
Abstract
Background
Neurofibroma is a rare benign tumor. The isolated presence of such lesions in the preperitoneal space with no evidence of systemic disease has never been reported in the literature.
Case presentation
A 29-year-old white man presented with a 12 months history of progressive abdominal distension. Clinical examination revealed a bulky hypogastric mass. Abdominal computed tomography and pelvic magnetic resonance imaging showed a large, well defined preperitoneal tumor measuring 18 x 17cm extending in the pelvis. A computed tomography guided biopsy was performed which revealed a neurofibroma. Exploratory laparotomy showed a well encapsulated elastic soft tumor in the preperitoneal space which measured 17 x 18cm and weighted 2 Kg. The tumor was completely excised. No recurrence occurred after one year of follow-up.
Conclusion
Solitary preperitoneal neurofibroma is an extremely rare benign tumor. Its clinical and radiological signs are nonspecific. Preoperative histology can be useful to guide the surgical approach which is the only curative treatment.
Similar content being viewed by others

Background
Neurofibroma is an uncommon benign tumor arising from the peripheral nerve sheaths. The isolated presence of neurofibromatous lesions in the preperitoneal space (between the parietal peritoneum and transversalis fascia), without any other sign of von Recklinghausen’s disease has rarely been reported [1]. Authors report an unusual case with a review of literature.
Case presentation
A 29-year-old white man, with no history of neurofibromatosis or other systemic disease, was complaining of insidious and progressive abdominal distension which appeared 12 months prior to admission. He had hypogastric abdominal pain and pollakiuries in the preceding five months. There were no vomiting or transit disorders, and no neurological symptoms.
On clinical examination, the patient was in good general condition with a normal temperature, a body mass index of 24 and no café au-lait spots or dermal neurofibromas on his body. The palpation of the abdomen revealed a well-defined, fixed and painless hypogastric mass, 20 cm in diameter. Digital rectal examination was normal.
All the standard hematological and serum biochemical data were normal.
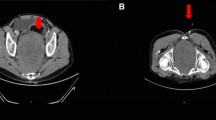
An abdomino-pelvic computed tomography (CT) scan (Figure 1) revealed a massive, inhomogeneous, hypodense, preperitoneal tumor, measuring 17 × 18cm extending in the pelvis, crushing both ureters and compressing the bladder. This mass was moderately enhanced with contrast. There were no signs of pelvic invasion or intraperitoneal effusion. Both kidneys were increased in size with dilation of renal cavities but the renal cortex was preserved.

Abdomino-pelvic computed tomography showed a massive, inhomogeneous, hypodense abdominal tumor.
Pelvic magnetic resonance imaging (Figure 2) revealed a large, well encapsulated preperitoneal tumor, slightly compressing the bladder and the pelvic ureters without invasion of the adjacent structures and without pelvic bone localization.

Pelvic magnetic resonance imaging showing a large, well encapsulated preperitoneal tumor.
A CT-guided biopsy was performed revealing a conjunctive tissue separated by a fuso-cellular proliferation compound of Schwann cells with regular nuclei immersed in a richly vascularized and myxoid fibrillar background. There was no evidence of malignancy within the tissue obtained. Immunohistochemically, the tumor cells were strongly positive for the S100 and the neurofilament proteins evoking a neurofibroma.
Surgical resection was decided by the multidisciplinary meeting for cancer care. Exploratory laparotomy revealed a well encapsulated elastic soft tumor in the preperitoneal space. Upon incision of the linea alba, there was a protrusion of the mass. It was easily cleaved before the opening of the anterior parietal peritoneum. At the end of excision, there was a peritoneal break due to size of the mass (Figure 3).

Intraoperative view showing the well encapsulated elastic soft tumor in the preperitoneal space.
The tumor measured 17 × 18 cm and weighted 2 Kg (Figure 4). It was completely excised without macroscopic capsule breaking. No lymph nodes or other palpable intra-abdominal masses were discovered.

Macroscopic appearance of the preperitoneal neurofibroma; size = 17x18cm, weight = 2 kg.
The histological and immuno-histochemical analysis of the surgical specimen confirmed the preoperative diagnosis (Figure 5). Thus, the tumor was diagnosed as a solitary neurofibroma. The post-operative course was uneventful. No adjuvant treatment was indicated. The patient remains well without any sign of tumor recurrence on a CT scan performed 12 months after surgery.

