
Abstract
Background
Molecular genetic testing for the 11p15-associated imprinting disorder Beckwith-Wiedemann syndrome (BWS) is challenging because of the molecular heterogeneity and complexity of the affected imprinted regions. An integrated molecular approach to analyze the epigenetic-genetic alterations is required for accurate diagnosis of BWS.
Case presentation: We reported a Chinese case with BWS detected by SNP array analysis and methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA). The genetic analysis showed a de novo duplication of 24 Mb at 11p15.5p14.3 is much longer than ever reported. MS-MLPA showed copy number changes with a peak height ratio value of 1.5 (three copies) at 11p15. The duplication of paternal origin with increase of methylation index of 0.68 at H19 and decreased methylation index of 0.37 at KCNQ1OT1.
Conclusion
Combined chromosome microarray analysis and methylation profiling provided reliable diagnosis for this paternally derived duplication of BWS. The phenotype associated with 11p15 duplications depends on the size, genetic content, parental inheritance and imprinting status. Identification of these rare duplications is crucial for genetic counselling.
Similar content being viewed by others

Introduction
Beckwith-Wiedemann syndrome (BWS) (OMIM#130,650) is an imprinting disorder characterized by variable presence of macroglossia and over-growth, abdominal wall defects, hypoglycemia and embryonal tumor predisposition [1, 2]. Although BWS might present prenatally or in adult life, it is most commonly diagnosed in the neonatal period or in early childhood, with an estimated prevalence of one affected child per 10,340 live births [1]. BWS is caused mainly by genetic or epigenetic defects within the evolutionary conserved imprinted gene cluster located at chromosome 11p15.5 region [3] which consists of two imprinted domains, H19/IGF2:IG-DMR(IC1) and KCNQ1OT1:TSS-DMR(IC2) [4, 5]. Among several causative alterations identified so far, gain of methylation (GOM) at H19/IGF2:IG-DMR (5–10%), loss of methylation (LOM) at KCNQ1OT1:TSS-DMR (50%) which leads to biallelic expression of KCNQ1OT1 and biallelic silencing of CDKN1C, and 11p15 paternal uniparental disomy (UPD) (20%), loss-of-function pathogenic variants of CDKN1C (5%), and 11p15.5 copy-number variants (CNVs, 1–4%) are isolated epi-mutations [6]. Although most cases are sporadic, about 5–10% of BWS patients have inheritance characteristics [7]. We investigated a patient with clinical features of BWS. Genetic analyses revealed that the patient harbored pUPD and copy number variation. This is a rare reported case of a patient with 24 Mb duplication, which is much longer than ever reported.
Materials and methods
Patient report
The female infant was the first-born baby to non-consanguineous, healthy, Chinese parents after 36 weeks and 6 days of gestation in Jiaxing maternal and Child Health Hospital. Her birth weight was 3790 g, and she exhibit edcolumnar head, collapse of nasal bridge, wide distance of eyes, right hand through the palm, mild tricuspid regurgitation (Opposite velocity: 2.88 M/S, PG:33.2 mmHg), patent ductus arteriosus, the right ventricle is markedly enlarged. Bilateral ventricle dilation (Left: body: 9.3 mm, Relief angle: 19.7 mm. Right: Body: 6.6 mm, Relief angle: 17.5 mm). Abdominal hydrocele (Left lower abdomen: 15 × 8 mm anechoic area. Right abdomen: 47 × 13 mm anechoic area)lower xiphoid process abdominal bulge, rectus abdominis separation, renal hypertrophy (Left kidney: 54 × 27 mm, Right kidney: 56 × 29 mm), Long QT syndrome, thickened pulmonary texture, enlarged heart shadow, pulmonary hypertension, expanded perineal anal junction, mixed echo of right lobe of liver. This research was approved by the Ethics Committee of the Jiaxing Maternity and Child Health Care Hospital.
Genetic analyses
DNA isolation
Sample was collected and genomic DNA was extracted from the peripheral blood of the patient. Isolation and purification of the genomic DNA were performed using a Qiagen DNeasy Tissue Kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany).
SNP array analysis
Single nucleotide polymorphism (SNP) array analysis was performed with CytoScan 750 K/HD array (Carlsbad, CA, USA) according to the manufacturer’s protocol. The microarray was used to investigate the CNVs and absence of heterozygosity (AOH) events.
