
Abstract
Background
Whereas duplications in 11p15.5 covering both imprinting centers (ICs) and their subordinated genes account for up to 1% of Beckwith–Wiedemann and Silver–Russell syndrome patients (BWS, SRS), the deletions in 11p15.5 reported so far only affect one of the ICs. In these cases, not only the size and gene content had an impact on the phenotype, but also the sex of the contributing parent influences the clinical signs of the deletion carrier.
Results
We here report on the first case with a heterozygous deletion within the maternal allele affecting genes which are regulated by both ICs in 11p15.5 in a BWS patient, and describe the molecular and clinical consequences in case of its maternal or paternal inheritance.
Conclusions
The identification of a unique deletion affecting both 11p15.5 imprinting domains in a BWS patient illustrates the complexity of the regulation mechanisms in these key imprinting regions.
Similar content being viewed by others

Background
The two differentially methylated imprinting control regions 1 and 2 (IC1, IC2) regulate the monoallelic and parent-of-origin dependent expression of a cluster of imprinted genes on chromosome 11p15.5. Whereas the paternally expressed IGF2 and the maternally expressed H19 genes are controlled by the telomeric and paternally methylated IC1 (H19/IGF2:IG-DMR), the maternally methylated and more centromeric IC2 (harboring the KCNQ1OT1:TSS-DMR) has an impact on the maternally expressed CDKN1C and the paternally expressed KCNQ1OT1 genes.
Disturbances of the two ICs (aberrant methylation/epimutations of one of the two ICs; uniparental disomies and copy number variants of both ICs) are associated with two congenital imprinting disorders, Beckwith–Wiedemann and Silver–Russell syndrome (BWS: OMIM130650; SRS: OMIM180860). These two disorders show opposite molecular defects as well as opposite growth phenotypes. BWS is an overgrowth disorder with an increased risk of developing embryonal tumors and several birth defects (e.g., macroglossia, abdominal wall defects, neonatal hypoglycemia, lateralized overgrowth) (for review: [1]). Molecularly, the majority of patients exhibit a loss of methylation (LOM) of the IC2 (40%), followed by paternal uniparental disomies of 11p15.5 and gain of methylation (GOM) of the IC1. SRS is a growth retardation syndrome characterized by relative macrocephaly, a typical facial gestalt, asymmetry, feeding difficulties, and other less constant features (for review: [2]). In up to 40% of patients, a LOM of the IC1 can be observed. In addition to (epi)genetic disturbances of the IC1 and/or IC2, pathogenic variants within the IGF2, CDKN1C and KCNQ1 genes have been reported in BWS and SRS (for review: [3,4,5]).
Deletions in 11p15.5 are rare [6,7,8,9,10], and losses affecting both imprinting regions have not yet been reported [8, 11,12,13]. In contrast, duplications of different sizes account for up to 1% of BWS and SRS patients, and they are either restricted to one of the two ICs, or affect both (for review: [13]).
We here report on the first case with a heterozygous deletion affecting the maternally inherited allele and genes regulated by both ICs in 11p15.5 in a BWS patient, and describe the molecular and clinical consequences in case of its maternal or paternal inheritance.
Results
In a patient referred for routine molecular genetic testing for BWS, an unusual deletion within 11p15.5 was identified. The patient was born preterm to healthy non-related German parents at 36 weeks of gestation because of premature labor. Birth weight was 3820 g (2.15 z) [14], length 53 cm (1.5 z). A polyhydramnion was documented. Hypoglycemia was mentioned for first the 6 months of life, but treatment was not required. An omphalocele required five surgical interventions. Neuropsychomotor development was normal.
At the age of 30 years, the patient sought for genetic counselling. His height was 190 cm (1.37z), weight 110 kg (2.73z), and head circumference 58.8 cm (1.32 z). His left leg was 3 cm larger than the other. Pits were present at both ears. Clinical scoring on the basis of the recently consented system for BWS [1] resulted in a score of 9 points, supporting the clinical diagnosis of BWS (according to that system, a score of ≥ 4 points corresponds to BWS). Tumor monitoring until the seventh year of life was negative, as was cardiological surveillance at the age of 28 years.
