRM1, SORCS3, KIF26B, SLCO3A1, KIF26B, LOC284930, SLIT3, NXPH1 and HLA-L).
Our placental epigenome data validated that the cadherin 13(CDH13) gene was hypermethylated in LGA as reported in a previous epigenome-wide association study [10] (our study: CDH13 methylation increased by 0.05 in LGA; the previous study: CDH13 methylation increased by 0.21 in LGA, according to the publication and communications with the corresponding author).
Pyrosequencing validation study
We sought to validate a few differentially methylated CpG sites with relatively large methylation differences between LGA and OGA groups in the epigenome-wide association analysis. The study subjects were an independent random sample of 47 pairs of LGA and OGA newborns matched by sex and gestational age from the Shanghai birth cohort (Additional file 3: Table S2). Women bearing a LGA fetus had higher pre-pregnancy BMI and were more likely to be have a cesarean section delivery, while other maternal characteristics were similar in LGA and OGA groups. As expected, average birth weight and birth length were substantially higher in LGA versus OGA newborns.
Three CpG sites were selected among the top 25 DMPs in the pyrosequencing validation study. The CpG site (cg11700298) annotated to cell adhesion molecule 2 (CADM2)] was selected because polymorphism in this gene has been associated with obesity and type 2 diabetes [11, 12]. We randomly selected 2 more CpG sites among the top 25 DMPs in the validation study. They were cg17650274 [annotated to gene visual system homeobox 1 (VSX1)] and cg17512353 [annotated to gene major histocompatibility complex, class I, L (HLA-L)]. The pyrosequencing validation vs. epigenome-wide study results for the 3 DMPs are shown in Additional file 3: Table S3; only one DMP (VSX1 gene) was validated in the pyrosequencing study.
GO and KEGG pathways
The GO analysis showed that DMPs were mostly enriched in axon development (biological processes), motile cilium (cellular components), metal-ion transmembrane transporter activity (molecular functions) (Additional file 2: Table S4). KEGG pathway analysis showed that DMPs might mainly be involved in 9 pathways including GnRH secretion, protein digestion and absorption (Additional file 2: Table S5). However, after correction for multiple tests, the results were not statistically significant for all pathways.
Ingenuity pathway analysis (IPA)
IPA identified 16 canonical pathways (Fisher’s exact crude P < 0.05, Additional file 2: Table S6). None of the pathways achieved statistical significance after Benjamini–Hochberg correction for multiple tests. Differentially methylated genes were most enriched in the G-Protein Coupled Receptor Signaling (9 genes, crude P < 0.05). There was also some evidence of enrichment (crude P < 0.05) in the Pentose Phosphate (Oxidative Branch) and Estrogen Biosynthesis pathways.
Differentially methylated regions (DMRs)
To reduce data dimensionality and identify differential methylations over gene areas, we analyzed the methylation data for differentially methylated regions (DMRs) by DMRcate and comb-p. The DMRcate program identified 135 DMRs at FDR < 5% between LGA and OGA groups (Additional file 2: Table S7), and these DMRs were annotated to 94 genes, while the other DMRs were located in the OpenSea areas. The comb-p program identified 31 DMRs with Sidak corrected P < 0.05 (Additional file 2: Table S8), 15 DMRs were annotated to genes, while the other DMRs were located in the OpenSea areas. Three DMRs were identified by both DMRcate and comb-p, and these loci were annotated to 3 genes (PRMT2, BACE1 and TRAK2; Table 4).
Correlations of DMPs with birth weight and cord blood biomarkers
Among the 543 DMPs, 494 CpG sites were correlated with birth weight z score (crude P < 0.05), and the correlations for 486 DMPs remained statistically significant after correction for multiple tests (Additional file 2: Table S9). These 486 DMPs were annotated to 286 genes. In LASSO regression identifying the most important DMPs, 25 DMPs were selected and could explain 80.2% of the variations in birth weight (z) (Additional file 2: Table S10). These 25 DMPs were annotated to 20 genes (QSOX1, FCHSD2, LOC101928162, ADGRB3, GCNT1, TAP1, MYO16, NAV1, ATP8A2, LBXCOR1, EN2, INCA1, CAMTA2, SORCS2, SLC4A4, RPA3, UMAD1,USP53, OR2L13 and NR3C2) (Additional file 2: Table S10).
Additional file 2: Table S11 presents the correlations of DMPs with cord blood insulin, proinsulin, C-peptide, IGF-II, IGF-I, leptin, total or HMW adiponectin. All these correlations did not reach statistical significance after Benjamini and Hochberg correction for multiple tests.