The locations of the CNVs and the UPD events were determined based on a human genome assembly from February 2009 (GRCH37/h19). OMIM genes and Ref-Seq genes were used to evaluate the CNVs identified in this study. The criteria used for interpreting whether a CNV was pathogenicor benign were according to the guidelines recommended by the American College of Medical Genetics (ACMG) [8].
MS-MLPA
Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) was carried out as a parallel test to confirm the variations observed by SNP array. The MS-MLPA probe (ME030 for BWS/SRS, MRC-Holland, Amsterdam, The Netherlands) was used to detect copy number changes and to analyze CpG island methylation statuses of the 11p15 region. The MS-MLPA procedure was performed according to the manufacturer’s instructions. The products were detected via capillary electrophoresis using an ABI-3100 genetic analyzer (Applied Biosystems) and were analyzed using Gene Marker software (SoftGenetics, Pa., USA).
Results
Identifying Pathogenic CNVs via SNP array analysis
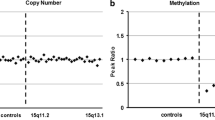
The SNP array result showed a 24,170 kb duplication at 11p15.5p14.3 including genes from DEAF1 to FANCF(arr[hg19]11p15.5p14.3 (230,680_24,400,276)×3) (Fig. 1a).

a The SNP array revealed a 24,170 kb duplication at (arr[hg19]11p15.5p14.3 (230,680_24,400,276)×3)). b MS-MLPA result of copy number change. c MS-MLPA result of methylation index
MS-MLPA Analysis
MS-MLPA analysis was used to validate the results observed with CMA. MS-MLPA for 11p15.5 showed copy number changes with a peak height ratio value of 1.5 (three copies) at 11p15 (Fig. 1b) in comparison with a ratio value of 1 (two copies) from a normal control. MS-MLPA indicated methylation index of 0.68 at IC1 and methylation index of 0.37 at IC2 (Fig. 1c). We indentified this patient with duplication of the BWS critical region.
Discussion
In this study, we report a Chinese BWS case with de novo paternally derived duplication and SNP-array test revealed a 24 Mb duplication at 11p15.5p14.3, involving 210 OMIM genes. It’s a pity that we did not perform a peripheral blood chromosome examination. A number of paternal reciprocal translocations associated with 11p15.5 duplications in the affected children have been reported [9,10,11,12,13]. It has been reported that patients with BWS due to a paternally inherited 11p15.5 duplication exhibit macroglossia, distinct craniofacial features, including prominent occiput and forehead, a round face with full cheeks, broad and flat nasal bridge, micrognathia, hypertelorism as well as deep set eyes with epicanthus.
Although our patient’s clinical phenotype fits well to this description, and the results from SNP array and MS-MLPA analysis fulfilled the diagnostic criteria for BWS, the 24 Mb duplication at 11p15.5p14.3 is much longer than ever reported [14, 15]. Qin Wang reported two Chinese cases with BWS, One case was a de novo paternal origin duplication spanning 896 Kb at 11p15.5. Case 2 was referred at 2-month old and the genetic analysis showed a de novo 228.8 Kb deletion at 11p15.5 telomeric end and a de novo duplication of 2.5 Mb at 11p15.4p15.5. Both duplications are paternal origin with gain of methylation at the imprinting center 1 [13]. In our case, the de novo duplication of 24 Mb is much longer and involving more genes.
We consider that most of the symptoms in our patient is caused or modulated by the duplication of these genes, including the OMIM genes H19, IGF2, TH, KCNQ1, STIMI and so on, involving 210 OMIM genes. H19 (103,280) plays a key role in the development of Beckwith-Wiedemann syndrome, Silver-Russell syndrome and it had been hypothesized that loss of H19 expression may be involved in Wilms tumorigenesis [16]. H19 is a developpmentally regulated gene with putative tumor suppressor activity. IGF2 (147,470) is a protein hormone involved in the regulation of cell proliferation, growth, migration, differentiation, and survival. It has been found that aberrant processing of pro-IGF2 by PCSK4 may be a cause of intrauterine growth restriction, a leading cause of perinatal mortality [17]. The expression of the IGF2 and H19 genes is imprinted. Although these neighboring genes share an enhancer, H19 is expressed only from the maternal allele, and IGF2 only from the paternally inherited allele. The region of paternal-specific methylation upstream of H19 appears to be the site of an epigenetic mark that is required for the imprinting of these genes. The KCNQ1OT1 (604,115) gene was expressed preferentially from the paternal allele [18], while KCNQ1 transcription is silent. In most patients with BWS, KCNQ1OT1 is abnormally expressed from both the paternal and maternal alleles. 21 of 36 (58%) BWS patients showed loss of maternal allele-specific methylation of a CpG island upstream of KCNQ1OT1. The authors determined that LOI of KCNQ1OT1 is the most common genetic alteration in BWS [19].
