
Abstract
Background
In India, multi-drug resistance in Salmonella enterica serovar Typhimurium poses a significant health threat. Indeed, S. Typhimurium has remained unknown for a large portion of its genome associated with various physiological functions including mechanism of drug resistance and virulence. The whole-genome sequence of a Salmonella strain obtained from feces of a patient with gastroenteritis in Odisha, India, was analyzed for understanding the disease association and underlying virulence mechanisms.
Results
The de novo assembly yielded 17 contigs and showed 99.9% similarity to S. enterica sub sp enterica strain LT2 and S. enteric subsp salamae strain DSM 9220. S. Typhimurium ms202 strain constitutes six known Salmonella pathogenicity islands and nine different phages. The comparative interpretation of pathogenic islands displayed the genes contained in SPI-1 and SPI-2 to be highly conserved. We identified sit ABCD cluster regulatory cascade in SPI-1. Multiple antimicrobial resistance genes were identified that directly implies antibiotic-resistant phenotype. Notably, seven unique genes were identified as "acquired antibiotic resistance". These data suggest that virulence in S. enterica Typhimurium ms202 is associated with SPI-1 and SPI-2. Further, we found several virulent genes encoding SPI regions belonging to type III secretion systems (T3SS) of bacteria were significantly upregulated in ms202 compared to control LT2. Moreover, all these genes were significantly downregulated in S. enterica Typhimurium ms202 as compared to control LT2 on adding Mn2+ exogenously.
Conclusions
Our study raises a vital concern about the potential diffusion of a novel multi-drug resistant S. enterica Typhimurium ms202. It justifies this clinical pathogen to demonstrate a higher degree survival due to higher expression of virulent genes and enhanced ability of metallic ion acquisition.
Similar content being viewed by others


Background
Salmonellosis, one of the primary causes of food-borne infections resulting from gram-negative enteropathogenic bacteria Salmonella spp, is a global threat to human health [1]. Besides typhoidal Salmonella that causes enteric fever in humans, the non-typhoidal Salmonella (NTS) results in acute/chronic gastroenteritis. Annually, the NTS epidemic reports ~ 93.8 million infection cases and ~ 155,000 deaths [2]. Indeed, NTS infections not only typically cause diarrhoea but also result in non-specific febrile illness that is clinically indistinguishable from other febrile illnesses [3].
Salmonella enterica subspecies enterica comprises more than 2600 serovars [4, 5] with unique somatic (O) and flagellar (H) antigenic formulae used to identify the serovar types [6]. ~ 53 serovars of Salmonella have been identified from India, and the numbers are still counting [7, 8]. Remarkably, among different serovars, Salmonella enterica Typhimurium (henceforth S. Typhimurium) and Salmonella enterica Enteritidis are lead pathogens responsible for causing gastroenteritis in humans [9, 10] typically involved in self-limiting diarrheal illness. Globally as well as in India, invasive diseases cause a high degree of mortality and morbidity. The typhoidal serovars cause invasive disease and are humanly restricted. However, S. Typhimurium, also associated with gastroenteritis in cattle, chicken and a wide range of hosts, is considered a relevant zoonotic pathogen [11]. Notably, the epidemiology of S. Typhimurium has remained unknown for a large portion of the genome associated with various physiological functions of the bacteria. Indeed, the systemic disease association and mechanism of transmission of virulent factors to other hosts have not been completely understood [12].
The Pathogenicity islands (PAIs) and Resistance islands (REIs) are key regulatory regions that play complementary roles in bacterial pathogenesis. The PAIs, including large gene clusters [10 to 100 kb], play a vital role in mediating pathogenicity through invasion into epithelial cells followed by replication and survival inside phagocytic cells [13, 14]. Interestingly, due to Direct Repeated Sequences, few PAIs can excise from and re-integrate into the bacterial chromosome. Indeed, Salmonella Pathogenicity Islands (SPIs) belonging to T3SS play a crucial role in host-cell interaction. Therefore, it is essential to gain detailed knowledge about serotype distribution and the SPI pattern to formulate appropriate therapeutic and intervention strategies against Salmonella induced diseases.
In the current study, we sequenced and annotated the whole genome of a multi-drug-resistant (MDR) Salmonella enterica subsp. enterica serovar Typhimurium ms202 (henceforth termed as S. enterica Typhimurium ms202) isolated from feces of a patient with gastroenteritis admitted to a hospital in Odisha, India. The in-depth genomic analysis of MDR S. Typhimurium and annotation elucidated the genomic features for the distribution of SPI regions, virulence factors and identified candidate genes for antimicrobial resistance. We found that several virulent genes encoding the T3SS were significantly upregulated in S. enterica Typhimurium ms202 as compared to control strain, suggesting a higher degree of virulence and fitness of this drug-resistant bacteria. We hypothesize that, due to greater degree of virulence, these bacteria probably utilize metallic ions more efficiently, as shown by a diminished expression of Mn2+ transporter genes in S. enterica Typhimurium ms202 with respect to control in the presence of Mn2+. The genome sequence data provides us a better insight into the mechanisms by which species or serovars have evolved and helps to know the status of antibiotic resistance patterns. The genetic divergence in S. Typhimurium reared under diverse geographical locations may help understand the genomic architecture of this newly isolated species.
Results
Identification of multi-drug resistant S. enterica Typhimurium ms202
The demographic details of 221 patients are shown in Additional file 1: Table S1. As shown in the workflow of 'methods' section, a total of 221 fecal samples was examined by microscopic analysis and biochemical assays to detect S. Typhimurium. The microscopic analysis revealed presence of nematode and cestodes in stool of 35 cases (15.8%) indicating presence of parasitic diarrhoea. Major parasites observed through microscopy included Strongyloides, Entamoeba, Giardia, Ascaris, Ankylostoma, Trichuris, Enterobius and Taenia. Biochemical analysis suggested that, nine fecal samples containing bacterial isolates showed an adverse urea reaction along with a positive Triple Sugar Iron (TSI) and citrate reaction identifying the isolates as Salmonella. To confirm the genus and species, all nine isolates were examined using VTEK 2 ID card, out of which 6 of them were identified as Salmonella enterica. It must be noted that, the remaining three bacterial isolates could not be recognized for any species, possibly due to sample handling error. Further, serotype analysis revealed four out of six strains as Salmonella enterica serovar Typhimurium. (The remaining two strains were identified as S. enterica Weltevreden). All four isolates of S. enterica serovar Typhimurium were subjected to antibiotic susceptibility test to examine drug resistance. The results showed three out of four to be resistant to more than three antibiotic groups considering these pathogens as multidrug-resistant. Of three MDR strains, one isolate (S. enterica Typhimurium ms202 in this manuscript) showing resistance to four antibiotic groups was selected for genome analysis study. The resistance pattern of the strain to antibiotics showed in Additional file 2: Table S2. Different antibiotics and antibiotic groups (in brackets) to which the strain was found resistant are as follows: Cefuroxime (Cephalosporin 2nd generation), Nalidixic acid and Ciprofloxacin (Quinolones), Amikacin and Gentamycin (Aminoglycosides) and Tigecycline (Tetracyclines).
Genome drafting of S. enterica Typhimurium ms202
Whole genome assembly and assessment
High-throughput sequencing generated a total of 4,894,062 bp with 318-depth coverage determined by accomplishing the Gap closure in the S. enterica Typhimurium ms202 entire genome sequence. The details on assembly are shown in Table-1. SOAPdenovo2 software suggested the most appropriate assembler in terms of genome completeness [15]. QC plots, GC content versus contig coverage showed a single cluster, indicating the absence of contamination in data. The total raw reads of 11,410,205 and the HQ read of 5,969,942 were recorded. All contigs were mapped to the reference genome successfully with slight variations in the alignments. An assembled genome was submitted to the Comprehensive Genome Analysis service.
