
Abstract
Background
Non-typhoidal Salmonella (NTS) is a major foodborne zoonotic pathogen worldwide. In the current study, Various NTS strains were isolated from (cows, milk and dairy products in addition to humans) in New Valley and Assiut Governorate, Egypt. NTS were firstly serotyped and tested by antibiotic sensitivity test. Secondly, some virulence genes and Antibiotic resistance genes have been identified by using PCR. Finally, Phylogenesis was performed depending on the invA gene, for two S. typhimurium isolates (one of animal origin and the other of human origin for evaluating zoonotic potential).
Results
Out of 800 examined samples, the total number of isolates was 87 (10.88%), which were classified into 13 serotypes, with the most prevalent being S. Typhimurium and S. enteritidis. Both bovine and human isolates showed the highest resistance to clindamycin and streptomycin, with 90.80% of the tested isolates exhibiting MDR. The occurrence of the invA gene was 100%, while 72.22%, 30.56%, and 94.44% of the examined strains were positive for stn, spvC, and hilA genes, respectively. Additionally, blaOXA-2 was detected in 16.67% (6/ 36) of the tested isolates, while blaCMY-1 was detected in 30.56% (11of 36) of the tested isolates. Phylogenesis revealed a high degree of similarity between the two isolates.
Conclusions
The high occurrence of MDR strains of NTS in both human and animal samples with high degree of genetic similarity, shows that cows, milk and milk product may be a valuable source of human infection with NTS and interfere with treatment procedures.
Similar content being viewed by others
Background
NTS is a major public health concern worldwide. Salmonella infections results in 93.8 million cases of gastroenteritis and 155,000 deaths annually [1]. Diarrhea, abdominal pain, and vomiting are the main characteristics of self-limiting gastroenteritis caused by NTS in individuals of all ages. However, severe invasive disease with complicated extraintestinal illness, bacteremia, and meningitis can be observed in children, the elderly, and immunocompromised patients [2, 3].
The main sources of human salmonellosis are contaminated food products of animal origin such as dairy products and direct contact with infected animals [4, 5]. Salmonella infection on cattle farms continues to be a major problem, causing economic losses and increasing the risk of transmission to humans. NTS shedding occurs in the feces of sick, recovered, and asymptomatic cattle, and the organism can survive for a long time in a favorable environment outside the host [6]. The presence of NTS in animal feces can increase intra-herd transmission, accidental spread to other herds, environmental contamination, and risk of human infection [7].
Many virulence genes have been linked to the pathogenicity of Salmonella, and the severity of infection depends mainly on the presence or absence of the invA, stn, hila, and spvC genes. These virulence genes are located either chromosomally or plasmidically and encodes products that help NTS interaction with the host cells. [8, 9]. The invA gene is a biomarker for NTS detection and play major role in invasion of epithelial cells. Whereas, stn gene encode for an enterotoxin production and spvC encodes for systemic infection in human and replication in animal [10,11,12].
The therapeutic and non-therapeutic use of various antibiotics in food-producing animals and their misuse in humans have led to the emergence of antimicrobial-resistant NTS. Consequently, the incidence of MDR Salmonella has increased in the recent decades. [13]. Plasmid genes can transfer antimicrobial resistance between bacteria, which can lead to the emergence of MDR strains. The global effort aims to limits antibiotic-resistant strains, as infection leads to a significant foodborne hazard to human [14,15,16].
Therefore, information on the Salmonella serotypes circulating in a given geographic area, virulence genes, and antimicrobial resistance is essential. The objectives of this study were to investigate the occurrence, distribution of different serotypes, virulence genes and antibiotic resistance of NTS in Assiut and New Valley Governorates.
Results
A total of 800 samples from different sources including raw milk, kareish cheese, Damietta cheese, yoghurt, ice cream, animal fecal swabs, human fecal swabs and hand swabs were examined for Salmonella. Out of the examined samples, 10.88% (87) were positive for Salmonella by conventional methods. These isolates were serotyped using "O" and "H" antisera and the results showed 13 different Salmonella serotypes including S. typhimurium, S. enteritidis, S. tsieve, S. infantis, S. larochelle, S. virchow, S. molade, S. haifa, S. shubra, S. alfort, S. essen, S. apeyeme and S. heidelberg (Tables 1 and 4).