Gene-specific correlations of placental DNA methylations and cord blood biomarkers
Additional file 3: Table S12 presents the gene-specific correlations of placental gene CpG sites and cord blood biomarkers. For INS-IGF2/IGF2AS/IGF2, methylation levels in four CpG sites (cg08014499, cg21728792, cg05203776 and cg22225943) were positively correlated with cord blood insulin (r = 0.26 to 0.28, crude P = 0.03 to 0.05), and in one CpG site positively correlated with cord blood C-peptide (cg17434309, r = 0.28, crude P = 0.03), proinsulin (cg17434309, r = 0.31, crude P = 0.02) and IGF-II (cg23889607, r = 0.30, crude P = 0.02). Methylation levels in four CpG sites (cg13928782, cg21574853, cg07096953 and cg20088847) were negatively correlated with cord blood IGF-II (r = − 0.30 to − 0.35, crude P = 0.007 to 0.024), and in one CpG site negatively correlated with cord blood proinsulin (cg13670288, r = − 0.26, crude P = 0.049). All these correlations, however, did not reach statistical significance after correction for multiple tests.
Several correlations between ADIPOQ gene methylation levels and cord blood total or HMW adiponectin remained statistically significant after correction for multiple tests. Total adiponectin was correlated with cg16126291 (r = − 0.35, adjusted P = 0.03), cg02235049 (r = − 0.32, adjusted P = 0.047), cg10681525 (r = − 0.40, adjusted P = 0.01) and cg18537894 (r = 0.40, adjusted P = 0.01). HMW adiponectin was correlated with cg16126291 (r = − 0.42, adjusted P = 0.01) and cg18537894 (r = 0.36, adjusted P = 0.036).
Discussion
Main findings
We observed 543 DMPs in placental DNA in LGA—an indicator of fetal overgrowth, and identified 25 DMPs annotated to 20 genes that could explain the majority of birth weight variations. We validated that the VSX1 gene was hypermethylated in fetal overgrowth in an independent pyrosequencing study sample, and confirmed a hypermethylated gene (CDH13) in fetal overgrowth reported in a previous epigenome-wide association study. Three DMRs were identified and annotated to three genes (PRMT2, BACE1 and TRAK2). We did not detect any specific significant pathway after correction for multiple tests. Placental gene methylation and fetal circulating hormone biomarkers were correlated for adiponectin, but not for leptin and fetal growth factors.
Data interpretation and comparisons with findings in previous studies
Our study is the largest in assessing genome-wide placental DNA methylations in LGA. A previous study in 27 LGA and 19 appropriate for gestational age (AGA) controls observed no gene methylation alterations in LGA in an epigenome-wide association analysis of cord tissue DNA methylations using the Infinium Human Methylation 450 K BeadChips [13]. Another epigenome-wide association study (5 LGA, 6 AGA controls) reported no differences in placental DNA methylation levels using reduced representation bisulfite sequencing [10]. However, a large study (n = 1023) assessing placental global DNA methylation by LC–MS/MS demonstrated that LGA displayed significantly higher global placental DNA methylation compared to AGA [14]. Another study (6 LGA and 6 AGA controls) reported that among 17,244 methylation variable positions, 705 were hypermethylated (> 1.7-fold) and 351 were hypomethylated (< 0.5-fold) in LGA in placental DNA methylations using the Infinium Human Methylation 850 K BeadChips [9]. These results are consistent with our data: Hypermethylated sites (n = 397) are more frequent than hypomethylated sites (n = 146) in LGA.
DMPs
We identified 486 DMPs in placental gene DNA comparing LGA versus controls. Many of these DMPs (165/486) demonstrated weak-to-moderate correlations (r: 0.25 to 0.45) with a fetal growth factor (IGF-I, IGF-II, insulin), leptin or adiponectin in cord blood in crude correlation analyses (Additional file 2: Table S11), but none reached statistical significance after correction for multiple tests. The placenta is a transient fetal organ, and its gene expression might affect fetal growth through influencing the function of the placenta, rather than through affecting fetal circulating levels of growth factors. This may explain the lack of correlation between placental IGF-I gene methylations and circulating/fetal IGF-I concentrations, because IGF-I is produced in fetal liver cells only. Gene methylation patterns are tissue-specific. A specific gene’s methylation pattern in the placenta might unlikely reflect the gene’s methylation pattern in other fetal tissues especially if the gene’s expression is tissue-specific. For genes that are expressed in multiple tissues, one might speculate that some methylation patterns may be shared across multiple tissues. It is unknown which gene’s specific methylation patterns in the placenta are present in other fetal tissues. The implications of placental gene methylation signatures for short- and long-term metabolic health in the offspring are largely unknown and remain to be understood. Emerging evidence suggests the prognostic value of placental gene methylations on long-term metabolic health in children: Placental lipoprotein lipase gene DNA methylation alterations have been correlated with fat mass in children at age 5 years [15].