Completeness of the genome annotation
The genome of S. enterica Typhimurium ms202 was annotated using the RAST tool kit (RASTtk) and was assigned a unique genome identifier: 90,371.4806. This genome is in the super kingdom bacteria and is annotated using genetic code 11 (Table 1). The annotation included 669 hypothetical proteins and 4,303 proteins with functional assignments, 83 transfer RNA (tRNA) genes and 10 ribosomal RNA (rRNA) genes (Table 2). 1,286 proteins with functional assignments and Enzyme Commission (EC) numbers, 1,052 with Gene Ontology (GO) assignments, 174 pseudogenes and 903 proteins were mapped to KEGG pathways. A circular graphical representation illustrated the distribution of the genome annotations (Fig. 1).

A circular graphical illustration depicting the genome annotations distribution. It includes, from outer to inner rings, the contigs, CDS on the forward strand, CDS on the reverse strand, RNA genes, CDS with homology to known antimicrobial resistance genes, CDS with homology to known virulence factors (yellow), GC content (pink), and GC skew (green)
Phylogenetic relationship of S. enterica Typhimurium ms202 with other bacterial species
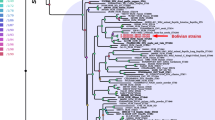
The phylogenetic analysis was carried out by concatenated sequence and multiple alignments using conserved sites phylogeny of the distinct Salmonella species and subspecies. The Neighbour-Joining method for the evolutionary history of alignment was inferred as shown in Fig. 2. However, this tree contains several branches with low bootstrap support (> 50%) and places the two sequenced Salmonella strains such as, Salmonella enterica subsp. arizonae serovar 62:z4,z23 and Salmonella enterica subsp. salamae strain DSM 9220. Further, elaboration of phylogram shows that there are two different clusters of which one cluster contains various S. enterica serovars and the other has Citrobacter freundii CFNIH1 1,333,848.3. The phylogenetic regions indicate that Salmonella bongori NCTC 12,419 218,493.5 is the last common ancestor, and the newly isolated strains evolved faster.

Phylogenetic position of S. enterica Typhimurium ms202 by neighbour-joining method for the evolutionary history of alignment. The tree contains several branches with low bootstrap support (> 50%)
Comparison of Amino Acid Identity (AAI) and Average Nucleotide Identity (ANI) between S. enterica Typhimurium ms202 and reference
The AAI profiler summarizes results for proteome-wide sequence search with two clear parameters obtained from pairwise genome comparisons and takes the average sequence identities of Salmonella's shared orthologous genes. The variations in the taxonomic groups indicated by colour (Fig. 3) suggested that the genome of S. enterica Typhimurium ms202 appears as a distinct clade with the Citrobacter genus. The ANI relationships between type assemblies and selection of the species boundaries are supported by 96% ANI cut-off. Further, we evaluated all taxa between taxonomy-matching (concordant) and taxonomy-non-matching (discordant) pairs selected from the taxonomically united 16S rRNA database and whole-genome assemblies. The genetic relatedness of newly isolated S. enterica Typhimurium ms202 was examined with two control strains such as, S. enterica subsp. enterica serovar Typhimurium_st_LT2 ('control 'LT2' in the current study) and S. enterica subsp. enterica serovar Typhimurium_st_SL1344 with (Table 3). Indeed, there was an evident distinction at 96% for most taxonomic groups observed between intra and inter-species relationship.

Average Aminoacid Identity (AAI)-profiler scatterplot for S. enterica Typhimurium ms202. The query proteome was obtained from NCBI genomes. The dot nearest coordinates (1, 1) is the query species and is closely related group which is distinct from other genera
Functional characterization and subsystem annotation of S. enterica Typhimurium ms202
The functional annotation of genomes was investigated. The number of protein-encoding genes associated with their active role is estimated using RAST-SEED viewer and results are shown in Fig. 4. A subsystem is a set of proteins that implements biological processes or structural complexes, and annotations include an analysis of the subsystems unique to the genome. Figure 4a represents a pie chart showing genes related to several physiological functions of bacteria. The complex processes include Metabolism (33.75%), followed by Protein processing (13.70%) and Stress response, Defense, and Virulence (13.38%). Further, we elaborated 'Metabolism' by dividing it into three sections as sequential metabolism (Fig. 4b and c) and genetic material metabolism (Fig. 4d). Most of the identified genes were supposed to be involved in carbohydrate metabolism and notably, many genes were predicted to show association with virulence and pathogenesis. The Multilocus Sequence Typing (MLST) method was used to identify seven matching genes. The length (in base pairs) of the alignment between the MLST allele referred to the database (DB) and the equivalent sequence in the input genome. (Additional file 3: Table S3).

Functional Characterization and subsystem annotation of S. enterica Typhimurium ms202. a A pie chart showing gene numbers for several biological processes/physiological functions of S. enterica Typhimurium ms202. b and c Genes involved in sequential metabolism, d Genes involved in genetic material metabolism
Phage analyses and region annotation
Bacteriophages are viruses that infect bacteria and are abundant in the natural environment. They are important in regulating bacterial growth and proliferation. Further, Phages are necessary for the transmission of genetic information. In the draft genome of S. enterica Typhimurium ms202, nine prophage regions were found, of which two are entire, five are partial, and the other two are contested; most likely relate to phage remnants (Fig. 5, Table 4) (Additional file 4: Table S4). These phage remains are almost of same length and G + C content. Future studies are directed to compare these identified bacteriophages with phages previously reported in other bacterial species.

Circular presentation for distribution of phage in S. enterica Typhimurium ms202genome island. 9 prophage regions have been identified, of which 2 regions are intact (score > 90), 5 regions are incomplete (score < 70), and 2 regions are questionable (score 70–90) likely corresponding to phage remnants
Distribution of SPIs in S. enterica Typhimurium ms202 genome and pathway analysis
The interpretation of virulence/resistance gene annotations in genomic islands was predicted. The whole-genome analysis revealed that S. enterica Typhimurium ms202 consists of six known Salmonella pathogenicity islands (SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, and SPI-11). The distribution of the genes in SPI regions has homology to known transporters, drug targets, and antibiotic resistance (Fig. 6) and the subset of genomic islands, which facilitates the horizontal transfer of genes encoding numerous resistance and virulence factors of T3SS. Irrespective of their origin, the gene content in SPI-1 to SPI-2 is highly conserved in all the reference strains selected from the PAIs database. The sit A to D genes clusters a regulatory cascade in SPI-1 promoting virulence of bacteria. Indeed, according to the PAIs database, a maximum number of genes are encoded at SPI-2 (~ 28) followed by SPI-1 (~ 12).

Distribution of SPIs in S. enterica Typhimurium ms202 genome island. Three methods such as IslandPath-DIMOB, Integrated and SIGI-HMM were utilized for virulence/resistance gene annotations. The grey colour circle represents GC content
Moreover, three unique genes such as sopD_2, serC, and ompF found in the SPI-5 region are mainly responsible for pathogenicity. Many SPIs are located near a tRNA gene, and differences are observed in their G + C content from the remaining part of the genome. Indeed, other virulent genes were also determined in SPI regions.
The KEGG analysis showed two common pathways associated with Salmonella pathogenesis: the pathway of invasion into epithelial cells and flagellar assembly. However, nine different pathways were identified in SPI-5 (Table 5). For further insights, protein–protein interaction was studied by the network and co-expression based annotations. The nodes in SPI-2 [40 nodes] with average local clustering coefficient (0.904, p-value: < 1.0e-16) showed the single cluster content of approximately 20 effectors. However, TtrR and TtrC have shown string characters (Fig. 7a). None of the co-expression profiling for TtrC and ssrB was found in the SPI-2 cluster (Additional file 5: Table S5). The two different network clusters are observed in the SPI-1 region gene content (Fig. 7b). A smaller number of co-expression was identified among the effector genes consisting of sitA, sitB, sitC, sitD. The nodes (22 nodes) with the average local clustering coefficient (0.821, p-value: < 1.0e-16) and the co-expression profiling showed the pathogenic property and the virulence factors. For instance, the GTPase-activating protein SptP (Salmonella protein tyrosine phosphatase), activates SPI-4 genes and weakly represses SPI-1 genes and cell invasion protein B that are required for invasion of bacteria to host cells (Additional file 6: Table S6).