An overall 87 Salmonella isolates tested for antibiotic susceptibility for 14 different antibiotics as shown in (Table 2). MDR was found in 90.80% of the tested isolates as illustrated in (Table 3).
Out of the 79 MDR NTS isolates, 36 isolates were tested by PCR for the presence of virulence genes including invA, stn, spvC and hilA genes. The occurrence of invA gene was 100% while, 72.22%, 30.56% and 94.44% of the examined stains were positive for stn, spvC and hilA genes, respectively (Table 4).
The same 36 isolates were investigated for the presence of β-lactamase antibiotic resistance genes including blaOXA-2 and blaCMY-1 genes. The blaOXA-2 was detected in 16.67% (6 out of 36) of the tested isolates while, the blaCMY-1 was detected in 30.56% (11out of 36) of the tested isolates (Table 5).
Phylogenetic analysis
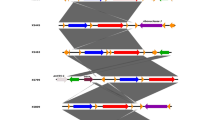
Collectively, sequencing and phylogenesis of two Salmonella isolates, with GenBank accession number op973126 and op973127, (one from animal and one from human) which based on invA gene in this study revealed high degree of similarity between the two isolates and also, between the two isolates and those retrieved from the GeneBank.
Discussion
Non-typhoidal Salmonella typically causes mild self-limiting gastroenteritis in the majority of people. The illness can also present as a febrile invasive disease, often without diarrhoea, with bacteraemia, meningitis, or focal infections that can be fatal if left untreated or improperly treated [17].
As illustrated in (Tables 1 and 6), the overall prevalence of NTS was 8.3% and 11.96% in humans and animals, respectively. The isolates recovered from all examined samples (87) were serotyped into 13 different serotypes, with differences in their distribution in different sample types. S. typhimurium and S. enteritidis were the predominant serotypes in animal and human isolates.
Many authors in previous studies agree with us in the opinion that the most common serotypes of salmonella in humans and animals were S. typhimurium and S. enteritidis [15, 18,19,20]. However, a previous study in Ethiopia demonstrated that the most common serovar were S. dublin and S. Virchow [21]. Additionally, S. newport and S. typhimurium were the most predominant in Colombia by (60.87%) and (17.4%) respectively [22].
NTS is the most common cause of acute gastroenteritis in humans. Antibiotic resistance has become a global issue as antibiotic use has increased [23]. Antimicrobial resistance refers to the ability of a microorganism to survive and reproduce in the presence of previously effective antibiotic doses. The results of antibiotic susceptibility testing are illustrated in (Table 2); the recovered NTS isolates showed 100% resistance to clindamycin, followed by streptomycin (98.85). These results are consistent with those obtained by many previous studies [15, 24] and It was observed that 100% of isolates from both animals and humans were resistant to clindamycin, although its use in large animals was limited. This demonstrated the cross-transmission of NTS between humans and animals (Fig. 1).

Phylogenetic tree of two S. typhimurium isolated from human and animal based on invA gene sequencing
The high levels of resistance observed in the present study may be due to the indiscriminate use of drugs for the treatment of both human and animal diseases as a result of self-administration of drugs without proper clinical examination and cessation of drug usage before the complete dose. There was also poor knowledge of withdrawal time of used drugs, although different antibiotic classes of drugs are used in animal health management and in human medicine, the selection of resistance to one drug class may lead to cross-resistance to another [13, 23, 25, 26].
In contrast, amikacin showed the highest level of NTS susceptibility, followed by cephalothin, gentamicin, and ciprofloxacin. Similarly, the NTs isolates that were recovered from retail food in Thailand remain sensitive to amikacin [14].
The global incidence of MDR in Salmonella has increased over the last few decades [27]. Infection of humans with MDR strains of Salmonella has been reported to be associated with an increased burden of morbidity, extended hospitalization, increased risk of invasive illness, and increased mortality compared to those infected with susceptible strains [28]. As shown in (Table 3), MDR was found in 90.80% of the tested isolates. 100% of S. typhimuruim, S. tsieve, S. heidelberg, S. Haifa, S. shubra, and S. alfort showed MDR, whereas 94.44%, 85.71%, 75%, 80%, 85.71%, and 50% of S. enteritidis, S. infantis, S. larochelle, S. Virchow, S. molade, and S. essen, respectively, showed MDR against the antibiotics used. Lower occurrence of MDR were observed among the tested Salmonella isolates by 70, 13.2, 50, and 47.06% [29,30,31]. The differences in MDR between studies may be due to different types of samples, different types of antibiotics tested, different strains of Salmonella, and the frequent development of resistant genes.