Chang reported a patient with a loss of heterozygosity in the region of chromosome 11p14.3 to 11p15.5. This region is similar to the case in this article. The results suggest that paternal uniparental isodisomy of chromosomal 11p15.5 is associated with the beta-thalassemia major in the patient [20]. In this case, the hemoglobin (Hb) of 117 g/L, RBCs of 3.17 × 10 × /L, may also be associated with the beta-thalassemia. Iourov demonstrated that shorter long contiguous stretches of homozygosity (LCSH) at chromosomes 7q21.3, 7q31.2, 11p15.5, and 15p11.2 occur with a frequency of about 5% in the children with intellectual disability, autism, congenital malformations and/or epilepsy. Consequently, this type of epigenetic mutations appears to be the most common one among children with neurodevelopmental diseases [21].
In this case, because the increase of paternal gene’s copy number, the H19 methylation index of 0.68 at IC1 is increased in comparison with normal control methylation index of 0.54 at IC1. And the KCNQ1OT1 methylation index of 0.37 at IC2 is decreased in comparison with normal control methylation index of 0.57 at IC2. A recent study in a serial of over 400 BWS cases also indicated that copy number changes in the 11p15.5 region contributed significantly to the etiology of the BWS [6]. Macchiaiolo M suggested that vascular tumors can also be associated with BWS[22]. In this case, the enlarged heart shadow, pulmonary hypertension, expanded perineal anal junction and mixed echo of right lobe of liver may also be with BWS. Papulino suggested there is an increased incidence of childhood tumor predisposition in BWS patients [9]. Cöktü [23] identified a group of 321 individuals with a molecularly confirmed diagnosis of BWS and analysed the cancer incidence. They confirmed an increased cancer risk in children with BWS. And suggested that the highest cancer risk is associated with UPDpat. So for this patient, the cancer risk may also increase, and physical examination should be performed rotinely for potential intellectual disability and the possible clinical effect involving the deleted genes. The phenotype associated with 11p15 duplications depends on the size, genetic content, parental inheritance and imprinting status. Identification of these rare duplications is crucial for genetic counselling. Furthermore, SNP arrays can be helpful in clarifying the molecular diagnosis in patient with BWS, especially to discriminate between pUPD and duplications [24].
References
Mussa A, Russo S, De Crescenzo A, et al. Prevalence of Beckwith-Wiedemann syndrome in North West of Italy. Am J Med Genet A. 2013;161:2481–6.
Brioude F, Kalish JM, Mussa A, et al. Expert consensus document: clinicaland molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat RevEndocrinol. 2018;14:229–49.
Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet. 2010;154C:343–54.
Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur JHum Genet. 2010;18:8–14.
Soejima H, Higashimoto K. Epigenetic and genetic alterations of the imprinting disorder Beckwith-Wiedemann syndrome and related disorders. J Hum Genet. 2013;58:402–9.
Baskin B, Choufani S, Chen YA, et al. High frequency of copy number variations (CNVs) in the chromosome 11p15 region in patients with Beckwith-Wiedemann syndrome. Hum Genet. 2014;133:321–30.
Ebinger C, Karch S, Moog U, et al. Frequency of KCNQ1 variants causing lossof methylation of Imprinting Centre 2 inBeckwith-Wiedemann syndrome. Clin Epigen. 2020;12(1):63–9.
Satoh Y, Nakadate H, Nakagawachi T, et al. Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic Wilms’ tumours. Br J Cancer. 2006;95:541–7.
