
Abstract
Background
The evolution of Alu elements has been ongoing in primate lineages and Alu insertion polymorphisms are widely used in phylogenetic and population genetics studies. Alu subfamilies in the squirrel monkey (Saimiri), a New World Monkey (NWM), were recently reported. Squirrel monkeys are commonly used in biomedical research and often require species identification. The purpose of this study was two-fold: 1) Perform locus-specific PCR analyses on recently integrated Alu insertions in Saimiri to determine their amplification dynamics, and 2) Identify a subset of Alu insertion polymorphisms with species informative allele frequency distributions between the Saimiri sciureus and Saimiri boliviensis groups.
Results
PCR analyses were performed on a DNA panel of 32 squirrel monkey individuals for 382 Alu insertion events ≤2% diverged from 46 different Alu subfamily consensus sequences, 25 Saimiri specific and 21 NWM specific Alu subfamilies. Of the 382 loci, 110 were polymorphic for presence / absence among squirrel monkey individuals, 35 elements from 14 different Saimiri specific Alu subfamilies and 75 elements from 19 different NWM specific Alu subfamilies (13 of 46 subfamilies analyzed did not contain polymorphic insertions). Of the 110 Alu insertion polymorphisms, 51 had species informative allele frequency distributions between Saimiri sciureus and Saimiri boliviensis groups.
Conclusions
This study confirms the evolution of Alu subfamilies in Saimiri and provides evidence for an ongoing and prolific expansion of these elements in Saimiri with many active subfamilies concurrently propagating. The subset of polymorphic Alu insertions with species informative allele frequency distribution between Saimiri sciureus and Saimiri boliviensis will be instructive for specimen identification and conservation biology.
Similar content being viewed by others
Background
Short interspersed elements (SINEs) have been key mobile elements in genomic studies and have helped researchers delve into the structure and history of the genomes which they reside [1,2,3,4,5,6]. SINEs, specifically Alu elements, have been extremely important in understanding genomic diversity, systematics and phylogenomics within primates [7,8,9,10,11,12,13]. They have been shown to shape the structure of primate genomes [14] and play an important role in phylogenetic studies of primates [11, 13,14,15,16,17,18,19,20,21]. Alu elements are non-autonomous, non-long terminal repeat retrotransposons found in primate genomes. They are commonly used for these analyses due to their primate specificity, small size (~ 300 base pairs) and unidirectional mode of evolution [22,23,24,25,26]. Since they are unidirectional insertions, they allow for confident inference that the ancestral state of an element is the absence of that element for each locus under examination [27].
The squirrel monkey (genus Saimiri) is a small forest dwelling neotropical primate native to Central and South America that belongs to the family Cebidae. Squirrel monkeys are commonly used in biomedical research [28,29,30] since they have similar immune systems to humans. In addition, squirrel monkeys are small and more easily handled compared to large Old World primates such as the rhesus macaque and chimpanzee. Some of the biomedical studies focus on infectious disease, gene expression, cancer treatments, reproductive physiology, and viruses [31,32,33,34,35,36,37]. Species differences with regard to disease susceptibility has largely been overlooked until recently [38].
Prior to 1984, squirrel monkeys were considered a single species, Saimiri sciureus, with many subspecies geographically separated [28]. In 1984, Hershkovitz published a detailed taxonomy of squirrel monkeys. Hershkovitz divided Saimiri into two major groups, Saimiri boliviensis and Saimiri sciureus [39]. The S. boliviensis group has one species that is subdivided into two subspecies, S. boliviensis boliviensis and S. peruviensis. The S. sciureus group consists of three species, S. sciureus, S. oerstedii and S. ustus, with the former two species harboring six subspecies [39, 40]. Subsequent [41, 42] studies using molecular and genetic data have generally supported this classification system. The samples used in this study represented both major groups as well as subspecies S. boliviensis peruviensis, S. oerstedii oerstedii, and S. sciureus macrodon (Additional file 1). Given recent nomenclature changes, it is not surprising that some tissue samples or specimens from older studies/stocks in natural science museums may be labeled simply as Saimiri, squirrel monkey, or S. sciureus. This does not mean the samples are necessarily mislabeled, but more likely represent incomplete identification due to limited availability regarding source animal data at the time of sampling. Studies to develop systems for Saimiri species identification have attempted to resolve this issue [40,41,42,43,44] by using various types of genetic markers. Therefore, having more nuclear autosomal genetic markers, especially those which are identical by descent, such as Alu element retrotransposon insertions would increase the number of species informative genetic markers.
Few studies have been conducted on mobile element dynamics within New World primates; however, the studies available have provided great insight into their genomes. Specifically, Alu elements have given a good representation of genome evolution within and between species. Alu elements have been used to confirm family relationships between New World monkeys (NWM) [10, 11, 43, 45]. New World monkeys have been shown to have platyrrhine specific Alu element subfamilies--AluTa7, AluTa10, and AluTa15 [10]. These subfamilies have amplified throughout the NWM lineage and have shown Cebus and Sapajus are sister taxa [46]. New world monkey specific subfamilies have also been used to investigate hybridization within the Saimiri lineage [47] and for use as identification markers [43].