Notably, 25 DMPs annotated to 20 genes (QSOX1, FCHSD2, LOC101928162, ADGRB3, GCNT1, TAP1, MYO16, NAV1, ATP8A2, LBXCOR1, EN2, INCA1, CAMTA2, SORCS2, SLC4A4, RPA3, UMAD1, USP53, OR2L13 and NR3C2) could explain over 80% of the birth weight variations. Some of these genes have been implicated in the regulation of glucose homeostasis, β cell function, adipose tissue or muscle growth. FCHSD2 and SLC4A4 have been associated with β cell function[16, 17], while USP53 has been related to adiposity homeostasis [18]. SLC4A4 knockout mice were protected from diet-induced metabolic stress and β cell dysfunction [17] Loss of FCHSD2 was associated with impaired insulin secretion in a human-derived β cell study [16]. Elevated USP53 gene RNA expression in adipose tissue has been associated with good weight control in obese subjects [18]. The QSOX1 gene has been implicated in cortical bone accrual and strength in mice [19]. The ADGRB3 gene may be involved in myoblast fusion in the muscle of vertebrates [20] and implicated in insulin secretion from pancreatic β-cells [21]. Adipocyte-specific overexpression of NR3C2 exacerbates metabolic syndrome in mice [22]. However, there have been no reports on whether these genes are correlated with fetal growth in humans or animals. Our data on placental gene methylations suggest the importance of these 20 genes for fetal growth, probably through their impacts on placental function that deserves further mechanistic studies. A lack of replicable findings in genome-wide association studies is a common problem in data interpretation concerning the robustness of positive findings. It is noteworthy that our data validated a hypermethylated gene (CDH13) reported in a previous epigenome-wide association study [10].
CDH13 (cadherin 13), a 95 kd glycoprotein, is an atypical member of the cadherin family of cell adhesion molecules [23]. CDH13 may serve as an adiponectin receptor and has been associated with plasma adiponectin and the risk of type 2 diabetes [24, 25]. CDH13 levels in adipose tissue and the circulation are decreased in obese mice and humans and are restored by weight loss in humans [26]. Therefore, CDH13 is considered as a marker of fat tissue plasticity that might reflect the health status of adipose tissue. We observed that CDH13 was hypermethylation in LGA, consistent with the results in a previous study [10]. Tyrberg and colleagues reported T-cadherin (CDH13) as a novel component of insulin granules, suggesting that it might contribute to the regulation of insulin secretion independently of adiponectin [27]. It would be interesting to determine whether altered placental CDH13 gene hypermethylation may be an epigenetic biomarker of the elevated risk of metabolic dysfunctional disorders in LGA subjects in long-term follow-up studies.
Placental VSX1 gene was observed to be hypermethylated in LGA in both the epigenome-wide association analysis and pyrosequencing validation study. We failed to confirm the other two DMPs identified from the epigenome-wide association analysis in the pyrosequencing validation study, underscoring the uncertain nature of genome-wide discovery research. Despite best efforts in accounting for potential confounding effects and biases, unmeasured confounding effects might weaken the power of genome-wide association studies in identifying the true differences.
The VSX1 (visual system homeobox 1) gene encodes a paired-like homeodomain transcription factor and is associated with eye development [28]. VSX1 gene variants may play an important role in the development of keratoconus [29]. Our data suggest hypermethylation of placental VSX1 gene in fetal overgrowth, and long-term follow-up studies are required to determine its potential significance as an epigenetic biomarker for metabolic health in later life.
DMRs
Three DMRs in LGA were identified in both DMRcate and comb-p analyses, which were annotated to three metabolic health relevant genes—BACE1, TRAK2 and PRMT2.
BACE1 (β-site APP-cleaving enzyme 1) is expressed in pancreas, liver and skeletal muscle [30]. BACE1 may play an important role in glucose metabolism; BACE1-deficient liver and skeletal muscle exhibit improved insulin sensitivity and glucose homeostasis in mice [31]. Neuronal human BACE1 knock-in in mice induced systemic diabetes [32]. High glucose levels might upregulate BACE1 expression via ROS generation in SK-N-MC cells [33]. Moreover, the BACE1 gene polymorphism has been associated with the risk of diabetes in PIMA Indians [34]. Thus, alterations in BACE1 levels may be involved in the pathophysiology of diabetes.
Trafficking protein kinesin binding 2 (TRAK2) is a regulator of protein and organelle trafficking through its role as a kinesin and dynein binding protein, and it may function in neuronal mitochondrial trafficking [35]. TRAK2 has been reported as a novel regulator of ATP-binding cassette, sub-family A member 1 (ABCA1) expression, cholesterol efflux and HDL biogenesis, and therefore, TRAK2 may be an important target in the treatment of cardiovascular disorders [36].
Protein arginine methyltransferase 2 (PRMT2)—a type I enzyme, contains a highly conserved catalytic Ado-Met binding domain and unique Src homology (SH) 3 domain that binds proteins with proline-rich motifs [37]. Genetic deletion of PRMT2 has been associated with a lean, leptin-hypersensitive “anti-diabetes-like” phenotype in mice [38].
Our results suggest altered methylations in placental PRMT2, BACE1 and TRAK2 genes in fetal overgrowth. It remains to be determined whether they could be promising epigenetic biomarkers of the increased risk of metabolic dysfunctional disorders in LGA subjects in later life.