The virulence of Salmonella is linked to a combination of chromosomal and plasmid factors. Of the 87 Salmonella-positive isolates, 36 isolates exhibiting MDR were tested by PCR for the presence of virulence genes, including invA, stn, spvC, and hilA. The occurrence of the invA gene was 100%, and 72.22%, 30.56%, and 94.44% of the examined strains were positive for stn, spvC, and hilA genes, respectively (Table 4).
The invA gene is considered a universal genetic marker identified in most Salmonella serovars [32]. Our results confirmed 100% occurrence of invA gene in the examined isolates, as recorded in many previous studies [15, 33, 34, 35]. However, only 67% of the examined samples were invA gene positive [5].
Data in (Table 4) illustrated that stn gene was found in 72.22% of the tested isolates. Similarly, the stn genes were detected in 71.7% of the tested isolates in Egypt [24]. On the other hand, the stn gene could be detected in all Salmonella isolates [34, 36, 37]. In contrast,
Of the 36 NTS isolates tested for the presence of virulence genes, 30.56% (11) were carrying the spvC gene (Table 4). Slightly higher percentages than ours were reported in Zambia and Burkina Faso by 24% and 58.8% respectively, [5, 38]. On the other hand the spvC gene could be detected in 100% of examined isolates in Egypt and Bangladesh [34, 36].
As shown in (Table 4), hilA were detected in 94.44% of the examined isolates. The presence of the hilA virulence gene in the examined NTS serovars was detected by lower percentage 66.6% [39]. Moreover, the hilA gene was detected in all examined NTS isolates [35, 40].β-Lactam antibiotics are widely used in many developing countries. This group of antibiotics belongs to a family of antibiotics with a β-lactam ring, such as penicillin and cephalosporins. Members of β-lactam antibiotics are inactivated by bacteria through the production of β-lactamases, which hydrolyze the β-lactam rings. Many genes such as blaCMY-1 and blaOXA-2 are responsible for this mechanism. The isolates were tested for the presence of these two β-lactamase antibiotic resistance genes by PCR. blaOXA-2 was detected in 16.67% (6 36) of the tested isolates, whereas blaCMY-1 was detected in 30.56% (11out 36) of the tested isolates (Table 5).
These genes were detected in Egypt with lower incidence rates where they were blaCMY-1 10.5%, followed by blaOXA-2 6.6% [41]. On the other hand, other authors failed to detect blaCMY-1 and blaCMY-2 in Salmonella isolates [42 and 43].
Collectively, sequencing and phylogenesis of two Salmonella isolates (one from animals and one from humans), which was based on the invA gene in this study as shown in Fig. 1, revealed a high degree of similarity between the two isolates, and those retrieved from GenBank. This finding was compatible with the results obtained by other authors and demonstrates the zoonotic cycling of Salmonella between animals and humans [44, 45]
Conclusion
There is a high prevalence of antibiotic-resistant NTS in the feces of cows, milk and its products, as well as in human stool. Genetic analysis showed a very high rate of convergence between both isolates, which indicates the danger of transmission of NTS between humans and animals, determines the possibility of NTS transmission to human via milk and milk products with difficulty in treatment, which reflects its significant impact on public health.
Materials
Ethical declaration
This research was conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of New Valley University in Egypt. Informed consent was obtained from human participants and/or their legal guardians after receiving detailed information about the aims of the study.
Study area and design
Samples were collected from different localities in the New Valley and Assiut Governorates, Egypt, between April 2020 and May 2021.
Sampling
to isolate Salmonella, from April 2020 to May 2021 from different localities in New Valley and Assiut Governorates, Egypt, 800 samples, including raw milk (80), milk products (kareish cheese (80), Damietta cheese (80), yoghurt (80), ice cream (80), animal fecal swabs (160), human stool swabs (170), and hand swabs (70) were collected aseptically in sterile and sealed containers, labeled, placed in an ice box at 4 °C, and transported to the laboratory for microbiological analysis.