The protein–protein network cluster and co-expression profiling of the genes from SPI-1 and SPI-2 regions of S. enterica Typhimurium ms202. A and C SPI-2; B and D SPI-1. The red squares inside C and D represent interaction scores
Annotation of antimicrobial resistance genes and virulence factors
The genes associated with virulence and pathogenicity were carried out using blast search with specific sources, and databases such as CARD, NDARO, VFDB, DrugBank were applied to avoid the limited list of identifiers and genes available in the database. The average number of genes were encoded with transporter (689), virulent factors (141) (Additional file 7: Table S7), drug target specific genes (530) (Additional file 8: Table S8), and antimicrobial resistance specific (Table 6).
The Genome Annotation Service in PATRIC uses a kmer-based method to detect antimicrobial resistance (AMR) gene variants. To each AMR gene, this service assigns functional annotation, comprehensive antibiotic resistance mechanism, different classes of drugs and occasionally, a specific antibiotic to which it confers resistance. It notes that the presence of AMR-related genes (even full length) in a given genome directly implies an antibiotic-resistant phenotype. It is essential to consider specific AMR mechanisms, especially regulator modulating expression, efflux pump conferring, and target in susceptible categories (Table 7). Noteworthy, seven genes such as fosA7, tet(A), sul1, aadA7, aac('6')-iaa, aadA7, and qacE were identified as the acquired antibiotic resistance genes in currently studied drug resistant strain. (Table 8).
Expression profile of virulence genes in S. enterica Typhimurium ms202
To examine if S. enterica Typhimurium ms202 has higher degree of virulence as compared to drug sensitive strain, we compared the expression profile of 9 different genes (Identified in the genome draft) associated with virulence of bacteria encoding SPI-1 (6 genes), SPI-2 (2 genes) and SPI-5 (1 gene) regions of the bacteria with that of control LT2. These genes were selected arbitrarily from the genome draft results, emphasizing SPI-1 encoding genes due to their vital role in the invasion and colonization of Salmonella in host gut. Specifically, four specific SPI-1 encoding genes such as, sitA, B, C and D are known to play a role in transporting ions such as Mn2+ and Fe2+ along with virulence function. Figure 8A shows that genes such as sitA, sitB, sitC, sitD, hilA and sopB encoding SPI-1 and ssaV under SPI-2 were increased by 12.1 fold, 212 fold, 14.4 fold, 7.3 fold, 7.4 fold, 11.3 fold and 14.3 fold respectively in S. enterica Typhimurium ms202 as compared to control LT2 indicating a significant upregulation of gene expression in the drug-resistant strain (P < 0.05). Although genes such as ompF (encoded by SPI-5) and ssaB (encoded by SPI-2) were upregulated by 5.5 fold and 4.9 fold, the difference was not statistically significant. An overall increase in virulent gene expression in S. enterica Typhimurium ms202as compared to control suggested a higher virulence potential of the MDR strain.

Expression profile of virulent genes in S. enterica Typhimurium ms202 under the influence of Mn2+. A Expression levels of nine different virulent genes encoding SPI1 (sit A, B, C, D, hilA, ompF and ssaV) and SPI2 (ssaB and sopB) regions of S. Typhimurium by qRT-PCR. Results are expressed as log2 fold change in S. enterica Typhimurium ms202 with respect to control LT2. 16 s rRNA gene was considered as a housekeeping gene in qRT-PCR. Error bars indicate Mean ± SE. The student's t-test determined statistical significance, and P < 0.05 was considered significant. B Expression levels of nine different virulent genes encoding SPI1 (sit A, B, C, D, hilA, ompF and ssaV) and SPI2 (ssaB and sopB) regions of S. Typhimurium by qRT-PCR after treatment of with 5uM MnCl2 compared to corresponding untreated controls. The 16 s rRNA gene was taken as the housekeeping gene in qRT-PCR. Error bars indicate Mean ± SE from three independent experiments. Statistical significance was determined by Student's t-test, and P < 0.05 was considered as significant
To understand if greater virulent potential in S. enterica Typhimurium ms202 results in an efficient utilization of metallic ions available in hosts, we examined the expression profile of all 9 genes in the drug resistant and control strain in the presence of exogenously added Mn2+ in culture media. Results shown in Fig. 8B revealed that sitA, B, C, and D (Mn2+ transporter genes) were downregulated in Mn2+ treated control LT2 than the untreated control. There was a 5.8 fold, 2.6 fold, 3.5 fold, and 3.7 fold decreased expression of these four genes in control LT2 exposed to Mn2+ compared to untreated ones. On the contrary, levels of hilA, ssaB, ssaV and sopB genes were significantly upregulated in treated control LT2 (P < 0.05). OmpF gene was found marginally increased (P > 0.05). Importantly, we found all 9 virulent genes significantly downregulated in the Mn2+ treated drug-resistant strain as compared to untreated S. enterica Typhimurium ms202. There was a 292.6 fold decreased expression of sitA in treated ms202 compared to untreated strain. In contrast, other genes such as sitB, C, D, hilA, ssrB, ssaV and sopB were downregulated by 167.5 fold, 113.4 fold, 270.6 fold, 263.7 fold, 193.5 fold, 214 fold and 35.3 fold in the drug-resistant strain. A marginal decrease in the ompF level (4.9 fold) was observed (P > 0.05). Ultimately, when compared between treated S. enterica Typhimurium ms202 vs treated LT2, a notably decreased expression of all 9 genes was observed in drug-resistant strain (P < 0.05).
Discussion
Studies using whole-genome sequence showed the serovar-level distribution in Salmonella enterica subsp enterica [16,17,18]. However, it has often been neglected and studies on in-depth pathway analysis regarding the NTS infections are rarely reported from India [19]. As noted earlier, S. Typhimurium is the most common pathogen for NTS infection, significantly contributing to gastroenteritis with a greater intensity of infection in immunocompromised hosts [20, 21]. As the resistance of Salmonella to multiple antibiotics has become a global issue disabling treatment options, detailed analysis of the genome of drug resistant strain is crucial to analyse new virulence factors and AMR genes. Accordingly, we were interested in exploring genome annotation of the multidrug-resistant clinical isolate, S. enterica Typhimurium ms202. Here, the de-novo genome assembly has shown better annotation with fewer gaps, genome coverage and better scaffold statistics. The study sheds light to identify the distributions of SPIs and the operon of virulent factors that might help to understand the pathogenic nature of this newly identified strain and information on antimicrobial-resistance [22].
In general, 16 s rRNA sequencing-based phylogenetic-tree analysis is the most frequently used method to classify organisms. In our phylogenetic tree analysis, we have identified that S. enterica Typhimurium ms202 has 99.9% similarity to S. enterica subsp. salamae strain DSM 9220 and Salmonella enterica subsp. enterica strain LT2. The study on comparative analysis indicated a close relationship among these bacteria [23] and suggested the association with disease in humans and animals [24]. Remarkably, S. enterica Typhimurium ms202 and S. enterica serovar Arizonae have also supported coalescing and sharing core serological properties, but the latter is not a common pathogen in humans and shows occasional disease association. From phylogenetic tree analysis, we find that S. enterica Typhimurium ms202 is far from S. bongori The distinct taxonomic grouping defines the genealogical species concept for S. enterica Typhimurium ms202 [25] and emphasizes the understanding of genetic diversity and phylogenicity among S. enterica serovars [18].