Pathways
The IPA results suggest the enrichment of three genes in the “Pentose Phosphate” and “Estrogen Biosynthesis” pathways. A smaller study (6 LGA and 6 controls) in cord blood DNA reported 27 genes enriched in “Diseases and Disorders” terms [39] without pathway analysis. There were no consistent findings between our GO and KEGG pathway results and those reported in a previous study [9]. To be noted, all pathways did not reach statistical significance after correction for multiple tests in our study. Previous studies did not adjust for multiple tests. Larger studies are warranted to clarify whether any specific pathway may be affected in placental gene methylations in LGA.
Associations with cord blood biomarkers
Placental gene methylations were correlated with fetal circulating (cord blood) hormone levels for adiponectin, but not for leptin and fetal growth factors after correction for multiple tests. Bouchard and colleagues reported that placental ADIPOQ gene methylation levels were negatively correlated with maternal circulating adiponectin concentrations [40]. We are unaware of any report on placental ADIPOQ methylation and cord blood adiponectin. In our data, total adiponectin was inversely correlated with ADIPOQ gene methylations at 3 CpG sites (cg16126291, cg02235049 and cg10681525), and HMW adiponectin was negatively correlated with methylations at 2 CpG sites (cg16126291 and cg02235049). Fetal/cord blood adiponectin levels are attributable to adiponectin secretion by both fetal adipose tissue (brown adipocytes) and vascular cells [41,42,43]. Adiponectin is highly expressed in vascular endothelial cells of fetal capillaries [41]. There is no evidence of maternal origin of adiponectin in fetal circulation [42]. It remains controversial whether the human placenta secrets adiponectin [44]. The placenta is a fetal tissue rich in blood vessels and capillaries which may explain the placental “production” of adiponectin. In our study, cord blood adiponectin concentrations were negatively correlated with multiple placental ADIPOQ gene methylation sites, suggesting the contribution of placenta-originated adiponectin in cord blood adiponectin.
Limitations
There were several study limitations. First, the sample size was relatively modest (but still the largest placenta epigenome study on LGA thus far). The study might be under-powered to detect small differences, and we could not rule out the possibility of false positive findings which may be present in all (epi)genome-wide association studies. This might explain the lack of consistent findings in previous (epi)genome-wide association studies and that only one of the three selected DMPs was validated in an independent sample. Large studies are warranted to validate the findings. Second, the DNA isolated from placental tissues comes from multiple cell types that might have not been adequately accounted for using the principal component analysis. This may limit the capacity to identify the true differences. Third, the study was limited to Chinese subjects. More studies in other ethnic groups are warranted to understand the generalizability of the study findings.
Conclusions
Fetal overgrowth appears to be associated with altered methylations in a large number of placental genes. Placental CDH13 and VSX1 genes are hypermethylated in fetal overgrowth. Long-term follow-up studies are required to determine whether these differentially methylated genes may be promising epigenetic biomarkers of the elevated risk of metabolic dysfunctional disorders in later life in subjects with fetal overgrowth. Placental ADIPOQ gene methylations and fetal circulating adiponectin levels were correlated, suggesting the contribution of placenta-originated adiponectin in cord blood adiponectin.
Methods
Study design
We conducted a nested case–control study in the Shanghai birth cohort (SBC) [45]. LGA was defined as birth weight > 90th percentile, according to the Chinese sex- and gestational age-specific birth weight standards [46]. Controls were optimal-for-gestational-age (OGA, birth weight 25th-75th percentiles) newborns. Each LGA was matched (1:1) to an OGA newborn by sex and gestational age (within 7 days) at delivery. The study subjects (30 pairs of LGA/OGA) were randomly sampled from all eligible LGA and OGA newborns in the SBC. All cases and controls were term births with normal Apgar score (> 7), and all mothers were free of severe chronic diseases before pregnancy (e.g., essential hypertension, type 1 or 2 diabetes), severe pregnancy complications (e.g., preeclampsia) or life-threatening conditions.
Placenta and cord blood samples
Trained research staff collected cord blood and placental tissue samples following standardized operating procedures. Each of the four placenta quadrants was sampled approximately 1.5 cm away from the umbilical cord insertion from the fetal side of the placenta. Fetal membranes and visible large vessels were removed, and phosphate-buffered saline was used to wash placenta samples before separating into maternal- and fetal-side samples. Placental and cord blood samples were kept at 4℃ in a refrigerator between 0 and 4 h before stored at − 80 °C in a freezer until DNA extraction. There were no reports of specimen handling protocol violations. Genomic DNA was extracted from fetal-side placental samples using DNeasy & Tissue Kit (Qiagen) following the manufacturer’s manual. Purity was examined by measuring the A260:A280 ratio (mean ± SD: 1.89 ± 0.02; range 1.85–1.98).
All collected maternal and cord blood samples (in EDTA tubes for plasma, in tubes without any coagulant for serum) were kept on ice, stored temporarily in a 4 °C refrigerator and centrifuged (4000 r/min for 10 min) within 2 h after the specimen collection. The separated serum and plasma samples were stored in multiple aliquots at − 80 °C until assays.