Microbiological analysis
Salmonella was detected using conventional culture-based methods according to ISO [46]. 1 ml from the sample was inoculated in 9 ml of buffered peptone water (HiMedia M1494I) and incubated at 37 °C for 24 h. 1 ml of the homogenate was added aseptically to 9 ml of Rappaport Vassilliadis R10 medium (RV) (HiMedia M1530) and incubated overnight at 42 °C. A loopful from the enriched broth was inoculated onto xylose lysine desoxycholate (XLD) agar (HiMedia M031I)and then incubated at 37 °C for 24 h. Typical colonies of salmonella (pink to red with or without black center) were picked and streaked onto nutrient agar slopes and incubated at 37 °C for 18–24 h for biochemical identification.
Biochemical identification was performed according to MacFaddin [47] using the indole production test, Simmons citrate test, urease test, triple sugar iron, and sugar fermentation tests, and then confirmed using API (analytical profile index).
Serotyping
The biochemically identified Salmonella isolates were serologically typed according to the Kauffman-White scheme [48] at the Faculty of Veterinary Medicine, Department of Food Hygiene and Control, Benha University, Egypt, using the slide agglutination technique for both somatic (O) and flagellar (H) antigens.
Antibiotic resistance (Antibiogramme)
According to the CLSI [49] Salmonella isolates were tested for the antimicrobial susceptibility by the single diffusion method against 14 different antibiotics including nalidixic acid (30 µg), ciprofloxacin (5 µg), tetracycline (30 µg), penicillin G(10 IU), clindamycin (10 µg), norocillin (25 µg), cephalothin (30 µg), streptomycin (10 µg), doxycycline (30 µg), kanamycin (30 µg), ampicillin (10 µg), amikacin (30 µg), gentamicin (10 µg) and sulphamethoxazol (25 µg).
Molecular detection of Salmonella virulence genes
Bacterial DNA was extracted Using GeneJET Genomic DNA Purification Kit (ThermoFisher, Cat. No. K0702 [50]. PCR was applied to amplify salmonella virulence genes invA, hilA, stn and spvC genes using specific primers as shown in (Table 7). PCR reaction mixture (25 µl) contained 2 µl of bacterial DNA, 2.5 µl of 10x Master Mix Green Master, Promega, USA) (containing 2.5 U of Taq DNA polymerase, 2.5 of 25 mM MgCl2 and 0.5 µl of 10mM dNTP mix), 0.5 µl of 1.2 µM primer mix (Applied Biosystem, USA) and 14.2 µl deionized water. The amplification done in Gradient Thermal Cycler (Veriti Applied Biosystem, USA). The PCR cycling protocol was applied as following: an initial denaturation at 94 °C for 2 min, followed by 30 cycles of denaturation at 94 °C for 45 sec, annealing at 53 °C for 1 min and extension at 72 °C for 1 min, followed by a final extension at 72 °C for 7 min. Finally, 5 µl of each amplicon was electrophoresed in 1.5 % agarose gel, stained with ethidium bromide and visualized and captured on UV transilluminator (UV, INC, UK). A 100 bp DNA ladder was used as a marker for PCR products.
Molecular detection of Salmonella B-lactamase resistance genes
PCR was applied to amplify salmonella virulence genes blaCMY-1 and blaOXA-2 genes using specific primers as shown in (Table 8).
Phylogenetic analysis
Sequencing and phylogenesis of two S. typhimurium isolates (one from animal and one from human) was performed using the invA gene. The invA gene sequences were aligned with CLUSTRAL W multiple sequence alignment program, version 1.83 of megalign module of lasergene DNAStar software pairwise, which was designed by [55] and phylogenetic analysis were done using maximum likelihood, neighbor joining and maximum parsimony in MEGA6 [56].
Availability of data and materials
The datasets used and/or analyzed in the current study were not publicly published to preserve the privacy of the participants but are available upon reasonable request from the corresponding author.