Family history was empty, there was no history of assisted reproduction. The parents were of normal heights (mother 168 cm, father 175 cm), as was the healthy sister (172 cm).
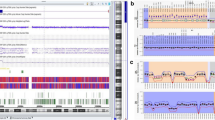
By methylation-specific multiplex ligation-dependent probe amplification assays (MS MLPA), a deletion affecting the KCNQ1 gene and a loss of methylation of the IC2 was detected (Fig. 1), whereas methylation of the IC1 was normal. SNP array analysis revealed a size of 591 kb, (arr[hg19] 11p15.5(2125923_2716862) × 1)), including the whole IGF2 gene, exons 1 to 12 of the KCNQ1 gene (NM_000218.2), the whole KCNQ1OT1 gene as well as further not imprinted genes (INS, TH, ASCL2, C11orf21, TSPAN32, CD81, TSSC4, TRPM5). In accordance with the MS MLPA results, neither the KCNQ1OT1:TSS-DMR nor the H19/IGF2:IG-DMR were affected.

Identification of a deletion affecting the maternal KCNQ1 allele by a methylation-specific MLPA analysis (assay ME030: upper panel: Copy number analysis, lower panel: methylation analysis) and b KCNQ1-specific MLPA (assay P114). The patient exhibits a deletion within the KCNQ1 gene, but the IC2 sequence itself was not affected. Nevertheless, hypomethylation of the IC2 could be observed. (The control range was based on five individuals of normal epigenotype)
MS-MLPA analyses of parental DNA samples targeting the 11p15.5 region gave normal copy number and methylation results.
Discussion
We report on the first deletion affecting parts of both imprinting domains in 11p15.5. Whereas recent reports describe patients in which only one of the two domains was affected and the disturbances had an impact on genes regulated by either the IC1 or the IC2, in our case, the disturbance of both regions has to be considered with respect to clinical significance and genetic counselling (Fig. 2).

Simplified (hypothetical) effects of the deletion in our patient on the regulation of the imprinting domain in 11p15.5. In the upper figure, the normal situation is shown, whereas the effects of deletions in the maternal (corresponding to our patient) or in the paternal allele are illustrated and described in the lower figures. (*consequences for the KCNQ1 isoform 2 which underlies genomic imprinting during embryogenesis; not to scale; arrows: expression of genes; filled lollipops: methylated ICs, empty lollipops: unmethylated ICs; green circles: enhancer elements; grey rhomb: CTCF; − suppression of expression, + enhancing of expression)
In our patient, the 11p11.5 is associated with a loss of methylation of the maternally methylated IC2. It can therefore be concluded that the maternal 11p15.5 allele is affected, but that it probably occurs de-novo as molecular analysis of parental DNA samples gave normal results. As molecular alterations resulting in a IC2 loss of methylation are associated with BWS features, the phenotype in our patient is attributable to the disturbance of the centromeric imprinting domain. The deletion affects the KCNQ1 and KCNQ1OT1 genes, but the IC2 itself is not deleted. However, as several reports on pathogenic variants causing an aberrant KCNQ1 transcript have already shown, only an intact KCNQ1 transcript on the maternal chromosome can drive across the IC2, and is thereby the prerequisite for the IC2 de-novo methylation in the oocyte [5, 15, 16]. Accordingly, in case the deletion affects the maternal allele the IC2 remains hypomethylated, a molecular finding which is characteristic for BWS. For the transmission of the deletion via the paternal allele it can hypothesized that the variant does not alter the methylation status of the IC2 as the paternal allele is per-se not methylated.