Genome-wide DNA methylation measurements
Prior to DNA isolation, placental tissue samples were homogenized for 1 min at 6000 rpm × 3 (5 min on ice in between intervals) in lysis buffer (180 μl buffer ATL with 20 μl proteinase K). Placental DNA was then isolated using the DNAeasy kit (Qiagen, UK, Catalog # 69,504) according to manufacturer’s instructions. DNA (500 ng) was treated with bisulfate using an EZ DNA Methylation Gold kit (Zymo Research, Irvine, CA, Catalog # D5006) according to the manufacturer’s instructions. DNA methylations were measured by Illumina Human Methylation EPIC BeadChip (Illumina, Inc., San Diego, CA, USA), which provides genome-wide coverage containing > 850,000 CpG methylation sites. The experiments followed the manufacturer’s protocol (https://emea.support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/infinium_assays/infinium_hd_methylation/infinium-hd-methylation-guide-15019519-01.pdf). Samples were randomly placed in different slides in the experiments.
Package “minfi” was used to import and preprocess raw methylation data. For all the study samples, the proportion of CpG sites with detection P value > 0.01 was less than 5%. Thus, all samples were included in subsequent data analyses. We excluded 2,677 CpG sites with detection P value > 0.01 in more than 5% of all the samples. Functional normalization was applied to remove between-array (unwanted) variations using control probes [47]. We further excluded 41,710 CpG sites with bead count < 3 in more than 5% of all the samples, 18,487 annotated to sex chromosomes, 83,460 SNPs inside the probe body, in the CpG interrogation site, or at the single nucleotide extension with a minor allele frequency of ≥ 0.05, 33,202 suspected cross-reactive sites [48], and 2301 non-CpG sites, leaving 684,022 CpG sites in subsequent data processing. Beta-mixture quantile dilation (BMIQ) was then applied to adjust for type 2 probe bias [49]. We filtered out sites with average methylation (β value) < 5% or > 95% (n = 183,267), as extreme β values tended to have low reproducibility [50], and small-to-moderate changes in methylation levels may not have much biologically significant implications at both extremes. A total of 500,755 CpG sites were retained in the final epigenome-wide association analysis. Potential bias due to different slides was adjusted for using the ComBat function in sva package [51]. The density plot did not reveal significant differences in beta value distributions between LGA and OGA (Additional file 1: Figure S1). Beta values were transformed to M values using lumi package in differential methylation analysis since M values had better statistical performance [52]. Beta values were presented for quantifying the differences in methylation levels between groups for data interpretation.
Pyrosequencing validation study
We sought to validate several DMPs identified in the genome-wide discovery analysis. The study subjects were an independent random sample of 47 pairs of LGA-OGA (control) subjects from the Shanghai birth cohort. Placental DNA was sodium-bisulfite treated using the EZ-96 DNA Methylation-Lighting Kit (Zymo Research, Irvine, CA, Catalog # D5006), and PCR-amplified with primers designed by PyroMark Assay Design software (version 2.0, Qiagen). All procedures were performed according to the manufacturer’s protocols. Pyrosequencing was performed using the PyroMark Q48 system (Qiagen), and cytosine methylation was quantified using the PyroMark Q241.010 software.
Biochemical assays
Cord serum insulin and insulin-like growth factor 1 (IGF-I) were measured by chemiluminescent assays (ADVIA Centaur and Immulite2000, SIEMENS, Germany). Cord plasma IGF-II was measured by an ELISA kit from R&D system (Minnesota, USA, catalog # DG200), and plasma C-peptide and proinsulin by ELISA kits from Mercodia system (Uppsala, Sweden. catalog # 10–1136-01 for C-peptide, catalog # 10–1118-01 for proinsulin), respectively. Plasma total and high-molecular-weight (HMW) adiponectin were measured by an ELISA kit from ALPCO (Salem, NH, USA, catalog # 47-ADPHU-E01), and plasma leptin by an ELISA kit from Invitrogen (Carlsbad, CA, USA, catalog # KAC2281), respectively. Maternal whole blood HbA1c was measured by high-performance liquid chromatography (BIO-RAD VARIANT II, California, USA). The detection limits were 3.5 pmol/l for insulin, 25 ng/ml for IGF-I, 1.88 pg/ml for IGF-II, 1.7 pmol/l for proinsulin, 25 pmol/L for C-peptide, 0.034 ng/mL for HMW and total adiponectin and 3.5 pg/mL for leptin, respectively. Intra-assay and inter-assay coefficients of variation were in the ranges of 2.0–6.5% for insulin and IGF-I, 5.0–8.6% for proinsulin, 0.4–13.5% for C-peptide and 2.4–9.3% for IGF-II, 6.9%–10.4% for leptin, total and HMW adiponectin, respectively. In all biomarker assays, the laboratory technicians were blinded to the clinical status (LGA or not) of study subjects.