References
Pal M, Alemu J, Mulu S, Karanfil O, Parmar B, Nayak J. Microbial and hygienic aspects of dry milk powder. Beverage Food World. 2016;43(7):28–31.
Tennant SM, MacLennan CA, Simon R, Martin LB, Khan MI. Nontyphoidal salmonella disease: current status of vaccine research and development. Vaccine. 2016;34(26):2907–10. https://doi.org/10.1016/j.vaccine.2016.03.072.
Wen SC, Best E, Nourse C. Non-typhoidal Salmonella infections in children: Review of literature and recommendations for management. J Paediatr Child Health. 2017;53(10):936–41. https://doi.org/10.1111/jpc.13585.
Eguale T, Engidawork E, Gebreyes WA, Asrat D, Alemayehu H, Medhin G, et al. Fecal prevalence, serotype distribution and antimicrobial resistance of Salmonellae in dairy cattle in central Ethiopia. BMC Microbiol. 2016;16(1):1–11. https://doi.org/10.1186/s12866-016-0638-2.
Nikiema ME, Kakou-Ngazoa S, Sylla A, Bako E, Addablah AYA, Ouoba JB, et al. Characterization of virulence factors of Salmonella isolated from human stools and street food in urban areas of Burkina Faso. BMC Microbiol. 2021;21(1):1–12. https://doi.org/10.1186/s12866-021-02398-6.
Muñoz-Vargas L, Pempek JA, Proudfoot K, Eastridge ML, Rajala-Schultz PJ, Wittum T, et al. The impact of overstocking and negative energy balance on quantitative measurement of non-typhoidal Salmonella in periparturient dairy cattle. Front Vet Sci. 2022. https://doi.org/10.3389/fvets.2022.779900.
Wells S, Fedorka-Cray P, Dargatz D, Ferris K, Green A. Fecal shedding of Salmonella spp. by dairy cows on farm and at cull cow markets. J Food Prot. 2001;64(1):3–11. https://doi.org/10.4315/0362-028x-64.1.3.
Bahramianfard H, Derakhshandeh A, Naziri Z, Khaltabadi FR. Prevalence, virulence factor and antimicrobial resistance analysis of Salmonella enteritidis from poultry and egg samples in Iran. BMC Vet Res. 2021;17(1):196. https://doi.org/10.1186/s12917-021-02900-2.
Ammar AM, Mohamed AA, Abd El-Hamid MI, El-Azzouny MM. Virulence genotypes of clinical SalmonellaSerovars from broilers in Egypt. J Infect Dev Ctries. 2016;10(04):337–46. https://doi.org/10.3855/jidc.7437.
Tawfiq S. Detection of gene stn in some non-typhoidal Salmonella spp. which isolated from patients with diarrhea. Tikrit J Pure Sci. 2019;24(1):29–35. https://doi.org/10.25130/tjps.24.2019.005.
Silva C, Puente JL, Calva E. Salmonella virulence plasmid: pathogenesis and ecology. Pathog Dis. 2017;75(6):ftx070. https://doi.org/10.1093/femspd/ftx070.
Abdel-Aziz NM. Detection of Salmonella species in chicken carcasses using genus specific primer belong to invA gene in Sohag city, Egypt. Vet World. 2016;9(10):1125. https://doi.org/10.14202/vetworld.2016.1125-1128.
Qamar A, Ismail T, Akhtar S. Prevalence and antibiotic resistance of Salmonella spp. South Punjab-Pakistan. Plos one. 2020;15(11):e0232382. https://doi.org/10.1371/journal.pone.0232382.
Kong-Ngoen T, Santajit S, Tunyong W, Pumirat P, Sookrung N, Chaicumpa W, et al. Antimicrobial resistance and virulence of non-typhoidal salmonella from retail foods marketed in Bangkok, Thailand. Foods. 2022;11(5):661. https://doi.org/10.3390/foods11050661.
Youssef RA, Abbas AM, El-Shehawi AM, Mabrouk MI, Aboshanab KM. Serotyping and antimicrobial resistance profile of enteric nontyphoidal salmonella recovered from febrile neutropenic patients and poultry in Egypt. Antibiotics (Basel). 2021. https://doi.org/10.3390/antibiotics10050493.