The previous study has shown that S. Typhimurium carried the 90 kb virulence plasmid [26]. However, the role of the virulence plasmid in pathogenesis is not clear [27]. It is suggested that phage remnants in the Salmonella genomes contribute towards both inter-species and inter-strain variability. Indeed, we didn't find any phage plasmid in S. enterica Typhimurium ms202. According to earlier reports, plasmids are often identified in S. Typhimurium and S. Enteritidis strains isolated from blood and other extra-intestinal sources than strains isolated from faeces [27,28,29]. The absence of phage plasmid in our MDR strain may be due to its isolation from feces. Moreover, a different framework is needed on the sequence, database, and tool limitations to overcome assessing the plasmid populations within S. enterica Typhimurium ms202.
MDR Salmonella has become a global threat, including India. Therefore, non-antibiotic approaches such as the use of bacteriophages (phages) has gained attention and may be effective in the treatment and prevention of infection caused by antibiotic-resistant bacteria. Salmonella consists of various phage receptors such as flagella, O-antigen, OmpC, FhuA, and TolC. The complex nature of lipopolysaccharide (LPS) of S. Typhimurium and the receptor diversity may block access of phage to some outer membrane proteins and receptors, which causes the host S. Typhimurium to become resistant to phage [30]. Herein, we identified nine phages and the genes mapping to NC_004745 and NC_031019 revealed the presence of a set of virulent factors translocated into the bacterial cytoplasm through the outer membrane [31, 32]. Further studies may insight the receptor interaction mechanisms between the phage and host that may lead to optimal phage therapy for protection against the MDR S. Typhimurium.
We summarized the distribution of the genes (kmer-based method) based on their antimicrobial resistance mechanisms. The maximum number of genes were identified in efflux pumps conferring antibiotic resistance and antibiotic targets in susceptible species categories. The efflux pumps play a vital role in drug resistance and serve other Salmonella functions [33]. Extensive studies on the AcrAB-ToIC system in Escherichia coli and Salmonella confer innate resistance to many antibiotics [34, 35]. Further evidence suggested that, S. Typhimurium isolate derived from a patient with ciprofloxacin treatment overproduced AcrAB [36]. We have also identified seven acquired antibiotic resistance genes. A previous study has reported that phage populations in S. enterica contribute to horizontal gene transfer, including virulence and virulence-related genes within the subspecies [37,38,39,40].
The objective of our study was to reveal the genome elements of the newly isolated S. enterica Typhimurium ms202 that may contribute to virulence and resistance. Prior reports suggested that SPI-1, SPI-2 encoding T3SS, SPI-3, SPI-4, SPI-5 and type VI secretion systems play an essential role in association with the host cell, invasion, and the intracellular lifestyle of the Salmonella [32, 41]. The SPI 1–5, 9, 13 and 14 are the core part of the genome, whereas SPI-6, 10–12, 16–19 are variable among genomes, and SPI-7, 8 and 15 are known to be absent in invasive non-typhoidal Salmonella [42]. In our case, the whole-genome comparison results revealed S. enterica Typhimurium ms202to harbour six pathogenicity islands (SPI-1 to SPI-5 and SPI-11). However, SPI-9, 13 and 14 were not detected. Indeed, the absence of a few SPIs has also been reported earlier. For example, Blondel et al. (2009) reported the lack of SPI-19 to 21 in both S. Typhimurium and S. Typhi [43]. Hence, these differences with previous reports may be due to different isolates and serotypes, methodologies, and SPI- host specificity [44, 45]. Our report has shown the significant diversity within the SPI-1, 2 and 5 in gene content. The sit A to sit D genes cluster regulates/triggers the growth and virulence of Salmonella Typhimurium by receiving specific environmental cues such as organic iron, calcium, and magnesium [46, 47].
SPI-1 plays a vital role in the colonization and invasion of Salmonella in the gut and the recruitment of neutrophils [44, 45]. We identified the IagA and hilA encoded in pathogenicity island SPI-1, that are associated with regulation of invasion and activate the expression of prgHIJK. The protein prgH is unique to the Salmonella SPI-1 and makes up the membrane-spanning needle complex with InvG, prgI, and prg K. This complex regulates the production of effector proteins from Salmonella into the host cell [48, 49]. Indeed, the central regulator of SPI-1 expression is correlated with the hilA transcription factor [50]. To gain further insights into the organization of the this MDR strain genomic SPI-2 region, we further analyzed the virulent proteins. We found that SPI-2 effector proteins such as sseB, sseD and sse F are essential for the appropriate localization of sseC and sseD on the cell surface of bacteria, causing translocation of effecter protein into the host cell and aggregation of the host endosomes, respectively (Fig. 9). Several studies have indicated that the protein encoded genes from ssa family members belong to T3SS involved in Salmonella's replication and intracellular survival in the host. In contrast, ssrA and ssrB are indispensable for the SPI-2 encoded virulence gene expression. The transcriptional regulator of sprB is primarily reported in SPI-4 and weakly represses SPI-1 too. This study not only has recapitulated reports from the previous investigations about the serotypes and distribution of genes in SPI regions but also verified their profiles using the database for S. enterica Typhimurium ms202 [19, 51, 52].

Mechanism of sequential activation of Salmonella infection and regulation pathways. HilA is the central regulator of SPI-1 and T3SS needle apparatus in association with the host cell. Salmonella invasion proteins (Sips), A–D mediate SPI-1 effectors secretion and translocation. After translocation into host cells, the Salmonella outer proteins (Sops) are involved in polymorphonuclear leukocyte influx. After crossing the epithelial barrier, T3SS encoded SPI-2 effector proteins are secreted into the cell's cytosol for gene expression. The pathogen first recognizes these influence factors in the distal gut lumen and later in the intra-macrophage environment
The survival of bacteria inside hosts is solely dependent on its virulence potential and ability to utilize host resources. Pathogenic drug-resistant bacteria are known to display a better degree of survival advantage inside hosts than a drug-sensitive strain and one obvious reason is inherent ability of the former to resist antibiotics. Salmonella Typhimurium acquires different micronutrients (Examples—Mn2+, Fe2+, Zn2+) from the host milieu through metal transporters, and these ions contribute to the virulence and survival of bacteria [53]. However, it is not known how drug-resistant Salmonella with a better survival strategy in hosts behaves to this physiological function (acquiring metal ions from the host) and we addressed the issue in the current study. [We deciphered that, probably this drug resistant bacterium is more efficient in acquiring metal ions (Mn2+) from an external source due to higher degree of virulence. The entire issue is addressed as follows.
It is known that, in the presence of Mn2+ in the host milieu, S. Typhimurium acquires these divalent ions and hilA is activated that further regulates other SPI-1 associated virulence genes [53]. However, when Mn2+ is absent in the host environment, the pathogen cannot acquire the required ions leading to the inactivation of hilA. In this case, Mn2+ transporter genes such as sit A, B, C and D modulate virulence and adaptation to stress response. They are found to be upregulated in bacteria and serve as SOS responses. In our study, we found a significant upregulation of 7 out of 9 different SPI-1, SPI-2 and SPI-5 genes in S. enterica Typhimurium ms202 strain as compared to control LT2, indicating a possible role of these genes in contributing to a higher degree of virulence of ms202 (Fig. 8A). However, future studies are directed to evaluate the degree of virulence of these bacteria by in vivo phenotype validation experiments.