Benign tumor cells immersed in a myxoid collagen background (A) with elongated and wavy nuclei (B).
Comments
Neurofibroma is a tumor of neural origin derived from the cells that constitute the nerve sheath. It’s an uncommon benign tumor found as a solitary tumor or as a partial manifestation of neurofibromatosis type I (NF1, also called von Recklinghausen’s disease) [2]. The cause of solitary neurofibroma is still unknown [3], it is defined by Marocchio et al. [4] as a hyperplastic hamartomatous malformation rather than neoplastic. NF1 is an autosomal dominant genetic syndrome caused by mutations in genes coding for neurofibromin [5], it is characterized by cutaneous manifestations as cafe-au-lait spots with a large number of nervous system tumors [6]. The hereditary factors and systemic symptoms present in the disseminated neurofibromas are absent in the solitary type [7].
This kind of neoplasms is known to mainly affect the cutaneous nerves of the trunk, neck, and head [8,9]. Rare cases in the deep organs or in the peritoneal cavity were also reported [10]. Concerning the retroperitoneal location, exceptional cases of benign nerve sheaths tumors were reported in large series of primitive retroperitoneal tumors [11,12]. To our knowledge, the presence of solitary neurofibroma in the preperitoneal space has never been reported in the literature.
Neurofibroma appears to affect adolescents and young adults without a gender preference. It grows along the peripheral nerves as a non-encapsulated focal mass with well-defined margins [8,9].
The clinical manifestations of solitary neurofibromas are not specific and change according to their locations, their effect on gastrointestinal motility and their possible impingement on contiguous structures, resulting in palpable masses, abdominal pain, and transit disorders due to extra-luminal pressure [13-15]. The deep seated neurofibroma on peripheral nerves and spinal roots frequently leads to neurological disability [16].
Intra pelvic tumors are often diagnosed at a late stage which is source of surgical resection difficulties [17].
Preoperative imaging is usually insufficient to establish the diagnosis with certainty, only the histology can do. In fact, the preoperative histological evidence can influence the treatment modifying the surgical approach, conservative versus aggressive [10].
Surgical removal is the only treatment option. Unlike our case, neurofibromas are difficult to manage surgically as they are extensively infiltrative and highly vascular [15,18].
Some authors posit that surgical resection is indicated only when the tumor causes pain, neurological deficiencies or when there is a strong suspicion of malignancy [19].
Solitary neurofibromas have a good prognosis, with rare instances of haemorrhage, malignant changes and local recurrences after excision [18,20].
Conclusion
The solitary preperitoneal neurofibroma is an extremely rare benign tumor arising from nerve sheaths. Its clinical and radiological signs are not specific. Preoperative histological diagnosis can be useful to help choosing the surgical approach, which is the gold-standard treatment option. Unlike other sites, tumor excision in the preperitoneal location remains without difficulties.
Consent
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
- CT:
-
Computed tomography
- NF1:
-
Neurofibromatosis type I
References
Al-Harake A, Chour M, Al Beteddini OS. Solitary intestinal neurofibroma with no associated systemic syndromes causing intussusception: Case report and literature review. Int J Surg Case Rep. 2013;4(7):629–32.
Ishikawa J, Kamidono S, Maeda S, Sugiyama T, Hara I, Takechi Y, et al. Solitary retroperitonea neurofibroma: a case report. Hinyokika Kiyo. 1989;35(7):1157–60.
De Raedt T, Maertens O, Chmara M, Brems H, Heyns I, Sciot R, et al. Somatic loss of wild type NF1 allele in neurofibromas: Comparison of NF1 microdeletion and non-microdeletion patient. Genes Chromosomes Cancer. 2006;45(10):893–904.
Marocchio LS, Oliveira DT, Pereira MC, Soares CT, Fleury RN. Sporadic and multiple neurofibromas in the head and neck region: a retrospective study of 33 years. Clin Oral Investig. 2007;11(2):165–9.
Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62(5):1573–7.
Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J. Harrison’s principles of internal medicine. 18th ed. New York: McGraw-Hill; 2012.
Depprich R, Singh DD, Reinecke P, Kübler NR, Handschel J. Solitary submucous neurofibroma of the mandible: review of the literature and report of a rare case. Head Face Med. 2009;5:24.
Lee YB, Lee JI, Park HJ, Cho BK. Solitary neurofibromas: does an uncommon site exist? Ann Dermatol. 2012;24(1):101–2.
Ameur A, Lezrek M, Jira H, el Alami M, Beddouch A, Abbar M. Neurofibrome rétropéritonéal solitaire géant. Prog Urol. 2002;12(3):465–8.
Van Sandick JW, van Coevorden F. Plexiform neurofibroma with intraspinal extension. J Am Coll Surg. 2002;195(4):572.
Pinson CW, ReMine SG, Fletcher WS, Braasch JW. Long term results with primary retropritoneal tumors. Arch Surg. 1989;124(10):1168–73.
Scanlan DB. Primary retroperitoneal tumors. J Urol. 1959;81(6):740–5.
Panteris V, Vassilakaki T, Vaitsis N, Elemenoglou I, Mylonakou I, Karamanolis DG. Solitary colonic neurofibroma in a patient with transient segmental colitis: case report. World J Gastroenterol. 2005;11(35):5573–6.
Carter JE, Laurini JA. Isolated intestinal neurofibromatous proliferations in the absence of associated systemic syndromes. World J Gastroenterol. 2008;14(42):6569–71.
Honeypalsinh HM, Girish S. An Unusual Presentation of Anterior Abdominal Wall Neurofibroma. Int J Biol Med Res. 2013;4(4):3731–2.
Blakely P, Louis DN, Short MP, MacCollin M. A clinical study of patients with multiple isolated neurofibromas. J Med Genet. 2001;38(7):485–8.
Feldenzer JA, McGauley JL, McGillicuddy JE. Sacral and presacral tumors: problems in diagnosis and management. Neurosurgery. 1989;25(6):884–91.
Jones RG, Kiatisevi P, Morris DC, Munk PL, Clarkson PW, Masri BA. Intravascular embolisation and surgical resection ofa giant neurofibroma with intratumoural haemorrhage. Br J Radiol. 2010;83(995):e225–9.
Kubiena H, Entner T, Schmidt M, Frey M. Peripheral neural sheath tumors (PNST) what a radiologist should know. Eur J Radiol. 2013;82(1):51–5.
Corbellini C, Vingiani A, Maffini F, Chiappa A, Bertani E, Andreoni B. Retroperitoneal pararenal isolated neurofibroma: report of a case and review of literature. Ecancermedicalscience. 2012;6:253.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NN and GA wrote the paper, ME and HE provided data; ME, FHE, and MRC revised the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Njoumi, N., Elabsi, M., Attolou, G. et al. Solitary preperitoneal neurofibroma: a case report. BMC Res Notes 8, 115 (2015). https://doi.org/10.1186/s13104-015-1098-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-015-1098-8