A detailed Alu subfamily analysis of Saimiri was recently reported by Baker et al. [48]. In that study on the evolution of Alu subfamilies in the Saimiri lineage [48], 108 Alu subfamilies within the genome [saiBol1], with 46 of those unique to the Saimiri lineage and the other 62 being NWM subfamilies [10, 49], were reported. These subfamilies were defined based on diagnostic nucleotide substitutions, insertions, or deletions that were exclusively shared. Nearly half of the Alu subfamilies included members that appeared to be relatively young insertion events (≤ 2% sequence divergence from their respective consensus sequence).
The purposes of this study were to identify polymorphic Alu insertions to examine population structure in Saimiri and to identify recently integrated insertions that might be informative for species identification. To accomplish these goals, we targeted recently integrated insertions and designed locus specific PCR primers for at least five Alu elements from every subfamily that was identified as ‘young’.
Methods
Alu element ascertainment
A data set of full length Alu elements from the Saimiri genome [saiBol1] was generated by using the Blat Table Browser. Alu full length elements plus 600 base pairs (bp) of flanking were obtained from the University of California Santa Cruz (UCSC) table browser. Full length elements are described as beginning within 4 bp of its respective consensus sequence and being ≥267 bp. Saimiri specific elements were RepeatMasked using an in-house installation of RepeatMasker [50] to determine the percent sequence divergence compared to their respective consensus sequences. Young elements, defined here as having a sequence divergence of ≤2% were retained for further analyses. We targeted at least five Alu elements for experimental validation from each Alu subfamily computationally determined to contain young elements.
Oligonucleotide primer design
Orthologous sequences to each respective Alu plus flanking were retrieved from human [hg38] and marmoset [calJac3] genomes using BLAT [51]. A multiple sequence alignment was created for each locus using BioEdit [52]. Oligonucleotide primers for polymerase chain reaction (PCR) were designed using Primer3 [53, 54] with the following adjustments: Tm range = 57–62, Max TmDifference =2, max poly x = 3, min Gc content = 40. All primers were ordered from Sigma Aldrich (Woodlands, TX). A list of PCR primers and genomic locations is available in Additional file 1.
DNA samples
A list of Saimiri samples and their source information is available in Additional file 1 (worksheet “squirrel monkey samples”). DNA samples from thirty-two (32) individuals were used in this study. Various tissue and DNA samples were obtained from multiple natural science museums and research centers. Labeled biomaterials were obtained for the following squirrel monkey species: Saimiri sciureus (10 samples), Saimiri sciureus sciureus (2 samples), Saimiri boliviensis (14 samples), Saimiri boliviensis peruviensis (3 samples), Saimiri oerstedii oerstedii (1 sample), Saimiri sciureus macrodon (1 sample), and Saimiri “species unknown” (1 sample). DNA from tissue samples were prepared using proteinase K digestion followed by phenol: chloroform extraction and ethanol precipitation [55]. Extracted DNA was stored in 10 mM Tris/0.1 mM EDTA (TLE) and quantified spectrophotometrically using an Eppendorf Biophotometer. The DNA panel and PCR format is shown in Additional file 1.
Polymerase chain reaction amplification
Polymerase chain reaction amplification was performed in 25 μL reactions that contained 25–50 ng of template DNA, 200 nM of each primer, 1.5 mM MgCl2, 10× PCR buffer, 0.2 mM deoxyribonucleotide triphosphates and 1 unit of Taq DNA polymerase. The polymerase chain reaction protocol is as follows: 95 °C for 1 min, 32 cycles of denaturation at 94 °C for 30 s, 30 s at the respective annealing temperature, and extension at 72 °C for 30 s, followed by a final extension step at 72 °C for 2 min. Gel electrophoresis was performed on a 2% agarose gel containing 0.2 μg/mL ethidium bromide for 60 min at 175 V. UV fluorescence was used to visualize the DNA fragments using a BioRad ChemiDoc XRS imaging system (Hercules, CA).
Alu insertion polymorphisms
Following gel electrophoresis, genotypic data were recorded for each allele as follows: an individual who was homozygous present for a given Alu locus was assigned the code 1, 1; homozygous absent, 0, 0; and heterozygous, 1, 0. This binomial data sheet was used to calculate the allele frequency for each Alu insertion for the panel of 32 squirrel monkeys to evaluate the polymorphism rate. Allele frequency calculations were also performed separately for S. sciureus and S. boliviensis groups in an effort to identify species informative markers.
DNA sequencing
PCR validation experiments identified certain ambiguous conditions that warranted further evaluation by chain termination DNA sequencing [56]. There were two basic categories; 1) gel electrophoresis revealed PCR amplicons for the predicted present / absent sizes plus a larger amplicon of unknown identity in some individuals, 2) to confirm a shared Alu insertion event among seemingly misidentified individuals. Sanger sequencing experiments were performed as follows: Four PCR fragments per locus were gel purified using a Wizard SV gel purification kit (Promega Corporation, Madison, WI, USA, catalog A9282) according to the manufacturer’s instructions with the following modification. The 50 μl elution step was performed twice, resulting in 100 μl, which was then dried in a SpeedVac (ThermoSavant SPD 111 V). The DNA was reconstituted in 30 μl TVLE (Tris Very Low EDTA; 10 mM Tris/ 0.05 mM EDTA) and 4 μl was used for chain termination cycle sequencing using BigDye Terminator v3.1. Cycle sequencing was performed under the following conditions: After initial denaturation at 95 °C for 2 min, 40 cycles at 95 °C for 10 s, 50 °C for 5 s, and 60 °C for 4 min were performed followed by a hold at 4 °C. Sequencing reactions were cleaned by standard ethanol precipitation to remove any unincorporated dye terminators and then stabilized in 15 μl Hi-Di Formamide (Life Technologies, Inc.). Capillary electrophoresis was performed on an ABI 3130xl Genetic Analyzer (Applied Biosystems, Inc., Foster City, CA). Sequence quality was evaluated using ABI software Sequence Scanner v.2.0. Sequencing results were then analyzed using BioEdit [52].