Statistical analysis
All analyses were conducted in R using the R studio (https://www.rstudio.com/). The association between LGA status and DNA methylation (M-value) in each CpG site was assessed by lmFit function in limma package. For placental cell types, we used the ReFACTor package to select the number of principal components and then included the selected components (k = 4) as the covariates in adjusting for cell-type heterogeneity in differential methylation position (DMP) analyses [53]. The selection of principal components was based on a score calculated as the -log of the ratio of two adjacent eigenvalues [the i-th eigenvalue to the (i-1)-th eigenvalue] in the principal components analysis. The number of principal components was chosen (k = 4) when the score was near 0. The comparisons were adjusted for important covariates including maternal age, pre-pregnancy BMI, and glycosylated hemoglobin levels (HbA1c) during the second and third trimesters of pregnancy. To minimize false discovery findings, p values were adjusted according to Benjamini and Hochberg’s method in correction for multiple tests. DMPs comparing LGA and control groups were selected at false discovery rate (FDR) < 5% and absolute methylation difference (delta beta) > 0.05 (to identify “true” DMPs with differences that are unlikely to be measurement errors). To identify the most important differentially methylated genes that are correlated with birth weight, the Least Absolute Shrinkage and Selection Operator (LASSO)-regression [54] was performed using the “glmnet” package.
To understand the functional roles of the DMPs, we performed Ingenuity Pathway Analysis (IPA) to annotate the significant canonical pathways and performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses using the missMethyl package [55].
At the region-level, differentially methylated regions (DMRs) were identified using the DMRcate package [56] and comb-p [57]. DMRcate package identifies the differentially methylated regions based on tunable kernel smoothing of the signal of methylation changes [56]. As recommended by the authors, a bandwidth of 1000 nucleosides and a scaling factor of 2 were used. Comb-p deals with autocorrelations in neighboring p values and reports region-based p values using Sidak correction for multiple tests. P value < 10–3 was set to start a region, and a distance of 200 bp was selected to extend the region in the presence of another P value < 10–3. Significant DMRs were selected at FDR < 5%.
Pearson partial correlations were used to examine the associations of DNA methylations with cord blood biomarkers (leptin, adiponectin, insulin, proinsulin, C-peptide, IGF-I and IGF-II) adjusting for gestational age at birth. First, we assessed the correlations with DNA methylation levels in the corresponding specific genes (LEP, ADIPOQ, IGF1, INS-IGF2). Of the 500,755 CpG sites, a total of 21 sites were annotated to LEP (chr7: 127,876,829–127,894,849), 12 sites to ADIPOQ (chr3: 186,559,147–186,575,325), 12 sites to IGF1 (chr1:102,795,843–102,875,301) and 119 sites to INS/INS-IGF2/IGF2 (chr11: 2,150,687–2,183,864). The total numbers of sites remained in the partial correlation analyses were 21 for LEP, 12 for ADIPOQ, 12 for IGF1 and 119 for INS/INS-IGF2/IGF2 genes, respectively. Benjamini–Hochberg’s method was used in calculating the p values in correction for multiple tests. Second, we assessed the correlations of LGA-associated DMPs with fetal growth (birth weight z score) and cord blood biomarkers. CpG sites with crude P values < 0.05 were presented along with the Benjamini–Hochberg corrected P values accounting for multiple tests.
Availability of data and materials
Access to the deidentified participant research data must be approved by the research ethics board on a case-by-case basis; please contact the corresponding authors (zcluo@lunenfeld.ca; jl@clin.au.dk) for assistance in data access request. The EWAS data on placental gene methylations are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE204977.
Abbreviations
- LGA:
-
Large-for-gestational-age
- OGA:
-
Optimal-for-gestational-age
- SBC:
-
Shanghai birth cohort
- DMP:
-
Differential methylation position
- DMR:
-
Differential methylation region
- DNA:
-
Deoxyribonucleic acid
- BMI:
-
Body mass index
- HbA1c:
-
Glycosylated hemoglobin A1c
- IGF-I:
-
Insulin-like growth factor I
- IGF-II:
-
Insulin-like growth factor II
- IPA:
-
Ingenuity pathway analysis
- FDR:
-
False discovery rate
References
Gluckman P, Hanson M. Living with the past: evolution, development, and patterns of disease. Science (New York, NY). 2004;305(5691):1733–6.
Calkins K, Devaskar S. Fetal origins of adult disease. Curr Probl Pediatr Adolesc Health Care. 2011;41(6):158–76.
Chiavaroli V, Marcovecchio M, de Giorgis T, Diesse L, Chiarelli F, Mohn A. Progression of cardio-metabolic risk factors in subjects born small and large for gestational age. PLoS ONE. 2014;9(8): e104278.
Ma H, Pd G. Early developmental conditioning of later health and disease: Physiology or pathophysiology? Physiol Rev. 2014;94(4):1027–76.
Henriksen T, Clausen T. The fetal origins hypothesis: placental insufficiency and inheritance versus maternal malnutrition in well-nourished populations. Acta Obstet Gynecol Scand. 2002.
Cn H, Dj B. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5–20.
Smith ZD, Mesisner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–20.