Our second important observation was a significant reduction in sit A, B, C and D genes in control LT2 in the presence of Mn2+ in M9 media as compared to ion deficient condition (Fig. 8B). These results are in accordance with previous studies by [54], which showed significant repression of sit ABCD operon of S. Typhimurium on the addition of Mn2+ to bacterial culture due to regulatory activity of mntR gene. During data analysis for S. enterica Typhimurium ms202, we found a similar trend (reduction of these four Mn2+ transporter genes in Mn2+ supplemented ms202 as compared to ion deficient condition), however to a greater extent. Importantly, we observed a significant fold reduction for all four sit genes in S. enterica Typhimurium ms202 compared to control strain LT2 when both types of bacteria were exposed to Mn2+. In this context, two possibilities can be discussed to explain higher fold reduction of genes in drug resistant stain on manganese exposure. First, reduction of gene expression may be due to the higher efficiency of S. enterica Typhimurium ms202 to acquire these divalent ions from external sources due to their higher virulent potential compared to control LT2 (as shown in Fig. 8A). Second, it is known that, the regulatory gene, mntR serves as a primary sensor for Mn2+ abundance leading to repression of sit ABCD genes in S. Typhimurium [54]. Therefore, mntR could be playing a regulatory role resulting in higher fold reduction of sit gene expression in Mn2+ supplemented S. enterica Typhimurium ms202, supposedly through an unknown mechanism that needs to be looked at in future.
Although our data showed an apparent down-regulation of four sit genes (possessing a role for both Mn2+ transport and virulence) in control LT2 on exposure to Mn2+. Four other genes, such as hilA, ssaB, ssaV and sopB (possessing a role in virulence but not in ion transport), showed a significant upregulation in bacteria upon exposure to Mn2+. Although the reason is not very clear to us, previous studies have indicated a firm requirement of different micronutrients by bacteria for their metabolism and virulence factor expression [47, 55]. Thus, it is assumed that adding Mn2+ to the culture might have contributed to the higher expression levels of few virulent genes, supposedly playing no role in ion transport. In contrast to these results, our observation showing a significant downregulation of these four specific genes (hilA, ssaB, ssaV and sopB) in Mn2+ exposed drug-resistant S. enterica Typhimurium ms202 compared to untreated bacteria is not properly understood.
Conclusions
This study sheds light on the genome draft of a new MDR Salmonella enterica serovar Typhimurium ms202 strain isolated from fecal sample of a patient with gastroenteritis and emphasizes the contribution of genetics architecture to antibiotic resistance and virulence. We found that the isolate commonly carried six SPIs, virulence operon, several MDR genes and phages. These genetic elements pose insight on the strain characteristics and allows further understanding of the diversity, host specificity of SPIs, and pathogenicity of S. enterica Typhimurium ms202. These features may promote a quicker and more accurate control of the food-borne illness outbreaks in eastern coastal areas of India. Further, the study also unravels a clue for a higher degree of virulence property as demonstrated by enhanced expression of several virulent genes. It is further observed that, due to a higher virulent property, this MDR bacterium showed an increased efficiency for acquiring Mn2+ ions from the external environment as indicated by a drastic reduction in Mn2+ transporter genes. Future study may be designed to find gene targets in drug resistant Salmonella using to which host derived molecules (as sensors) may activate causing reduction of virulent features of drug-resistant bacteria ultimately resulting in decreased pathogenesis.
Methods
Study design

The overall design of our study is mentioned as a workflow shown above. During a study period from September 2018 to December 2019, fecal samples could be collected from 221 patients (aged 1–80 years of age) suffering from acute gastroenteritis (Subjects with loose motion > 3 times or one large voluminous stool in 24 h, with/without dysentery) admitted in SCB medical college, Cuttack, India and Pradyumna Bal Memorial Hospitals (KIMS), Bhubaneswar, India and. The study only included in-house patients and OPD cases were excluded as sample collection was not possible from later case due to their temporary stay in hospitals.—Among 221 study subjects, 6 of them had concurrent complication such as acute renal failure presenting anuria or oliguria with altered mental status due to electrolyte deficiency. Upon admission into hospital, symptomatic treatment (drugs for oral rehydration, probiotics etc..) was initiated for patients and all subjects were undergone routine microscopic and biochemical analysis in feces. Patients demonstrating bacteria in stool samples were undergone treatment with appropriate antibiotics after analysing the test reports. All study aspects were performed following National Ethics Regulations and were approved by Institutional Review Boards of the hospitals taken under the study.
Identification of S. Typhimurium in feces of patients
Stool samples from patients suffering from gastroenteritis were collected in Cary-Blair transport media (M202, Hi Media), and were brought to laboratory within 2–3 h of collection.. To detect Salmonella, procedure by [56] was followed. Briefly, feces were first pre-enriched in Selenite Cysteine Selective enrichment broth (LQ070, HiMedia) for 18–24 h at 35 °C. This process enables the detection of Salmonella in the non-acute stages of illness and further inhibits the growth of competing non-Salmonella faecal coliforms and enterococci. After appropriate growth, bacteria were sub-cultured two times on plate with XLT4 selective agar (M1147, HiMedia); first sub-cultured after 6–8 h and then after 18–24 h of incubation at 35 °C. Salmonella was identified by examining a black centred colony on the agar plate surrounded by a yellow-coloured halo. These colonies were further inoculated in the Triple Sugar Iron agar (TSI) (MM021, HiMedia), Citrate agar (M728, HiMedia) and Urea agar (M112S, HiMedia). After overnight incubation, organisms demonstrating an adverse urea reaction and a positive TSI and citrate reaction were considered Salmonella. These isolates were further analyzed using VTK 2 ID card (Biomeriuex, Germany) in VITEK II COMPACT instrument to confirm Salmonella enterica. To identify serovar ''Typhimurium', S. enterica isolates were sent to Central Research Institute, Kasauli, Himachal Pradesh, for serotype analysis. The collected isolates were stored at − 80 °C with 30% glycerol in phosphate-buffered saline until further use.
Antimicrobial susceptibility test for S. Typhimurium
The resistance pattern of Salmonella Typhimurium to several antibiotics was examined using VITEK II COMPACT AST card (a device to examine antibiotic susceptibility of bacteria belonging to Gram -ve Enterobacteriaceae family; Biomereux, France) in VITEK II COMPACT instrument. Bacteria showing resistance to three or more than three groups of antibiotics were considered MDR strains. One MDR isolate (S. enterica Typhimurium ms202) was used for current genome analysis study that showed resistance to the highest numbers (four) of antibiotic groups.
De novo genome sequencing, quality control and pre-processing of FASTQ files
According to the manufacturer's instructions, genomic DNA extraction was performed from overnight cultures using QIAamp 96 DNA QIAcube HT Kit (Qiagen, Gaithersburg, MD). The DNA integrity was determined by agarose gel electrophoresis and the A260/280 ratio used for purity check. Followed by the usage of Illumina TruSeq DNA LT library kit was used for preparing the genomic library. Whole-genome sequencing was conducted utilizing the Illumina MiSeq system (Illumina, San Diego, CA) using the SBS sequencing kit V3.0, with a 2 × 100 paired-end option.
Downstream analysis requires Quality control and pre-processing of FastQ files to obtain clean data. The FastQC program [14] developed in C++ containing multi-threading support, is used for quality control and features for data-filtering. The quality of the raw reads was checked using FastQC (0.11.5), followed by trimming of adapters and low-quality bases with a Phred quality score of less than 20. Briefly, Trimmomatic [57] (k-mer 41) were applied to remove the low-quality reads (average quality scores < 20) and adapter sequences and the clean reads assembled.
Primary assembly of the raw data validation
The purpose of a sequence assembler is to create lengthy continuous sections of sequence (contigs) from several reads. Contigs are sometimes arranged and positioned in reference to one another to build scaffolds. The high quality (HQ) filtered reads from sample were assembled using the SPAdes assembler with the default parameters [58]. Furthermore, three distinct assemblers, ALLPATHS-LG, ABySS, and SOAPdenovo2 were used and an assembly assessment was done, despite the fact that the assembly methodology differed in approaches.