The deletion also affects the noncoding RNA KCNQ1OT1 which is transcribed only from the paternal allele, and suppresses the expression of the paternal CDKN1C copy, a negative regulator of cell proliferation. If KCNQ1OT1 is partly deleted on the maternal allele, this should not impact CDKN1C expression as the maternal KCNQ1OT1 is silenced whereas CDKN1C is expressed (Fig. 2). However, the deletion might affect CDKN1C expression as an enhancer motif for its expression has been suggested in this region [17]. Thus, a (slight) decrease of CDKN1C expression can be postulated and might contribute to the overgrowth phenotype in our patient. In case the deletion affects the paternal allele, expression of KCNQ1OT1 is suppressed, and the overdose of CDKN1C results in a growth retardation phenotype [10].
Independent of the sex of the transmitting parent, the deletion of the first 12 exons of the KCNQ1 gene predisposes for Long QT 1 syndrome. Therefore, carriers of truncating KCNQ1 variants should be monitored cardiologically despite the variable penetrance of LQTS [18] which is confirmed by the negative cardiological examination results in our patient.
In the telomeric imprinting domain of our patient, the coding sequencing of the IGF2 gene is deleted, but the H19/IGF2:IG-DMR is not affected and shows a normal methylation pattern.
As IGF2 is transcribed from the paternal allele only, the deletion on the maternal chromosome 11p15.5 in our patient does not have an impact on his phenotype. The situation would change in case the paternal allele is affected, here the lack of IGF2 should result in a growth retardation phenotype [3]. Children of our patient therefore have a chance of 50% to inherit the deletion and to be growth retarded due to the decreased expression of IGF2 and increase of CDKN1C expression. The genomic sequence of H19 gene is not affected by the deletion, but an alteration of its expression cannot be excluded as the chromatin structure of the region might be changed by the alteration. Finally, an altered expression of the other not imprinted genes within the deleted region cannot be precluded. However, evidences for their clinical relevance in patients with deletions or duplications in this region have not yet been reported.
Conclusions
We report on a unique BWS patient with an alteration affecting both 11p15.5 imprinting domains, and thereby confirm the complexity of the regulation mechanisms in these key imprinting regions. Due to the different clinical consequences of 11p15.5 disturbances and the impact of the sex of the contributing parent, their precise size and genomic content has to be determined.
Materials and methods
Genomic DNA of the patient was isolated from peripheral blood lymphocytes by simple salting out. Due to the BWS phenotype, the IC1 and IC2 in 11p15.5 were analyzed by two commercially available methylation-specific multiplex ligation-dependent probe amplification kits (MS MLPA) (ME030-C3 and ME034-A1, MRC Holland, Amsterdam/NL). The result was confirmed by another MLPA assay targeting the KCNQ1 gene (assay P114-B3) (Fig. 1). Further characterization was conducted by SNP array analysis (CytoScan® HD Array (Affymetrix, Santa Clara/CA, USA)).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BWS:
-
Beckwith–Wiedemann syndrome
- IC1:
-
Imprinting center region 1
- IC2:
-
Imprinting center region 2
- GOM:
-
Gain of methylation
- LOM:
-
Loss of methylation
- MS MLPA:
-
Methylation-specific multiplex ligation-dependent probe amplification
- SRS:
-
Silver–Russell syndrome
References
Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, et al. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018;14(4):229–49.
Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM, Salem J, Bliek J, et al. Diagnosis and management of Silver–Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017;13(2):105–24.
Masunaga Y, Inoue T, Yamoto K, Fujisawa Y, Sato Y, Kawashima-Sonoyama Y, et al. IGF2 mutations. J Clin Endocrinol Metab. 2020;105(1):116–25.
Eggermann T, Binder G, Brioude F, Maher ER, Lapunzina P, Cubellis MV, et al. CDKN1C mutations: two sides of the same coin. Trends Mol Med. 2014;20(11):614–22.
Essinger C, Karch S, Moog U, Fekete G, Lengyel A, Pinti E, et al. Frequency of KCNQ1 variants causing loss of methylation of Imprinting Centre 2 in Beckwith–Wiedemann syndrome. Clin Epigenet. 2020;12(1):63.