WHO. 2018. www.who.int/mediacentre/factsheets/fs139/en/print.html.In. Accessed 20 Feb 2018.
Marchello CS, Birkhold M, Crump JA, Martin LB, Ansah MO, Breghi G, et al. Complications and mortality of non-typhoidal salmonella invasive disease: a global systematic review and meta-analysis. Lancet Infect Dis. 2022. https://doi.org/10.1016/s1473-3099(21)00615-0.
Yang J, Meng L, Liu X, Ma L, Wang W. Detection of non-typhoidal salmonella gastroenteritis in a tertiary children’s hospital in China. Jundishapur J Microbiol. 2019. https://doi.org/10.5812/jjm.84400.
Shen H, Chen H, Ou Y, Huang T, Chen S, Zhou L, et al. Prevalence, serotypes, and antimicrobial resistance of Salmonella isolates from patients with diarrhea in Shenzhen, China. BMC Microbiol. 2020;20(1):1–10. https://doi.org/10.1186/s12866-020-01886-5.
Orabi A, Armanious W, Radwan IA, Girh ZM, Hammad E, Diab MS, et al. Genetic correlation of virulent salmonella serovars (Extended Spectrum β-Lactamases) isolated from broiler chickens and human: a public health concern. Pathogens. 2022;11(10):1196. https://doi.org/10.3390/pathogens11101196.
Ketema L, Ketema Z, Kiflu B, Alemayehu H, Terefe Y, Ibrahim M, et al. Prevalence and antimicrobial susceptibility profile of Salmonella serovars isolated from slaughtered cattle in Addis Ababa, Ethiopia. BioMed Res Int. 2018. https://doi.org/10.1155/2018/9794869.
Castañeda-Salazar R, del Pilar Pulido-Villamarín A, Ángel-Rodríguez GL, Zafra-Alba CA, Oliver-Espinosa OJ. Isolation and identification of Salmonella spp. in raw milk from dairy herds in Colombia. Brazilian J Vet Res Animal Sci. 2021;58:e172805. https://doi.org/10.11606/issn.1678-4456.bjvras.2021.172805.
Wu L, Luo Y, Shi G, Li Z. Prevalence, clinical characteristics and changes of antibiotic resistance in children with nontyphoidal salmonella infections from 2009–2018 in Chongqing, China. Infect Drug Resist. 2021;14:1403–13. https://doi.org/10.2147/IDR.S301318.
Abd-Elghany SM, Fathy TM, Zakaria AI, Imre K, Morar A, Herman V, et al. Prevalence of multidrug-resistant Salmonella enterica serovars in buffalo meat in Egypt. Foods (Basel, Switzerland). 2022;11(18):2924. https://doi.org/10.3390/foods11182924.
Diab MS, Zaki RS, Ibrahim NA, Abd El Hafez MS. Prevalence of multidrug resistance non-typhoidal salmonellae isolated from layer farms and humans in Egypt. World Vet J. 2019;9(4):280–8. https://doi.org/10.36380/scil.2019.wvj35.
Andoh LA, Ahmed S, Olsen JE, Obiri-Danso K, Newman MJ, Opintan JA, et al. Prevalence and characterization of Salmonella among humans in Ghana. Trop Med Health. 2017;45(1):3.
Wright JG, Tengelsen LA, Smith KE, Bender JB, Frank RK, Grendon JH, et al. Multidrug-resistant Salmonella Typhimurium in four animal facilities. Emerg Infect Dis. 2005;11(8):1235. https://doi.org/10.3201/eid1108.050111.
Varma JK, Mølbak K, Barrett TJ, Beebe JL, Jones TF, Rabatsky-Ehr T, et al. Antimicrobial-resistant nontyphoidal Salmonella is associated with excess bloodstream infections and hospitalizations. J Infect Dis. 2005;191(4):554–61. https://doi.org/10.1086/427263.
Eguale T. Non-typhoidal Salmonella serovars in poultry farms in central Ethiopia: prevalence and antimicrobial resistance. BMC Vet Res. 2018;14(1):217. https://doi.org/10.1186/s12917-018-1539-4.