Structure analysis
Population structure analyses were performed using Structure 2.3.4 software [57]. Using genotype data from unlinked markers, this software performs a model-based clustering method to infer the population structure. For our initial analysis, the information regarding the origin of the samples was omitted. The analyses were performed under the admixture model which assumes that individuals may have mixed ancestry. The settings used to determine the estimated number of populations (K) were as follows: K ranging from 1 to 7 and 10,000 burnin for 100,000 MCMC at 3 iterations. The most likely value of K was calculated to be three based on the “estimated ln probability” scores generated by Structure. Sometimes Structure detects the upper most K value. Therefore, we used Structure Harvester [58] to assess all of the likelihood values for K = 1 to 7 and determine the most likely number of population clusters. K = 2 was determined to be the best fit for the data set. Structure was then run using the following settings: K (projected number of populations) = 2; 100,000 burnin for 1 million MCMC at 5 iterations. The data from 5 iterations were averaged to generate the final data set. The final graph was generated in Excel. For comparison, a second Structure analysis was performed for K = 3 with the same parameters.
Results
Recently integrated Alu insertions
Based on a recent analysis of the genome data from Baker et al. 2017 [48], and data generated from RepeatMasker [50], we retained 48 Alu subfamilies in the [saiBol1] genome that contained members that were less than 2 % diverged from their respective consensus sequence. Elements that are less than 2% diverged from their consensus sequence are considered to be relatively young, as they have not accrued many mutations since their insertion [59, 60]. The data were organized in excel and sorted based on the number of elements per subfamily in various divergence categories (0.0, 0.5, 1.0, 1.5, and 2.0). These data can be found in Additional file 2. Table 1 displays the number of insertions in each divergence category. The elements descended in correlation to the divergence categories with the most elements being 2% diverged followed by 1.5, 1.0, 0.5, and 0.0% diverged. There were a total of 4184 young Alu elements identified in the genome having ≤2% sequence divergence from their respective consensus sequence.
In this study, we targeted at least N = 5 young insertions from each Alu subfamily computationally determined to contain young elements. We successfully performed PCR validation experiments on 382 Alu insertion events having ≤2% sequence divergence from their respective consensus sequence (Table 2) and (Additional file 1, worksheets “PCR primers & coordinates” and “genotypes”). These loci represented 46 Alu subfamilies, 25 from Saimiri specific subfamilies [48] and 21 from NWM specific subfamilies [10, 49]. On a DNA panel of 32 squirrel monkey individuals (Additional file 1, worksheet “PCR format”), 272 of the 382 loci were homozygous present for the Alu insertion and 110 were polymorphic for insertion presence/ absence. The number of loci analyzed per subfamily and insertion presence/absence data are listed in Table 2 and Additional file 1, worksheets “PCR primers & coordinates” and “genotypes”. The number of Alu insertions in each of the percent divergence bins from 0.0 to 2.0 is shown in Table 1. Table 1 illustrates that many insertions with very low sequence divergence have already reached very high allele frequency among squirrel monkey species (fixed present in our panel), while concurrently Alu insertions from all five divergence bins have elements that remain polymorphic in the population.
The dataset of polymorphic insertions included three loci with homozygous absent genotypes (0, 0) for the target Alu insertion in all 32 squirrel monkey individuals: L-21071-subfam11, L-38701-subfam32 and L-19471-Ta15. These Alu elements were ascertained from the reference genome [saiBol1] of S. boliviensis but the DNA for that reference individual was not available and therefore not included on our test panel. This very low allele frequency (near zero) is indicative of very recent insertion events. These results confirm the previously reported Alu subfamily network analysis [48] showing the existence of many young subfamilies. These data provide evidence for a prolific expansion of young Alu elements in the Saimiri lineage currently polymorphic between species.
Sanger sequencing validation
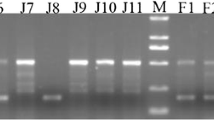
During PCR validation experiments certain ambiguous conditions occurred that warranted further evaluation by traditional Sanger sequencing [56]. These conditions had two basic categories. One, gel electrophoresis revealed PCR amplicons for the predicted present / absent sizes plus a larger amplicon in some individuals. This occurred for three loci and the details are outlined in Additional file 1, Worksheet “Table S1”. DNA sequencing of the larger amplicon determined that the loci contained more than one Alu element or an extra Alu element between the original PCR primers. These non-reference (not present in the [saiBol1] genome) Alu insertions appeared to be polymorphic across the various species in the DNA panel. The genotypes for these three extra Alu polymorphisms are recorded with the locus ID –“Alu-2” in the genotypes worksheet of Additional file 1. The second category which required Sanger sequencing was to confirm a shared Alu insertion event among seemingly misidentified individuals. Specifically, when genotype data for individuals labeled S. sciureus, and believed to be common squirrel monkeys, matched more closely to the S. boliviensis group, sequencing was warranted. An example of this is shown in Fig. 1. Forty-five loci from the dataset of 382 matched this condition. We sequenced 28 of the 45 and it was determined that all amplicons except one were the same Alu element identified in the reference genome. Only one amplicon was a near parallel insertion (Locus 16089, individual UWBM# 75531).