Robins J, Marsit C, Padbury J, Sharma S. Endocrine disruptors, environmental oxygen, epigenetics and pregnancy. Front Biosci (Elite Ed). 2011;3:690–700.
Shen Z, Tang Y, Song Y, Shen W, Zou C. Differences of DNA methylation patterns in the placenta of large for gestational age infant. Medicine. 2020;99(39): e22389.
Chen P, et al. Prenatal growth patterns and birthweight are associated with differential DNA methylation and gene expression of cardiometabolic risk genes in human placentas: a discovery-based approach. Reprod Sci (Thousand Oaks, Calif). 2018;25(4):523–39.
Miranda-Lora A, et al. Exploring single nucleotide polymorphisms previously related to obesity and metabolic traits in pediatric-onset type 2 diabetes. Acta Diabetol. 2017;54(7):653–62.
Yan X, et al. Cadm2 regulates body weight and energy homeostasis in mice. Mol Metab. 2018;8:180–8.
Chiavaroli V, et al. Infants born large-for-gestational-age display slower growth in early infancy, but no epigenetic changes at birth. Sci Rep. 2015;5:14540.
Dwi Putra S, et al. Being born large for gestational age is associated with increased global placental DNA methylation. Sci Rep. 2020;10(1):927.
Gagné-Ouellet V, et al. Placental lipoprotein lipase DNA methylation alterations are associated with gestational diabetes and body composition at 5 years of age. Epigenetics. 2017;12(8):616–25.
Hu M, et al. Chromatin 3D interaction analysis of the STARD10 locus unveils FCHSD2 as a regulator of insulin secretion. Cell Rep. 2021;34(5): 108703.
Brown M, et al. Electrogenic sodium bicarbonate cotransporter NBCe1 regulates pancreatic β cell function in type 2 diabetes. J Clin Investig. 2021. https://doi.org/10.1172/JCI142365.
Bolton J, et al. Molecular biomarkers for weight control in obese individuals subjected to a multiphase dietary intervention. J Clin Endocrinol Metab. 2017;102(8):2751–61.
Al-Barghouthi BM, et al. Systems genetics in diversity outbred mice inform BMD GWAS and identify determinants of bone strength. Nat Commun. 2021;12(1):3408.
Hamoud N, Tran V, Croteau LP, Kania A, Côté JF. G-protein coupled receptor BAI3 promotes myoblast fusion in vertebrates. Proc Natl Acad Sci USA. 2014;111(10):3745–50.
Gupta R, et al. Complement 1q-like-3 protein inhibits insulin secretion from pancreatic β-cells via the cell adhesion G protein-coupled receptor BAI3. J Biol Chem. 2018;293(47):18086–98.
Nguyen Ding Cat A, et al. Adipocyte-specific mineralocorticoid receptor overexpression in mice is associated with metabolic syndrome and vascular dysfunction: role of redox-sensitive PKG-1 and rho kinase. Diabetes. 2016;65(8):2392–403.
Ranscht B, Dours-Zimmermann M. T-cadherin, a novel cadherin cell adhesion molecule in the nervous system lacks the conserved cytoplasmic region. Neuron. 1991;7(3):391–402.
Chung C, et al. A genome-wide association study reveals a quantitative trait locus of adiponectin on CDH13 that predicts cardiometabolic outcomes. Diabetes. 2011;60(9):2417–23.
Morisaki H, et al. CDH13 gene coding T-cadherin influences variations in plasma adiponectin levels in the Japanese population. Hum Mutat. 2012;33(2):402–10.
Göddeke S, et al. CDH13 abundance interferes with adipocyte differentiation and is a novel biomarker for adipose tissue health. Int J Obes (Lond). 2018;42(5):1039–50.
Tyrberg B, et al. T-cadherin (Cdh13) in association with pancreatic β-cell granules contributes to second phase insulin secretion. Islets. 2011;3(6):327–37.
Guan T, Wang X, Zheng LB, Wu HJ, Yao YF. Analysis of the VSX1 gene in sporadic keratoconus patients from China. BMC Ophthalmol. 2017;17(1):173.
Barbaro V, et al. Expression of VSX1 in human corneal keratocytes during differentiation into myofibroblasts in response to wound healing. Invest Ophthalmol Vis Sci. 2006;47(12):5243–50.
Huse JT, et al. Endoproteolysis of beta-secretase (beta-site amyloid precursor protein-cleaving enzyme) within its catalytic domain. A potential mechanism for regulation. J Biol Chem. 2003;278(19):17141–9.
Meakin PJ, et al. Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem J. 2012;441(1):285–96.
Plucinska K, et al. Neuronal human BACE1 knockin induces systemic diabetes in mice. Diabetologia. 2016;59(7):1513–23.
Lee HJ, et al. High glucose upregulates BACE1-mediated Abeta production through ROS-dependent HIF-1alpha and LXRalpha/ABCA1-regulated lipid raft reorganization in SK-N-MC cells. Sci Rep. 2016;6:36746.