RNAmmer 1.2 4 was used to predict the closest reference check based on the 16 s rRNA gene sequence from the generated main genome assembly [59]. The projected 16 s rRNA gene sequence was used to find the closest genome reference for the newly generated genome assembly using EzBiocloud. The assembled genome FASTA file was used as a reference, and the filtered HQ reads from sample were aligned with respective assembled genome using the aligner Bowtie2 (v 2.2.2) to validate the percentage of HQ reads used to make the respective draft genome assembly.
Assembly of the final genome draft and prediction of phylogeny position
The assembled primary genome FASTA and the closest reference for the assembled primary genome were aligned with MAUVE aligner version 2.4.0 using the progressive algorithm with default parameters. The contigs of the primary assembled genome are rearranged using the web-based tool MeDuSa for ordering and orientating assembled genome contigs FASTA file into scaffold with the default web interface parameters. [MeDuSa uses information obtained from a set of (draft or closed) genomes from related organisms to determine the correct order and orientation of the contigs]. This process results in a total length of 2.4 Gb (contig N50: 48.8 kb and scaffold 12.8 Mb). The projected 16S rRNA sequence was aligned to the NCBI 16S rRNA database, and the top 50 aligned hits were used to construct a tree. All sequences were concatenated, and several alignments were made through the use of conserved locations. MAFFT version 7 was used to create the phylogeny. The scale reflects the predicted number of substitutions per site based on the best-fitting GTR2 + F0 + R2 (binary) model.
Prediction of Average Nucleotide Identity (ANI) and Average Amino Acid Identity (AAI)
An Average Nucleotide Identity (ANI) above 95% between two genomes indicates that they belong to the same species. ANI was calculated with the nearest reference genome for the assembled genome of the sample using ChunLab's online Average Nucleotide Identity (ANI) calculator.
One-sided and bidirectional AAI profiles were generated for a query proteome (protein sequences in FASTA format). To extract homologous proteins from Uniprot, SANSparallel. Taxonomic information was employed obtained via the DictServer. Each query protein was matched to all database species with the highest bitscore. One-sided AAI profiles are based on a many-to-one mapping from query proteins to database species target proteins. Multiplicity was defined as the number of query proteins showing a match in the target species divided by the number of different target proteins. Bidirectional AAI profiles are based on a one-to-one mapping; in which we eliminate the match of a query protein to a database protein if a better score match occurs for either sequence. AAI is the average of all matches between the query proteome and a database species, with each query protein having a weight of one. SANSparallel reports that query proteins with no matches have zero weight.
Draft for gene prediction, annotation, and functional characterization
RAST is a fully automated service that provides high-quality genome annotations for annotating complete or nearly complete bacterial and archaeal genomes across the whole phylogenetic tree. The draft genome was subjected to RAST annotation server (Rapid Annotation using Subsystem Technology) for gene prediction & annotation [60] with annotation parameters such as Genetic code = 11, E Value cut-off for selection 0 pinned Metricd CDSs = 1e–20.
Plasmid and phage screening
A tool, PlasmidFinder 2.0.8, was used to identify the plasmid sequence, if any, in the ms202 draft genome.—Sequences of phages were screened in the draft genome using the tool PHASTER (PHAge Search Tool Enhanced Release) [61].
Identification of antimicrobial resistance genotypes and genes for virulence
We compiled a list of genes associated with drug resistance and virulence identified from earlier reports in the NCBI database, Perl scripts to perform local BLASTn with ResFinder (https://cge.cbs.dtu.dk/services/ResFinder) and AMR Finder Plus with the default settings. Besides, the predicted protein sequence from the assembled genome was screened individually for antibiotic resistance genes using the Web portal—RGI 5.1.1, CARD 3.1.0. The sequences showing < 100% identity or sequence length were further examined by additional BLAST analysis.
Detection of SPI regions in the genome of S. enterica serovar Typhimurium ms202
The genes in the genomic island use strategies for genomic comparison analysis associated with specific and valuable functions in bacteria. The SPIs were detected from the assembled scaffold using Pathogenicity Island (PAI) database and bioinformatics pipeline. In brief, the sequences of all the 23 SPIs were subjected to the BLAST search of the assembled genomes versus the SPI local database [62]. Further, IslandViewer4 integrates the Islander curated database to identify unique regions in the related organism's genomes and reveals genes showing features for virulence, pathogenicity, drug resistance, and homologous factors.—The IslandPath-DIMOB, Integrated, SIGI-HMM methods were used for annotations of virulence/resistance genes.
Protein–protein interaction cluster and co-expression profiling
Kyoto Encyclopedia of Genes and Genomes (KEGG) was used for pathway mapping (www.genome.jp/kegg). DAVID's Expression Analysis Systematic Explorer (EASE) tool was used to investigate and represent functional groups. The Interaction networks and the co-expression profiling were carried out using STRING online software [63]. All associations in STRING are provided with a probabilistic confidence score representing a functional linkage among the proteins. We have selected proteins from the SPI regions whose genes are observed to be correlated in the expression.
Bacteria and growth conditions
Salmonella enterica Typhimurium MDR strain, ms202 and Salmonella enterica Typhimurium strain LT2 (hereafter termed as 'control LT2 'strain', ATCC, 700,720) were grown in M9 minimal media (20% 5X M9 salts) (G013, HiMedia), or M9 supplemented with 5 µM MnCl2. [This concentration of MnCl2 selected following study by [64]. 1% of overnight culture inoculum of bacterial strains were then sub-cultured in minimal media and grown at 37℃ and 150 rpm. The absorbance at 600 nm was monitored at every 30 min interval using UV–Vis spectrophotometer till optical density (O.D.) = 0.6 was achieved.
RNA Isolation from S. enterica Typhimurium ms202
Both LT2 and S. enterica Typhimurium ms202 strains attending log-phase of growth in M9 minimal medium (G013, HiMedia) or M9 supplemented with Mn2+ were collected. RNA was isolated following the methodology by [65]. RNA was quantified using a Colibri microvolume spectrometer (Titertek Berthold) and normalized for cDNA synthesis. cDNA synthesis was carried out using the Verso cDNA synthesis kit (Thermo Scientific, catalog no. AB-1453/A).
qRT-PCR
qRT-PCR was performed following the protocol reported by [65]. Briefly, synthesized cDNA was used as the template and DyNAmo ColorFlash SYBR green qPCR kit (Thermo Scientific, catalog no. F416-L) in a Realplex4 ep gradient Mastercycler (Eppendorf). The annealing temperature was set at 56 °C. The primers used for qRT-PCR analysis are listed in Additional file 9: Table S9. For forward and reverse sequences of few primers, references [65 and 66] were followed. The 16S rRNA gene was used as the housekeeping gene in this study. Relative gene expression was calculated by the threshold cycle (ΔΔCT) method. Genes with a log2 fold change of ≥ 1.5 were considered upregulated, and those with a log2 fold change of ≤ -1.5 were considered downregulated. Experiments were carried out in biological triplicates with statistical analyses (t-test) to validate the fold change in expression of the genes. P < 0.05 was considered statistically significant.
Availability of data and materials
Not applicable.
References
Hernandez-Reyes C, Schikora A. Salmonella, a cross-kingdom pathogen infecting humans and plants. FEMS Microbiol Lett. 2013;343(1):1–7.
Majowicz SE, Musto J, Scallan E, Angulo FJ, Kirk M, O’Brien SJ, et al. The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis. 2010;50(6):882–9.
GBDN-TSID Collaborators. The global burden of non-typhoidal salmonella invasive disease: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Infect Dis. 2019;19(12):1312–24.
Das S, Ray S, Ryan D, Sahu B, Suar M. Identification of a novel gene in ROD9 island of Salmonella Enteritidis involved in the alteration of virulence-associated genes expression. Virulence. 2018;9(1):348–62.