Gel Image of Polymorphic Locus 35154 (JH378108:33053451–33054957). This image displays a polymorphic locus in the Saimiri genome [saiBol1]. Lanes: 1- 100 bp ladder, 2- TLE (Negative control), 3- Human (HeLa), 4-Callithrix jacchus (Common marmoset), 5–16 Saimiri sciureus (Common squirrel monkey), 17–32 Saimiri boliviensis (Bolivian squirrel monkey), 33–35 Saimiri boliviensis peruviensis (Peruvian squirrel monkey), 36- Saimiri oerstedii oerstedii (Panamanian red back squirrel monkey), 37- Saimiri sciureus macrodon, 38-Saimiri sp. The presence of the Alu element is indicated by the ~ 655 bp band and the absence by the ~ 346 bp band. Species with multiple individuals are grouped together by colored brackets (Orange- Common squirrel monkey, Blue- Bolivian squirrel monkey, Green-Peruvian squirrel monkey). Lanes 7(UWBM# 75531) and 12(MVZ Mamm 193661) share an insertion with the Bolivian squirrel monkeys whom are either homozygous present or heterozygous for the insertion (lanes 17–32). Lane 38 (species unknown) is heterozygous for the insertion
Population structure
Following PCR and gel electrophoresis, genotypes for the 32 squirrel monkey individuals were recorded in an excel spreadsheet as follows: homozygous absent for the reference Alu insertion, (0, 0), homozygous present for the target Alu insertion (1, 1) and heterozygous as (1, 0) (Additional file 1, worksheet “genotypes”). During genotype analysis we identified 24 loci (of 382) with > 25% missing data due to poor PCR (highlighted in tan in the genotype spreadsheet; Additional file 1). Most of these (21) were homozygous present for the insertion and would not influence population structure, but 3 were from the polymorphic dataset. These were omitted from the population structure analysis. Also, the samples from the KCCMR S. boliviensis breeding colony included two known sibling pairs, one sibling from each sibling pair was omitted from the Structure analysis [57].
To determine the value of K (where K equals the number of population clusters) with the highest likelihood, initially K was set from 1 to 7. The initial burn-in period was set at 10,000 iterations and followed by a run-length of 100,000 MCMC and repeated three times. The most likely value of K was calculated to be three based on the “estimated ln prob. of data” scores generated by Structure. The authors of Structure indicate that this method is generally accurate with small data sets, but acknowledge it is still an estimate of K. Therefore, we also employed the Delta K method by Evanno [61] implemented using Structure Harvester [58]. The Delta K method is widely accepted in the literature as an accurate estimate of the true K. Here, the Delta K was calculated to be K = 2. The structure results for K = 2 are shown in Fig. 2. In general, Cluster 1 contains individuals previously labeled as common squirrel monkeys and Cluster 2 contains individuals previously labeled as Bolivian squirrel monkeys. However, there is a large amount of admixture in some individuals (a mixture of Cluster 1 & 2). These admixed appearing individuals were previously labeled as common squirrel monkey (UWBM # 75531 & MVZ Mamm 193661), Bolivian squirrel monkey (LSUMZ M-4970, MVZ Mamm 196088), Peruvian squirrel monkey (3526, 2291, KB17911), Ecuadorian squirrel monkey (KB17915) and species unknown (MVZ Mamm 196089). The results of this Structure analysis are generally consistent with the geographic ranges of the Saimiri species and subspecies. Maps of the geographic ranges can be found in Hershkovitz 1984 and Chiou et al. 2011 [39, 40]. Sample KB7456 is the only member of the S. oerstedii species on our panel. This species is the Panamanian squirrel monkey located in Central America. The geographic range of S. oerstedii is closer to the S. sciureus group than to the S. boliviensis group [39, 40] and Structure assigns this individual to Cluster 1. The geographic “locality” provided for sample MVZ Mamm 193661 (clusters with Bolivian) is the Acre region of Brazil (listed in Additional file 1, worksheet “Squirrel monkey samples”) and it is labeled Saimiri sciureus ssp. the Acre region is consistent with the geographic range of S. sciureus macrodon and borders the region of S. boliviensis peruviensis [39]. S. sciureus macrodon are the Ecuadorian squirrel monkeys native to Peru. Therefore, we can interpret these results as meaning that MVZ Mamm 193661 has an incomplete identification, rather than being misclassified. MVZ Mamm 193685 was also labeled as Saimiri sciureus ssp. the geographic locality provided for this sample is the Penedo region of Brazil, consistent with the geographic range of Saimiri sciureus sciureus, and consistent with the Structure assignment to Cluster 1. MVZ Mamm 196089 is labeled Saimiri sp., indicating the species is not known. The geographic locality listed for this sample is the Sao Jose region of Brazil, the same locality as reported for MVZ Mamm 196088, and consistent with the geographic range of S. boliviensis. Therefore, we can infer that this dataset accurately captures the majority of the geographic population structure among Saimiri species.