Smith AC, Suzuki M, Thompson R, et al. (2012) Maternal gametic transmission of translocations or inversions of human chromosome 11p15.5 results in regional DNA hypermethylation and down regulation of CDKN1C expression. Genomics. 2012;99(1):25–35.
Krajewska-Walasek M, Gutkowska A, Mospinek-Krasnopolska M, et al. A new case of Beckwith-Wiedemann syndrome with an 11p15 duplication of paternal origin [46, XY,-21,+der(21), t(11;21)(p15.2;q22.3)pat]. Acta Genet Med Gemellol. 1996;45:245–50.
Slavotinek A, Gaunt L, Donnai D. Paternally inherited duplications of 11p15.5 and Beckwith-Wiedemann syndrome. J Med Genet. 1997;34:819–26.
Heide S, Chantot-Bastaraud S, Keren B, et al. Chromosomal rearrangements in the 11p15 imprinted region: 17 new 11p15.5 duplications with associated phenotypes and putative functional consequences. J Med Genet. 2018;55:205–13.
Wang Q, Geng Q, Zhou Q, et al. De novo paternal origin duplication of chromosome 11p15.5: report of two Chinese cases with Beckwith-Wiedemann syndrome. Mol Cytogen. 2017;10:46–52.
Lekszas C, Nanda I, Vona B, et al. Unbalanced segregation of a paternal t(9;11)(p24.3;p15.4) translocation causing familial Beckwith- Wiedemann syndrome: a case report. BMC Med Genom. 2019;12:83.
Bachmann N, Crazzolara R, Bohne F, et al. Novel deletion in 11p15.5 imprinting center region 1 in a patient with Beckwith-Wiedemann syndrome provides insight into distal enhancer regulation and tumorigenesis. Pediatr Blood Cancer. 2017;64:643–50.
Thorvaldsen JL, Duran KL, Bartolomei MS. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 1998;12:3693–702.
Calvello M, Tabano S, Colapietro P, et al. Quantitative DNA methylation analysis improves epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. Epigenetics. 2013;8(10):1053–60.
Vals MA, Kahre T, Mee P, et al. Familial 13-Mb 11p15.5p15.4 duplication in three generations causing silver-Russell and Beckwith-Wiedemann syndromes. Mol Syndromol. 2015;6(3):147–51.
Soejima H, Higashimoto K. Epigenetic and genetic alterations of the imprinting disorder Beckwith-Wiedemann syndrome and related disorders. J Hum Genet. 2013;58(7):402–9.
Chang JG, Tsai WC, Chong IW, et al. Beta-thalassemia major evolution from Beta-thalassemia minor is associated with paternal uniparental isodisomy of chromosome 11p15. Haematologica. 2008;93(6):913–6.
Iourov IY, Vorsanova SG, Korostelev SA, et al. Long contiguous stretches of homozygosity spanning shortly the imprinted loci are associated with intellectual disability, autism and/or epilepsy. Mol Cytogen. 2015;8:77–85.
Macchiaiolo M, Markowich AH, Diociaiuti A, et al. Diffuse infantile hepatic hemangiomas in a patient with Beckwith-Wiedemann syndrome: a new association? Am J Med Genet A. 2020;182(8):1972–6.
Cöktü S, Spix C, Kaiser M, et al. Cancer incidence and spectrum among children with genetically confirmed Beckwith-Wiedemann spectrum in Germany: a retrospective cohort study. Br J Cancer. 2020;123(4):619–23.
Keren B, Chantot-Bastaraud S, Brioude F, et al. SNP arrays in Beckwith-Wiedemann syndrome: an improved diagnostic strategy. Eur J Med Genet. 2013;56:546–50.
Acknowledgements
The authors appreciate the family to take part in this study and permission for the publication of the report.
Funding
This work was supported by Zhejiang Provincial Natural Science Foundation of China (LY20C110003), The Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (2020KY962). The Lin He’s Academician Workstation of New Medicine and Clinical Translation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors have no competing financial interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiang, H., Ping, Z., Wang, J. et al. A Beckwith-Wiedemann syndrome case with de novo 24 Mb duplication of chromosome 11p15.5p14.3. Mol Cytogenet 14, 14 (2021). https://doi.org/10.1186/s13039-021-00532-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-021-00532-7