Niemitz EL, DeBaun MR, Fallon J, Murakami K, Kugoh H, Oshimura M, et al. Microdeletion of LIT1 in familial Beckwith–Wiedemann syndrome. Am J Hum Genet. 2004;75(5):844–9.
Algar E, Dagar V, Sebaj M, Pachter N. An 11p15 imprinting centre region 2 deletion in a family with Beckwith Wiedemann syndrome provides insights into imprinting control at CDKN1C. PLoS ONE. 2011;6(12):e29034.
Cytrynbaum C, Chong K, Hannig V, Choufani S, Shuman C, Steele L, et al. Genomic imbalance in the centromeric 11p15 imprinting center in three families: further evidence of a role for IC2 as a cause of Russell–Silver syndrome. Am J Med Genet A. 2016;170(10):2731–9.
Gurrieri F, Zollino M, Oliva A, Pascali V, Orteschi D, Pietrobono R, et al. Mild Beckwith–Wiedemann and severe long-QT syndrome due to deletion of the imprinting center 2 on chromosome 11p. Eur J Hum Genet. 2013;21(9):965–9.
De Crescenzo A, Sparago A, Cerrato F, Palumbo O, Carella M, Miceli M, et al. Paternal deletion of the 11p15.5 centromeric-imprinting control region is associated with alteration of imprinted gene expression and recurrent severe intrauterine growth restriction. J Med Genet. 2013;50(2):99–103.
Begemann M, Spengler S, Gogiel M, Grasshoff U, Bonin M, Betz RC, et al. Clinical significance of copy number variations in the 11p15.5 imprinting control regions: new cases and review of the literature. J Med Genet. 2012;49(9):547–53.
Sparago A, Cerrato F, Riccio A. Is ZFP57 binding to H19/IGF2:IG-DMR affected in Silver–Russell syndrome? Clin Epigenet. 2018;10:23.
Heide S, Chantot-Bastaraud S, Keren B, Harbison MD, Azzi S, Rossignol S, et al. Chromosomal rearrangements in the 11p15 imprinted region: 17 new 11p15.5 duplications with associated phenotypes and putative functional consequences. J Med Genet. 2018;55(3):205–13.
Kuczmarski RJ, Ogden CL, Guo SS, Grummer-Strawn LM, Flegal KM, Mei Z, et al. 2000 CDC Growth Charts for the United States: methods and development. Vital Health Stat 11. 2002;246:1–190.
Beygo J, Burger J, Strom TM, Kaya S, Buiting K. Disruption of KCNQ1 prevents methylation of the ICR2 and supports the hypothesis that its transcription is necessary for imprint establishment. Eur J Hum Genet. 2019;27(6):903–8.
Valente FM, Sparago A, Freschi A, Hill-Harfe K, Maas SM, Frints SGM, et al. Transcription alterations of KCNQ1 associated with imprinted methylation defects in the Beckwith–Wiedemann locus. Genet Med. 2019;21(8):1808–20.
Cerrato F, De Crescenzo A, Riccio A. Looking for CDKN1C enhancers. Eur J Hum Genet. 2014;22(4):442–3.
Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294(23):2975–80.
Acknowledgements
The group is funded by the Deutsche Forschungsgemeinschaft (DFG, EG110/15-1).
Funding
Open Access funding enabled and organized by Projekt DEAL. TE was supported by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation—EG110/15-1).
Author information
Authors and Affiliations
Contributions
TE planned the study and wrote the draft. LP examined the patients and supported writing. MB overviewed the molecular data. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was reviewed by the Ethics Committee of the Medical Faculty of Aachen (EK303-18). All individuals and caregivers gave written informed consent before participation. The study was performed in accordance with the Declaration of Helsinki of the World Medical Association.
Consent for publication
Not applicable.
Competing interests
The other authors have nothing to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Eggermann, T., Begemann, M. & Pfeiffer, L. Unusual deletion of the maternal 11p15 allele in Beckwith–Wiedemann syndrome with an impact on both imprinting domains. Clin Epigenet 13, 30 (2021). https://doi.org/10.1186/s13148-021-01020-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-021-01020-w