Population Structure analysis based on 110 Alu insertion polymorphisms and 32 squirrel monkey individuals for K = 2. The percent assignment of each individual to K = 2 clusters is shown on the Y-axis. The ID numbers and species names are shown on the X-axis. K = 2 captures the population structure of the two Saimiri groups, S. sciureus and S. boliviensis, and is consistent with the geographic origins of these samples
However, the original “estimated ln prob. of data” scores generated by Structure suggested that K = 3 was likely. In an effort to make sure our interpretations of the data were accurate, we also tested K = 3 (Fig. 3 and Additional file 2, sheet K = 3 Table). In Fig. 3, the samples from JAV (DNA originally from KCCMR) and KCCMR appear to be isolated and more genetically similar. If the dataset is analyzed using K = 3, the third cluster is formed by isolating the ten members in the dataset from the KCCMR S. boliviensis captive breeding colony into its own cluster (shown in gray in Fig. 3), the remaining individuals segregate into the other two clusters similar to their respective assignments in the K = 2 analysis (orange and blue). To further investigate this observation we analyzed the Fst values for K = 2 and 3 for all of the clusters (Table 3). When K = 2 Fst values are similar, which implies the populations share genetic diversity. When K = 3 two clusters have similar values and one cluster has an extremely low value of 0.3391. A value of 0.3391 would imply that the individuals in Cluster 3 may be sharing genetic material through high levels of inbreeding and appears to be an isolated group in Fig. 3. While K = 2 captures the primary geographic origins of the Saimiri populations, K = 3 is also reasonable as it reveals genetic evidence of inbreeding among members of a captive colony.

Population Structure analysis based on 110 Alu insertion polymorphisms and 32 squirrel monkey individuals for K = 3. The percent assignment of each individual to K = 3 clusters is shown on the Y-axis. The ID numbers and species names are shown on the X-axis. K = 3 captures the population structure of the two Saimiri groups, S. sciureus and S. boliviensis while also detecting the genetic isolation of members of a captive breeding colony within the S. boliviensis samples
Species informative Alu polymorphisms
Within the dataset of 110 polymorphic Alu insertions, there were 51 with species informative allele frequency distribution between Saimiri sciureus and Saimiri boliviensis. A locus was categorized as species informative if it was present at a high frequency in one species and generally absent in the other. These are listed in Table 4 and are highlighted in green in Additional file 1, Worksheets “PCR primers & coordinates” and “Genotypes”. The 14 members of S. boliviensis have a group allele frequency of 80–100% whereas the 12 samples labeled S. sciureus have a group allele frequency of 0–20% on average (Table 4). If we omit samples 75531 and 193661 from the S. sciureus group due to the Structure data (described above) showing that these two samples justifiably clustered more closely with the S. boliviensis group, then the group allele frequency in the S. sciureus group drops to near zero (0.5% on average) (Table 4). These 51 Alu insertion events represent 26 different Alu subfamilies, 10 Saimiri lineage specific subfamilies reported in Baker et al. 2017 [48] and 12 NWM Alu subfamilies discovered previously in Marmoset [49].
Discussion
An analysis of a large number of Alu insertions from many different Alu subfamilies, and a diverse DNA panel of squirrel monkeys, allowed us to determine the Alu insertion diversity in the Saimiri lineage. This suggests that many different Alu subfamilies were active in Saimiri and generated new Alu insertions. These data also support the stealth model of Alu amplification [62] in which relatively older Alu subfamilies are still producing new copies. In this case, the AluTa subfamily [63] is estimated to have originated about 15 MYA).
However, this study also has limitations, considering only one Saimiri species has a sequenced genome, S. boliviensis. The Alu elements in this study were ascertained from the reference genome [saiBol1] of a Bolivian squirrel monkey. The allele frequency data for the polymorphic insertions reflect the inherent single genome frequency spectrum ascertainment bias. Within the dataset of 51 polymorphic Alu insertions with species informative allele frequency distribution between Saimiri sciureus and Saimiri boliviensis samples, the S. boliviensis group has a relatively high allele frequency (~ 80–100%) whereas the S. sciureus group has a very low allele frequency (near zero) (Table 4). However, the three new polymorphic Alu insertions discovered during Sanger sequencing appear to be S. sciureus derived, rather than S. boliviensis derived (Additional file 1, genotypes worksheet). As more whole genome sequence data become available for Saimiri species, the frequency spectrum limitation due to ascertainment from a single reference genome will diminish. Thus, a more comprehensive assessment of Alu mobilization dynamics among Saimiri species will be attainable.
Prior to 1984, squirrel monkeys were considered a single species, named Saimiri sciureus, with many subspecies geographically separated [28]. Therefore, it is not surprising that some archival tissue samples from natural science museums or specimens from older studies may have typically been labeled simply as Saimiri, squirrel monkey, or S. sciureus. This does not mean they are necessarily mislabeled, but more than likely represent incomplete identification due to limited availability regarding source animal data at the time of sampling. Although we have no direct confirmation that this occurred with some of the samples in our DNA panel, the genetic diversity evidence from the Structure analysis in this study suggests it is likely. Individuals UWBM#75531, MVZ Mamm 193661 and MVZ 196089 in particular had ambiguous amplicons in 45 different Alu loci. Based on our Sanger sequencing, geographic locality and the Structure data, we believe these individuals may have previously been “under-classified” and they are most closely related to the Ecuadorian squirrel monkeys, S. sciureus macrodon, or the Peruvian squirrel monkeys S. boliviensis peruviensis. Considering there were only three Peruvian squirrel monkeys and one Ecuadorian squirrel monkey on the DNA panel, a larger sample size with more whole genome sequence data would be required for the identification of the exact species of these individuals.