Baier L, et al. Positional cloning of an obesity/diabetes susceptibility gene(s) on chromosome 11 in Pima Indians. Ann NY Acad Sci. 2002;967:258–64.
van Spronsen M, et al. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77(3):485–502.
Lake N, et al. TRAK2, a novel regulator of ABCA1 expression, cholesterol efflux and HDL biogenesis. Eur Heart J. 2017;38(48):3579–87.
Scott H, Antonarakis S, Lalioti M, Rossier C, Silver P, Henry M. Identification and characterization of two putative human arginine methyltransferases (HRMT1L1 and HRMT1L2). Genomics. 1998;48(3):330–40.
Iwasaki H, et al. Disruption of protein arginine N-methyltransferase 2 regulates leptin signaling and produces leanness in vivo through loss of STAT3 methylation. Circ Res. 2010;107(8):992–1001.
Lin X, et al. Altered DNA methylation in neonates born large-for-gestational-age is associated with cardiometabolic risk in children. Oncotarget. 2016;7(52):86511–21.
Bouchard L, Hivert M, Guay S, St-Pierre J, Perron P, Brisson D. Placental adiponectin gene DNA methylation levels are associated with mothers’ blood glucose concentration. Diabetes. 2012;61(5):1272–80.
Pinar H, et al. High molecular mass multimer complexes and vascular expression contribute to high adiponectin in the fetus. J Clin Endocrinol Metab. 2008;93(7):2885–90.
Mazaki-Tovi S, et al. Determining the source of fetal adiponectin. J Reprod Med. 2007;52(9):774–8.
Viengchareun S, Zennaro MC, Pascual-Le Tallec L, Lombes M. Brown adipocytes are novel sites of expression and regulation of adiponectin and resistin. FEBS Lett. 2002;532(3):345–50.
Chen J, et al. Secretion of adiponectin by human placenta: differential modulation of adiponectin and its receptors by cytokines. Diabetologia. 2006;49(6):1292–302.
Zhang J, et al. Cohort profile: the Shanghai birth cohort. Int J Epidemiol. 2019;48(1):21-g.
Zhu L, et al. [Chinese neonatal birth weight curve for different gestational age]. Zhonghua er ke za zhi = Chinese journal of pediatrics. 2015;53(2):97–103.
Fortin J-P, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15(11):1–17.
Pidsley R, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17(1):1–17.
Teschendorff AE, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–96.
Logue MW, et al. The correlation of methylation levels measured using Illumina 450K and EPIC BeadChips in blood samples. Epigenomics. 2017;9(11):1363–71.
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–3.
Du P, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010;11(1):1–9.
Rahmani E, et al. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods. 2016;13(5):443.
Tibshirani R. Regression shrinkage and selection via the lasso: a retrospective. J R Stat Soc Ser B (Stat Methodol). 2011;73(3):267–88.
Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics (Oxford, England). 2016;32(2):286–8.
Peters T, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6.
Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28(22):2986–8.
Acknowledgements
We gratefully acknowledged all research staff who had contributed to patient recruitment and data collection in the Shanghai birth cohort. ZC Luo is the guarantor of this work taking full responsibility for the work as a whole including the study design, access to data and the decision to submit and publish the manuscript.
Funding
Funding was supported by research grants from the Ministry of Science and Technology of China (2019YFA0802501, 2017YFE0124700), the Shanghai Municipal Science and Technology Commission (21410713500, 19410713500), the Shanghai Municipal Health Commission (2020CXJQ01, 2019Y0157), the National Natural Science Foundation of China (82073570, 81903323, 81761128035, 81930095, 82125032 and 81961128023), the National Human Genetic Resources Sharing Service Platform (2005DKA21300) and the Canadian Institutes of Health Research (155955, 158616). The funders have no role in all aspects of the study, including study design, data collection and analysis, the preparation of the manuscript and the decision for publication.
Author information
Authors and Affiliations
Consortia
Contributions
FL, JGF, FO, JZ, JL and ZCL conceived the study. MNY, RH, WJW, TZ, YD, YJX, VM, GDZ, XL, HH, FF, FL, JGF, JZ, FO, LB, JL and ZCL contributed to the acquisition of research data and/or data interpretation. MNY, RH and VM conducted the literature review. MNY and RH conducted data analysis and drafted the manuscript. All authors contributed to revising the article critically for important intellectual content and approved the final version for publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the research ethics committees of Xinhua Hospital (the coordination center, reference number M2013-010) and all participating hospitals. Written informed consent was obtained from all study participants.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing conflicts.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Figure S1.
Density plots of beta values in LGA and OGA (control) groups.
Additional file 2.
Appendix Tables S1, S4, S5, S6, S7, S8, S9, S10 and S11.
Additional file 3.
Appendix Tables S2, S3 and S12.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, MN., Huang, R., Zheng, T. et al. Genome-wide placental DNA methylations in fetal overgrowth and associations with leptin, adiponectin and fetal growth factors. Clin Epigenet 14, 192 (2022). https://doi.org/10.1186/s13148-022-01412-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-022-01412-6