Mechesso AF, Moon DC, Kim S-J, Song H-J, Kang HY, Na SH, et al. Nationwide surveillance on serotype distribution and antimicrobial resistance profiles of non-typhoidal Salmonella serovars isolated from food-producing animals in South Korea. Int J Food Microbiol. 2020;335:108893. https://doi.org/10.1016/j.ijfoodmicro.2020.108893.
Gong B, Li H, Feng Y, Zeng S, Zhuo Z, Luo J, et al. Prevalence, serotype distribution and antimicrobial resistance of non-typhoidal salmonella in hospitalized patients in Conghua District of Guangzhou, China. Front Cell Infect Microbiol. 2022;12:805384. https://doi.org/10.3389/fcimb.2022.805384.
El-Baz AH, El-Sherbini M, Abdelkhalek A, Al-Ashmawy MA. Prevalence and molecular characterization of Salmonella serovars in milk and cheese in Mansoura city, Egypt. J Adv Vet Animal Res. 2017;4(1):45–51. https://doi.org/10.5455/javar.2017.d189.
Elkenany R, Elsayed MM, Zakaria AI, El-Sayed SA-E-S, Rizk MA. Antimicrobial resistance profiles and virulence genotyping of Salmonella enterica serovars recovered from broiler chickens and chicken carcasses in Egypt. BMC Vet Res. 2019;15(1):1–9. https://doi.org/10.1186/s12917-019-1867-z.
Sabry MA, Abdel-Moein KA, Abdel-Kader F, Hamza E. Extended-spectrum β-lactamase-producing Salmonella serovars among healthy and diseased chickens and their public health implication. J Glob Antimicrob Resist. 2020;22:742–8. https://doi.org/10.1016/j.jgar.2020.06.019.
Siddiky NA, Sarker S, Khan SR, Begum R, Kabir E, Karim R, et al. Virulence and antimicrobial resistance profiles of Salmonella enterica serovars isolated from chicken at wet markets in Dhaka, Bangladesh. Microorganisms. 2021;9:952. https://doi.org/10.1371/journal.pone.0254465.
Elsayed SM, Moustafa AM, Abo-Sakaya RY, Ali AR. Prevalence and molecular characterization of Salmonella serovars isolated from diarrheic cattle and buffalo-calves. Zagazig Vet J. 2020;48(3):273–83.
Jassim AA, Al-Gburi NM. Virulence genes and antimicrobial resistance of Salmonella isolated from milk in Wasit Province, Iraq. Plant Arch. 2020;20(1):2033–9.
Mubita CM, Muma BJ, Nalubamba K, Pandey GS, Samui K, Munyeme M, et al. Characterization of non-typhoid Salmonellae isolated from domestic animals and wildlife from selected areas of Zambia. Sci Afr. 2020;8:e00345.
Allam SA, Mostafa NY, Kirrella GA, Eleiwa NZ, El-Magd MA. Molecular detection of invA and hilA virulent genes in Salmonella serovars isolated from fresh water fish. Slov Vet Res. 2019;56:693–8. https://doi.org/10.26873/SVR-809-2019.
Farahani NN, Jazi FM, Nikmanesh B, Asadolahi P, Kalani BS, Amirmozafari N. Prevalence and antibiotic susceptibility patterns of salmonella and shigella species isolated from pediatric diarrhea in Tehran. Arch Pediatr Infect Dis. 2018;6(4):57328.
Elshebrawy HA, Mahros MA, Abd-Elghany SM, Elgazzar MM, Hayashidani H, Sallam KI. Prevalence and molecular characterization of multidrug-resistant and β-lactamase producing Salmonella enterica serovars isolated from duck, pigeon, and quail carcasses in Mansoura, Egypt. LWT. 2021;149:111834.
Hasman H, Mevius D, Veldman K, Olesen I, Aarestrup FM. β-Lactamases among extended-spectrum β-lactamase (ESBL)-resistant Salmonella from poultry, poultry products and human patients in The Netherlands. J Antimicrob Chemother. 2005;56(1):115–21.
Archambault M, Petrov P, Hendriksen RS, Asseva G, Bangtrakulnonth A, Hasman H, et al. Molecular characterization and occurrence of extended-spectrum β-lactamase resistance genes among Salmonella enterica serovar Corvallis from Thailand, Bulgaria, and Denmark. Microb Drug Resist. 2006;12(3):192–8. https://doi.org/10.1089/mdr.2006.12.192.