Conclusions
Many different Alu subfamilies were active in the Saimiri genome producing a large number of young polymorphic insertions. These young polymorphic Alu insertions provide a valuable resource for species identification and population structure within Saimiri. This dataset may prove useful to natural science museums that may contain archival tissue samples labeled simply as “Saimiri” or “squirrel monkey” due to limited data available about the source animal at the time of sampling. Some of these samples may now be further classified at the species level and possibly even at the subspecies level, with this dataset. Future whole genome sequencing studies will further elucidate these findings.
Abbreviations
- Bp:
-
Base pair
- NWM:
-
New World Monkey
- PCR:
-
Polymerase chain reaction
- SINE:
-
Short interspersed element
- UCSC:
-
University of California Santa Cruz
References
Wang W, Kirkness EF. Short interspersed elements (SINEs) are a major source of canine genomic diversity. Genome Res. 2005;15(12):1798–808.
Lenoir A, Lavie L, Prieto JL, Goubely C, Cote JC, Pelissier T, Deragon JM. The evolutionary origin and genomic organization of SINEs in Arabidopsis Thaliana. Mol Biol Evol. 2001;18(12):2315–22.
Ray DA, Pagan HJ, Platt RN 2nd, Kroll AR, Schaack S, Stevens RD. Differential SINE evolution in vesper and non-vesper bats. Mob DNA. 2015;6:10.
Seibt KM, Wenke T, Muders K, Truberg B, Schmidt T. Short interspersed nuclear elements (SINEs) are abundant in Solanaceae and have a family-specific impact on gene structure and genome organization. Plant J. 2016;86(3):268–85.
Shedlock AM, Takahashi K, Okada N. SINEs of speciation: tracking lineages with retroposons. Trends Ecol Evol. 2004;19(10):545–53.
Takahashi K, Terai Y, Nishida M, Okada N. A novel family of short interspersed repetitive elements (SINEs) from cichlids: the patterns of insertion of SINEs at orthologous loci support the proposed monophyly of four major groups of cichlid fishes in Lake Tanganyika. Mol Biol Evol. 1998;15(4):391–407.
Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10(10):691–703.
Konkel MK, Walker JA, Batzer MA. LINEs and SINEs of primate evolution. Evol Anthropol. 2010;19(6):236–49.
McLain AT, Carman GW, Fullerton ML, Beckstrom TO, Gensler W, Meyer TJ, Faulk C, Batzer MA. Analysis of western lowland gorilla (Gorilla Gorilla Gorilla) specific Alu repeats. Mob DNA. 2013;4(1):26.
Ray DA, Batzer MA. Tracking Alu evolution in new world primates. BMC Evol Biol. 2005;5:51.
Ray DA, Xing J, Hedges DJ, Hall MA, Laborde ME, Anders BA, White BR, Stoilova N, Fowlkes JD, Landry KE, et al. Alu insertion loci and platyrrhine primate phylogeny. Mol Phylogenet Evol. 2005;35(1):117–26.
Shedlock AM, Okada N. SINE insertions: powerful tools for molecular systematics. BioEssays. 2000;22(2):148–60.
Xing J, Wang H, Han K, Ray DA, Huang CH, Chemnick LG, Stewart CB, Disotell TR, Ryder OA, Batzer MA. A mobile element based phylogeny of old world monkeys. Mol Phylogenet Evol. 2005;37(3):872–80.
Li J, Han K, Xing J, Kim HS, Rogers J, Ryder OA, Disotell T, Yue B, Batzer MA. Phylogeny of the macaques (Cercopithecidae: Macaca) based on Alu elements. Gene. 2009;448(2):242–9.
Hartig G, Churakov G, Warren WC, Brosius J, Makalowski W, Schmitz J. Retrophylogenomics place tarsiers on the evolutionary branch of anthropoids. Sci Rep. 2013;3:1756.
McLain AT, Meyer TJ, Faulk C, Herke SW, Oldenburg JM, Bourgeois MG, Abshire CF, Roos C, Batzer MA. An alu-based phylogeny of lemurs (infraorder: Lemuriformes). PLoS One. 2012;7(8):e44035.
Meyer TJ, McLain AT, Oldenburg JM, Faulk C, Bourgeois MG, Conlin EM, Mootnick AR, de Jong PJ, Roos C, Carbone L, et al. An Alu-based phylogeny of gibbons (hylobatidae). Mol Biol Evol. 2012;29(11):3441–50.
Roos C, Schmitz J, Zischler H. Primate jumping genes elucidate strepsirrhine phylogeny. Proc Natl Acad Sci U S A. 2004;101(29):10650–4.
Salem AH, Ray DA, Xing J, Callinan PA, Myers JS, Hedges DJ, Garber RK, Witherspoon DJ, Jorde LB, Batzer MA. Alu elements and hominid phylogenetics. Proc Natl Acad Sci U S A. 2003;100(22):12787–91.