Saleh S, Van Puyvelde S, Staes A, Timmerman E, Barbe B, Jacobs J, et al. Salmonella Typhi, Paratyphi A, Enteritidis and Typhimurium core proteomes reveal differentially expressed proteins linked to the cell surface and pathogenicity. PLoS Negl Trop Dis. 2019;13(5): e0007416.
Grimont PA, Weill F-XJWccfr, Salmonella ro. Antigenic formulae of the Salmonella serovars. 2007;9:1-166
Singh B, Singh V, Agarwal M, Sharma G, Chandra M. Haemolysins of Salmonella, their role in pathogenesis and subtyping of Salmonella serovars. Indian J Exp Biol. 2004;42(3):303.
Mir IA, Kashyap SK, Maherchandani SJAPJoTB. Isolation, serotype diversity and antibiogram of Salmonella enterica isolated from different species of poultry in India. Asian Pac J Trop Biomed. 2015;5(7):561–7.
Rabsch W, Andrews HL, Kingsley RA, Prager R, Tschape H, Adams LG, et al. Salmonella enterica serotype Typhimurium and its host-adapted variants. Infect Immun. 2002;70(5):2249–55.
Carden S, Okoro C, Dougan G, Monack DJP. Non-typhoidal Salmonella Typhimurium ST313 isolates that cause bacteremia in humans stimulate less inflammasome activation than ST19 isolates associated with gastroenteritis. Pathogens Dis. 2015. https://doi.org/10.1093/femspd/ftu023.
Wemyss MA, Pearson JS. Host cell death responses to non-typhoidal salmonella infection. Front Immunol. 2019;10:1758.
Jacob JJ, Solaimalai D, Muthuirulandi Sethuvel DP, Rachel T, Jeslin P, Anandan S, et al. A nineteen-year report of serotype and antimicrobial susceptibility of enteric non-typhoidal Salmonella from humans in Southern India: changing facades of taxonomy and resistance trend. Gut Pathog. 2020;12:49.
Hensel M. Salmonella pathogenicity island 2. Mol Microbiol. 2000;36(5):1015–23.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90.
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaSci. 2012;1(1):2047-217X-1–18.
Trujillo S, Keys CE, Brown EW. Evaluation of the taxonomic utility of six-enzyme pulsed-field gel electrophoresis in reconstructing Salmonella subspecies phylogeny. Infect Genet Evol. 2011;11(1):92–102.
Desai PT, Porwollik S, Long F, Cheng P, Wollam A, Bhonagiri-Palsikar V, et al. Evolutionary Genomics of Salmonella enterica Subspecies. mBio. 2013;4(2):e00579.
Timme RE, Pettengill JB, Allard MW, Strain E, Barrangou R, Wehnes C, et al. Phylogenetic diversity of the enteric pathogen Salmonella enterica subsp enterica inferred from genome-wide reference-free SNP characters. Genome Biol Evol. 2013;5(11):2109–23.
Shahane V, Muley V, Kagal A, Bharadwaj R. Non-typhoid salmonellosis: emerging infection in Pune? Indian J Med Microbiol. 2007;25(2):173–4.
Hohmann EL. Nontyphoidal salmonellosis. Clin Infect Dis. 2001;32(2):263–9.
Laupland KB, Schonheyder HC, Kennedy KJ, Lyytikainen O, Valiquette L, Galbraith J, et al. Salmonella enterica bacteraemia: a multi-national population-based cohort study. BMC Infect Dis. 2010;10:95.
Hsu CH, Li C, Hoffmann M, McDermott P, Abbott J, Ayers S, et al. Comparative genomic analysis of virulence, antimicrobial resistance, and plasmid profiles of Salmonella Dublin isolated from sick cattle, retail beef, and humans in the United States. Microb Drug Resist. 2019;25(8):1238–49.
Porwollik S, Wong RM, McClelland M. Evolutionary genomics of Salmonella: gene acquisitions revealed by microarray analysis. Proc Natl Acad Sci USA. 2002;99(13):8956–61.
Chan K, Baker S, Kim CC, Detweiler CS, Dougan G, Falkow S. Genomic comparison of Salmonella enterica serovars and Salmonella bongori by use of an S. enterica serovar typhimurium DNA microarray. J Bacteriol. 2003;185(2):553–63.
Baum D, Shaw K, Hoch P, Stephenson A. Experimental and molecular approaches to plant biosystematics. Syst Bot. 1995;20(4):560–73.
Ahmer BM, Tran M, Heffron FJJ. The virulence plasmid of Salmonella typhimurium is self-transmissible. J Bacteriol. 1999;181(4):1364–8.
Rotger R, Casadesus J. The virulence plasmids of Salmonella. Int Microbiol. 1999;2(3):177–84.
Montenegro MA, Morelli G, Helmuth R. Heteroduplex analysis of Salmonella virulence plasmids and their prevalence in isolates of defined sources. Microb Pathog. 1991;11(6):391–7.
Fierer J, Krause M, Tauxe R, Guiney D. Salmonella typhimurium bacteremia: association with the virulence plasmid. J Infect Dis. 1992;166(3):639–42.
Shin H, Lee JH, Kim H, Choi Y, Heu S, Ryu S. Receptor diversity and host interaction of bacteriophages infecting Salmonella enterica serovar Typhimurium. PLoS ONE. 2012;7(8): e43392.
Yoon H, Ansong C, Adkins JN, Heffron F. Discovery of Salmonella virulence factors translocated via outer membrane vesicles to murine macrophages. Infect Immun. 2011;79(6):2182–92.
Giner-Lamia J, Vinuesa P, Betancor L, Silva C, Bisio J, Soleto L, et al. Genome analysis of Salmonella enterica subsp. diarizonae isolates from invasive human infections reveals enrichment of virulence-related functions in lineage ST1256. BMC Genomics. 2019;20(1):99.
Li XZ, Nikaido H. Efflux-mediated drug resistance in bacteria: an update. Drugs. 2009;69(12):1555–623.
Buckley AM, Webber MA, Cooles S, Randall LP, La Ragione RM, Woodward MJ, et al. The AcrAB-TolC efflux system of Salmonella enterica serovar Typhimurium plays a role in pathogenesis. Cell Microbiol. 2006;8(5):847–56.
Murakami S. Multidrug efflux transporter, AcrB–the pumping mechanism. Curr Opin Struct Biol. 2008;18(4):459–65.
Quinn T, O’Mahony R, Baird AW, Drudy D, Whyte P, Fanning S. Multi-drug resistance in Salmonella enterica: efflux mechanisms and their relationships with the development of chromosomal resistance gene clusters. Curr Drug Targets. 2006;7(7):849–60.
Hardt WD, Urlaub H, Galan JE. A substrate of the centisome 63 type III protein secretion system of Salmonella typhimurium is encoded by a cryptic bacteriophage. Proc Natl Acad Sci USA. 1998;95(5):2574–9.
Figueroa-Bossi N, Uzzau S, Maloriol D, Bossi L. Variable assortment of prophages provides a transferable repertoire of pathogenic determinants in Salmonella. Mol Microbiol. 2001;39(2):260–71.
Switt AI, Sulakvelidze A, Wiedmann M, Kropinski AM, Wishart DS, Poppe C, et al. Salmonella phages and prophages: genomics, taxonomy, and applied aspects. Methods Mol Biol. 2015;1225:237–87.
Worley J, Meng J, Allard MW, Brown EW, Timme RE. Salmonella enterica phylogeny based on whole-genome sequencing reveals two new clades and novel patterns of horizontally acquired genetic elements. mBio. 2018;9(6):e02303.
Hensel M. Evolution of pathogenicity islands of Salmonella enterica. Int J Med Microbiol. 2004;294(2–3):95–102.