Eguale T, Asrat D, Alemayehu H, Nana I, Gebreyes WA, Gunn JS, et al. Phenotypic and genotypic characterization of temporally related nontyphoidal Salmonella strains isolated from humans and food animals in central Ethiopia. Zoonoses Public Health. 2018;65(7):766–76. https://doi.org/10.1111/zph.12490.
Ahmed LM, Sayed AS, Abd ElKader H, Faddan NHA, Al Hosary AAT. Phylogenetic analysis of Salmonella species isolated from cows, buffaloes, and humans based on gyrB gene sequences. Trop Anim Health Prod. 2020;52(3):1487–92. https://doi.org/10.1007/s11250-019-02155-y.
Iso I. 6579–1: 2017 Microbiology of the food chain–Horizontal method for the detection, enumeration and serotyping of Salmonella-Part 1: Detection of Salmonella spp. Geneva: International Organization for Standardization; 2017.
MacFaddin J. Biochemical tests for identification of medical bacteria, williams and wilkins. Philadelphia: PA; 2000. p. 113.
Grimont PA, Weill F-X. Antigenic formulae of the Salmonella serovars. WHO collaborating centre for reference and research on Salmonella. Geneva: WHO; 2007. p. 1–166.
CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Fifth Informational Supplement CLSI document M100–S25. Wayne, PA: Clinical and Laboratory Standards Institute; 2016.
Joseph S, David WR. The condensed protocols from molecular cloning: a laboratory manual. Cold Spring Harb. 2006;3:800
Nayak R, Stewart T, Wang R-F, Lin J, Cerniglia C, Kenney P. Genetic diversity and virulence gene determinants of antibiotic-resistant Salmonella isolated from preharvest turkey production sources. Int J Food Microbiol. 2004;91(1):51–62. https://doi.org/10.1016/s0168-1605(03)00330-1.
Murugkar H, Rahman H, Dutta P. Distribution of virulence genes in Salmonella serovars isolated from man & animals. Indian J Med Res. 2003;117:66.
Swamy SC, Barnhart HM, Lee MD, Dreesen DW. Virulence determinants invA and spvC in salmonellae isolated from poultry products, wastewater, and human sources. Appl Environ Microbiol. 1996;62(10):3768–71.
Cardona-Castro N, Restrepo-Pineda E, Correa-Ochoa M. Detection of hilA gene sequences in serovars of Salmonella enterica sufigbspecies enterica. Mem Inst Oswaldo Cruz. 2002;97:1153–6. https://doi.org/10.1590/s0074-02762002000800016.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 60. Mol Biol Evol. 2013;30:2725–9. https://doi.org/10.1093/molbev/mst197.
Acknowledgements
The authors would like to thank the staff members of Department of Animal Hygiene and Zoonoses, faculty of veterinary medicine in New Valley University for their valuable corporation in this study.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
All authors collaborated in work planning, experimental design, measurement of parameters, and writing of the manuscript. MSD, AST, MA, and SAS conceived and designed the experiments. MSD, AST, MA measured the parameters. SAS and AST statistically analyzed the data. SAS, AST and MSD wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research was conducted according to the guidelines of the Institutional Animal Care and Use Committee of New Valley University, Egypt (. All farm owners included in the study were informed of all the study procedures and aims, and permission to collect animal samples was obtained from them verbally. All methods were performed in accordance with the ARRIVE guidelines for the reporting of animal experiments (https://arriveguidelines.org). In addition, a comprehensive discussion was conducted with each worker, and they received detailed information about the aims of the study. Informed consent was obtained from the human participants and/or their legal guardian after receiving detailed information about the aims of the study. The study was conducted in accordance with the Declaration of Helsinki for medical research involving human subjects. The animal and human experiments were approved by the ethics committee of New Valley University, Egypt.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Diab, M.S., Thabet, A.S., Elsalam, M.A. et al. Detection of Virulence and β-lactamase resistance genes of non-typhoidal Salmonella isolates from human and animal origin in Egypt "one health concern". Gut Pathog 15, 16 (2023). https://doi.org/10.1186/s13099-023-00542-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13099-023-00542-3