Schmitz J, Noll A, Raabe CA, Churakov G, Voss R, Kiefmann M, Rozhdestvensky T, Brosius J, Baertsch R, Clawson H, et al. Genome sequence of the basal haplorrhine primate Tarsius Syrichta reveals unusual insertions. Nat Commun. 2016;7:12997.
Schmitz J, Roos C, Zischler H. Primate phylogeny: molecular evidence from retroposons. Cytogenet Genome Res. 2005;108(1–3):26–37.
Batzer MA, Deininger PL. A human-specific subfamily of Alu sequences. Genomics. 1991;9(3):481–7.
Batzer MA, Stoneking M, Alegria-Hartman M, Bazan H, Kass DH, Shaikh TH, Novick GE, Ioannou PA, Scheer WD, Herrera RJ, et al. African origin of human-specific polymorphic Alu insertions. Proc Natl Acad Sci U S A. 1994;91(25):12288–92.
Perna NT, Batzer MA, Deininger PL, Stoneking M. Alu insertion polymorphism: a new type of marker for human population studies. Hum Biol. 1992;64(5):641–8.
Ryan SC, Dugaiczyk A. Newly arisen DNA repeats in primate phylogeny. Proc Natl Acad Sci U S A. 1989;86(23):9360–4.
Stoneking M, Fontius JJ, Clifford SL, Soodyall H, Arcot SS, Saha N, Jenkins T, Tahir MA, Deininger PL, Batzer MA. Alu insertion polymorphisms and human evolution: evidence for a larger population size in Africa. Genome Res. 1997;7(11):1061–71.
Ray DA, Xing J, Salem AH, Batzer MA. SINEs of a nearly perfect character. Syst Biol. 2006;55(6):928–35.
Abee CR. Squirrel monkey (Saimiri spp.) research and resources. ILAR J. 2000;41(1):2–9.
Galland GG. Role of the squirrel monkey in parasitic disease research. ILAR J. 2000;41(1):37–43.
Vanchiere JA, Ruiz JC, Brady AG, Kuehl TJ, Williams LE, Baze WB, Wilkerson GK, Nehete PN, McClure GB, Rogers DL, et al. Experimental Zika virus infection of Neotropical primates. Am J Trop Med Hyg. 2018;98(1):173–7.
Boyne JR, Colgan KJ, Whitehouse A. Herpesvirus saimiri ORF57: a post-transcriptional regulatory protein. Front Biosci. 2008;13:2928–38.
Cazalla D, Yario T, Steitz JA. Down-regulation of a host microRNA by a herpesvirus saimiri noncoding RNA. Science (New York, NY). 2010;328(5985):1563–6.
Jung JU, Choi JK, Ensser A, Biesinger B. Herpesvirus saimiri as a model for gammaherpesvirus oncogenesis. Semin Cancer Biol. 1999;9(3):231–9.
Rogers DL, McClure GB, Ruiz JC, Abee CR, Vanchiere JA. Endemic viruses of squirrel monkeys (Saimiri spp.). Comp Med. 2015;65(3):232–40.
Stevenson AJ, Frolova-Jones E, Hall KT, Kinsey SE, Markham AF, Whitehouse A, Meredith DM. A herpesvirus saimiri-based gene therapy vector with potential for use in cancer immunotherapy. Cancer Gene Ther. 2000;7(7):1077–85.
Tardif SD, Abee CR, Mansfield KG. Workshop summary: neotropical primates in biomedical research. ILAR J. 2011;52(3):386–92.
Walker ML, Anderson DC, Herndon JG, Walker LC. Ovarian aging in squirrel monkeys (Saimiri sciureus). Reproduction (Cambridge, England). 2009;138(5):793–9.
Ward JM, Vallender EJ. The resurgence and genetic implications of new world primates in biomedical research. Trends Genet. 2012;28(12):586–91.
Hershkovitz P. Taxonomy of squirrel monkeys genus Saimiri (Cebidae, platyrrhini): a preliminary report with description of a hitherto unnamed form. Am J Primatol. 1984;7(2):155–210.
Chiou KL, Pozzi L, Lynch Alfaro JW, Di Fiore A. Pleistocene diversification of living squirrel monkeys (Saimiri spp.) inferred from complete mitochondrial genome sequences. Mol Phylogenet Evol. 2011;59(3):736–45.
Boinski S, Cropp SJ. Disparate data sets resolve squirrel monkey (Saimiri) taxonomy: implications for behavioral ecology and biomedical usage. Int J Primatol. 1999;20(2):237–56.
Lavergne A, Ruiz-Garcia M, Catzeflis F, Lacote S, Contamin H, Mercereau-Puijalon O, Lacoste V, de Thoisy B. Phylogeny and phylogeography of squirrel monkeys (genus Saimiri) based on cytochrome b genetic analysis. Am J Primatol. 2010;72(3):242–53.
Osterholz M, Vermeer J, Walter L, Roos C. A PCR-based marker to simply identify Saimiri Sciureus and S. Boliviensis Boliviensis. Am J Primatol. 2008;70(12):1177–80.
Vandeberg JL, Williams-Blangero S, Moore CM, Cheng M-L, Abee CR. Genetic relationships among three squirrel monkey types: implications for taxonomy, biomedical research, and captive breeding. Am J Primatol. 1990;22(2):101–11.
Singer SS, Schmitz J, Schwiegk C, Zischler H. Molecular cladistic markers in new world monkey phylogeny (Platyrrhini, primates). Mol Phylogenet Evol. 2003;26(3):490–501.