Suez J, Porwollik S, Dagan A, Marzel A, Schorr YI, Desai PT, et al. Virulence gene profiling and pathogenicity characterization of non-typhoidal Salmonella accounted for invasive disease in humans. PLoS ONE. 2013;8(3): e58449.
Blondel CJ, Jimenez JC, Contreras I, Santiviago CA. Comparative genomic analysis uncovers 3 novel loci encoding type six secretion systems differentially distributed in Salmonella serotypes. BMC Genomics. 2009;10:354.
Raffatellu M, Wilson RP, Chessa D, Andrews-Polymenis H, Tran QT, Lawhon S, et al. SipA, SopA, SopB, SopD, and SopE2 contribute to Salmonella enterica serotype typhimurium invasion of epithelial cells. Infect Immun. 2005;73(1):146–54.
Boyen F, Pasmans F, Van Immerseel F, Morgan E, Adriaensen C, Hernalsteens J-P, et al. Salmonella Typhimurium SPI-1 genes promote intestinal but not tonsillar colonization in pigs. Microb Infect. 2006;8(14–15):2899–907.
Das C, Dutta A, Rajasingh H, Mande SSJG. Understanding the sequential activation of Type III and Type VI secretion systems in Salmonella typhimurium using Boolean modeling. Mol Microbiol. 2013;5(1):1–12.
Tan Z, Chekabab SM, Yu H, Yin X, Diarra MS, Yang C, et al. Growth and virulence of salmonella typhimurium mutants deficient in iron uptake. ACS Omega. 2019;4(8):13218–30.
Lunelli M, Hurwitz R, Lambers J, Kolbe M. Crystal structure of PrgI-SipD: insight into a secretion competent state of the type three secretion system needle tip and its interaction with host ligands. PLoS Pathog. 2011;7(8): e1002163.
Lou L, Zhang P, Piao R, Wang Y. Salmonella pathogenicity island 1 (SPI-1) and its complex regulatory network. Front Cell Infect Microbiol. 2019;9:270.
Ellermeier CD, Ellermeier JR, Slauch JMJM. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol Microbiol. 2005;57(3):691–705.
Mather AE, Phuong TLT, Gao Y, Clare S, Mukhopadhyay S, Goulding DA, et al. New variant of multidrug-resistant Salmonella enterica serovar Typhimurium associated with invasive disease in immunocompromised patients in Vietnam. mBio. 2018;9(5):e01056-e1118.
Carroll LM, Huisman JS, Wiedmann MJS. Twentieth-century emergence of antimicrobial resistant human-and bovine-associated Salmonella enterica serotype Typhimurium lineages in New York State. Sci Rep. 2020;10(1):1–15.
Boyer E, Bergevin I, Malo D, Gros P, Cellier MF. Acquisition of Mn(II) in addition to Fe(II) is required for full virulence of Salmonella enterica serovar Typhimurium. Infect Immun. 2002;70(11):6032–42.
Ikeda JS, Janakiraman A, Kehres DG, Maguire ME, Slauch JM. Transcriptional regulation of sitABCD of Salmonella enterica serovar Typhimurium by MntR and Fur. J Bacteriol. 2005;187(3):912–22.
Waldron KJ, Robinson NJJNRM. How do bacterial cells ensure that metalloproteins get the correct metal. Nat Rev Microbiol. 2009;7(1):25–35.
Schönenbrücher V, Mallinson ET, Bülte MJIjofm. A comparison of standard cultural methods for the detection of foodborne Salmonella species including three new chromogenic plating media. Int J Food Microbiol. 2008;123(1–2):61–6.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DWJN. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–8.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9(1):1–15.
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44(W1):W16-21.
Yoon SH, Park YK, Kim JF. PAIDB v2.0: exploration and analysis of pathogenicity and resistance islands. Nucleic Acids Res. 2015;43:D624-30.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Martin JE, Waters LS, Storz G, Imlay JAJP. The Escherichia coli small protein MntS and exporter MntP optimize the intracellular concentration of manganese. PLoS Genet. 2015;11(3): e1004977.
Arunima A, Swain SK, Patra SD, Das S, Mohakud NK, Misra N, et al. Role of OB-fold protein ydei in stress response and virulence of Salmonella enterica Serovar Enteritidis. J Bacteriol. 2020;203(1): e00237.
Balasubramanian R, Im J, Lee JS, Jeon HJ, Mogeni OD, Kim JH, et al. The global burden and epidemiology of invasive non-typhoidal Salmonella infections. Hum Vaccin Immunother. 2019;15(6):1421–6.
Acknowledgements
The authors acknowledge Department of Biotechnology (Centre of Excellence and Innovation in Biotechnology), Government of India, for providing scientific assistance (Project Grant no. BT/MED/30/SP19662/2018). Further, the authors also acknowledge SCB Medical College, Odisha, India; Kalinga Institute of Medical Sciences, Bhubaneswar, India, for examining faecal samples of diarrheal patients to identify multidrug-resistant S. Typhimurium. Besides, the School of Biotechnology, KIIT University and Technology Business Incubator (TBI), Bhubaneswar, India, are highly acknowledged for providing laboratory facilities to carry out experiments.
Funding
The preparation of this manuscript is not supported by any funding from central fund of the institution or project.
Author information
Authors and Affiliations
Contributions
MS conceived the idea and designed the study along with data curation, project administration, Review & Editing. NKM, RKP, BRS performed the sample collection, conducted the primary data arrangements. SDP carried out the formal analysis, in vitro experimental study. NM, GSK involved in Resources, Investigation, Methodology, Data curtain. MG performed Methodology, Software, Validation, Visualization, and writing. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Ethical clearance for using fecal samples of patients with gastroenteritis were approved by Institutional Review Boards of the hospitals taken under the study.
Consent for publication
All authors in this manuscript have thoroughly read and agreed to the content and have given their consent for publication.
Competing interests
Authors have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Socio-Demographic details of patients with acute gastroenteritis.(n=221) admitted in SCB Medical college, Cuttack, Odisha, India and KIMS, Bhubaneswar, Odisha, India.
Additional file 2: Table S2.
S. enterica Typhimurium ms202 showing resistance to four antibiotic groups. Antibiotic groups are shown yellow highlighted as mentioned in VTEK AST Card (AST N280, Biomereux)
Additional file 3:
Table S3. Multi locus Sequence Typing of the genes in S. enterica Typhimurium ms202
Additional file 4: Table S4.
Details of Phages identified in S. enterica Typhimurium ms202. A total of 9 different regions and each region containing different numbers of CDS are shown.
Additional file 5:
Table S5. The interaction networks and the co-expression details of the SPI-2 cluster proteins in S. enterica Typhimurium ms202
Additional file 6: Table S6.
The interaction networks and the co-expression details of the SPI-1 cluster proteins in S. enterica Typhimurium ms202
Additional file 7:
Table S7. The whole-genome annotation for virulent genes for S. enterica Typhimurium ms202. Genome – Salmonella enterica subsp. enterica serovar Typhimurium ms202; Genome ID - 90371.4806, Accession - 90371.4806.con.0001, Annotation – PATRIC, Feature Type – CDS.
Additional file 8:
Table S8. The whole-genome annotation for drug target specific genes for S. enterica Typhimurium ms202. Genome – Salmonella enterica subsp. enterica serovar Typhimurium MDR; Genome ID - 90371.4806, Accession - 90371.4806.con.0001, Annotation – PATRIC, Feature Type – CDS
Additional file 9: Table S9.
List of primers used for qRT-PCR
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mohakud, N.K., Panda, R.K., Patra, S.D. et al. Genome analysis and virulence gene expression profile of a multi drug resistant Salmonella enterica serovar Typhimurium ms202. Gut Pathog 14, 28 (2022). https://doi.org/10.1186/s13099-022-00498-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13099-022-00498-w