Martins AM Jr, Amorim N, Carneiro JC, de Mello Affonso PR, Sampaio I, Schneider H. Alu elements and the phylogeny of capuchin (Cebus and Sapajus) monkeys. Am J Primatol. 2015;77(4):368–75.
Carneiro J, ESJ DS Jr, Sampaio I, Pissinatti A, Hrbek T, Rezende Messias M, Rohe F, Farias I, Boubli J, Schneider H. Phylogeny of the titi monkeys of the Callicebus Moloch group (Pitheciidae, primates). Am J Primatol. 2016;78(9):904–13.
Baker JN, Walker JA, Vanchiere JA, Phillippe KR, St Romain CP, Gonzalez-Quiroga P, Denham MW, Mierl JR, Konkel MK, Batzer MA. Evolution of Alu subfamily structure in the Saimiri lineage of new world monkeys. Genome Biol Evol. 2017;9(9):2365–76.
Consortium MGSA. The common marmoset genome provides insight into primate biology and evolution. Nat Genet. 2014;46(8):850–7.
RepeatMasker Open-4.0 [http://www.repeatmasker.org]. Accessed Feb 2018.
Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64.
Hall TA. BioEdit:a user friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics (Oxford, England). 2007;23(10):1289–91.
Untergasser AC, Koressaar T, Ye J, Faircloth B, Rozen S. Primer 3--new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115.
Strauss W. Current protocols in molecular biology. New York: Wiley; 1998.
Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–7.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–87.
Earl D, vonHoldt B. Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4(2):359–61.
Bennett EA, Keller H, Mills RE, Schmidt S, Moran JV, Weichenrieder O, Devine SE. Active Alu retrotransposons in the human genome. Genome Res. 2008;18(12):1875–83.
Konkel MK, Walker JA, Hotard AB, Ranck MC, Fontenot CC, Storer J, Stewart C, Marth GT, Batzer MA. Sequence analysis and characterization of active human Alu subfamilies based on the 1000 genomes pilot project. Genome Biol Evol. 2015;7(9):2608–22.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Han K, Xing J, Wang H, Hedges DJ, Garber RK, Cordaux R, Batzer MA. Under the genomic radar: the stealth model of Alu amplification. Genome Res. 2005;15(5):655–64.
Ray DA. SINEs of progress: mobile element applications to molecular ecology. Mol Ecol. 2007;16(1):19–33.
Acknowledgements
The authors would like to thank all the members of the Batzer laboratory for their help with experiments and constructive criticism of the manuscript. The squirrel monkey genome assembly (Saimiri boliviensis) is provided with the following acknowledgements: We acknowledge the Broad Institute (Cambridge, MA) for the [saiBol1] sequencing and assembly. We also acknowledge Hiram Clawson, Chin Li, Brian Raney, Pauline Fujita, Luvina Guruvadoo, Steve Heitner, Brooke Rhead, Greg Roe, and Donna Karolchik for the UCSC squirrel monkey genome browser/initial annotations. This research was supported by the National Institutes of Health R01 GM59290 (M.A.B). The authors also wish to thank the following people and institutions for their generous donation of samples: Dr. Frederick H. Sheldon, Curator, and Donna Dittmann of the Louisiana State University Museum of Natural Science Collection of Genetic Resources; Michale E. Keeling Center for Comparative Medicine and Research, The University of Texas MD Anderson Cancer Center, Bastrop, TX; San Diego Zoo Global Biomaterials Review Group, San Diego Zoo Institute for Conservation Research; Sharon Birks, Genetics Resources Collections Manager at the Burke Museum of Natural History and Culture, University of Washington; Kristof Zyskowski, Collection Manager at the Peabody Museum of Natural History, Yale University; Christopher C. Conroy, Curator, Mammals Collection at the Museum of Vertebrate Zoology, University of California – Berkeley, and Dr. John A. Vanchiere, Chief, Pediatric Infectious Diseases, Louisiana State University Health Sciences Center – Shreveport.
Funding
This work was supported by National Institutes of Health Grant RO1 GM59290 (MAB).
Availability of data and materials
All DNA samples, genotype, divergence percentages data are available as part of the Additional Materials.
Author information
Authors and Affiliations
Contributions
JNB performed all computational analyses. JNB and JAW performed analysis of Saimiri genomic data, sequencing data, structure data, and created resulting figures and tables. JNB and MWD designed primers, performed PCR and gel electrophoresis/imaging. CDLIII performed PCR and gel electrophoresis/imaging. JNB and JAW designed the research and wrote the paper. MAB helped design the research, provided analytical tools and made final edits to the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
An excel file containing worksheets for PCR primers & coordinates, Squirrel monkey samples, PCR format, and genotype data for each locus. Table S1 details the loci sequenced. (XLSX 503 kb)
Additional file 2:
An excel file showing Alu element subfamilies in the [saiBol1] genome with 0% to 2% divergence from their respective consensus sequences and the number of members per divergence category. A separate worksheet shows the numerical values for K = 3 Structure analysis. (XLSX 19 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Baker, J.N., Walker, J.A., Denham, M.W. et al. Recently integrated Alu insertions in the squirrel monkey (Saimiri) lineage and application for population analyses. Mobile DNA 9, 9 (2018). https://doi.org/10.1186/s13100-018-0114-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13100-018-0114-7