
Abstract
Background
Synovial tissue fibrosis is common in advanced OA with features including the presence of stress fiber-positive myofibroblasts and deposition of cross-linked collagen type-I. Proteoglycan-4 (PRG4) is a mucinous glycoprotein secreted by synovial fibroblasts and is a major component of synovial fluid. PRG4 is a ligand of the CD44 receptor. Our objective was to examine the role of PRG4-CD44 interaction in regulating synovial tissue fibrosis in vitro and in vivo.
Methods
OA synoviocytes were treated with TGF-β ± PRG4 for 24 h and α-SMA content was determined using immunofluorescence. Rhodamine-labeled rhPRG4 was incubated with OA synoviocytes ± anti-CD44 or isotype control antibodies and cellular uptake of rhPRG4 was determined following a 30-min incubation and α-SMA expression following a 24-h incubation. HEK-TGF-β cells were treated with TGF-β ± rhPRG4 and Smad3 phosphorylation was determined using immunofluorescence and TGF-β/Smad pathway activation was determined colorimetrically. We probed for stress fibers and focal adhesions (FAs) in TGF-β-treated murine fibroblasts and fibroblast migration was quantified ± rhPRG4. Synovial expression of fibrotic markers: α-SMA, collagen type-I, and PLOD2 in Prg4 gene-trap (Prg4GT) and recombined Prg4GTR animals were studied at 2 and 9 months of age. Synovial expression of α-SMA and PLOD2 was determined in 2-month-old Prg4GT/GT&Cd44−/− and Prg4GTR/GTR&Cd44−/− animals.
Results
PRG4 reduced α-SMA content in OA synoviocytes (p < 0.001). rhPRG4 was internalized by OA synoviocytes via CD44 and CD44 neutralization attenuated rhPRG4’s antifibrotic effect (p < 0.05). rhPRG4 reduced pSmad3 signal in HEK-TGF-β cells (p < 0.001) and TGF-β/Smad pathway activation (p < 0.001). rhPRG4 reduced the number of stress fiber-positive myofibroblasts, FAs mean size, and cell migration in TGF-β-treated NIH3T3 fibroblasts (p < 0.05). rhPRG4 inhibited fibroblast migration in a macrophage and fibroblast co-culture model without altering active or total TGF-β levels. Synovial tissues of 9-month-old Prg4GT/GT animals had higher α-SMA, collagen type-I, and PLOD2 (p < 0.001) content and Prg4 re-expression reduced these markers (p < 0.01). Prg4 re-expression also reduced α-SMA and PLOD2 staining in CD44-deficient mice.
Conclusion
PRG4 is an endogenous antifibrotic modulator in the joint and its effect on myofibroblast formation is partially mediated by CD44, but CD44 is not required to demonstrate an antifibrotic effect in vivo.
Similar content being viewed by others


Background
Osteoarthritis (OA) is the most prevalent type of musculoskeletal disorder in patients 60 years or older [1,2,3]. OA is a leading cause of pain and disability and patients with symptomatic knee OA are at an increased risk of all-cause mortality [4,5,6]. The etiology of OA is complex where joint trauma, obesity, and genetics, among other risk factors, contribute to disease development [7,8,9,10]. OA affects the whole joint with manifestations that include degeneration of articular cartilage and the meniscus, abnormal bone remodeling, and synovial inflammation [11,12,13]. Macroscopic evidence of synovial inflammation or synovitis is a common finding in up to 74% of patients with knee OA of different grades and 95% of patients with moderate to severe OA [14,15,16]. OA synovitis is likely caused by an innate immune response, mediated by the toll-like receptors 2 and 4 (TLR2 and TLR4) in the synovium, and resultant expression of inflammatory cytokines, chemokines, and matrix-degrading enzymes [17,18,19,20,21]. The extent of synovitis is a strong predictor of OA progression across multiple studies, and therefore treating synovial inflammation is a potentially important target for therapeutic intervention, especially during the early stage of OA [14, 22,23,24,25,26,27].
The normal synovium is composed of a cellular intimal layer and a subintimal connective tissue layer [14, 28]. The intimal layer is usually 1–4 cells in thickness with two types of cells: type A macrophages and type B synovial fibroblasts, with macrophages making up a smaller percentage of cells in the intimal layer of the healthy synovium [28]. Synovial fibroblasts synthesize hyaluronic acid (HA), a glycosaminoglycan, and proteoglycan-4 (PRG4), a mucinous glycoprotein, which are major components of synovial fluid (SF) with important roles in joint lubrication [29, 30]. In synovitis, synovial membranes become hyperplastic with increased accumulation of macrophages and other inflammatory cells, along with subintimal fibrosis and angiogenesis [13, 14, 31]. Synovial fibrosis can be considered a maladaptive healing response to chronic synovitis which is characterized by the upregulation of collagen type-I expression, fibroblast transition to a myofibroblast-like phenotype through de novo expression of alpha smooth muscle actin (α-SMA) and enhanced migratory behavior, and upregulation of procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 (PLOD2) leading to accumulation of cross-linked collagen in the synovium [32,33,34]. These pathological changes are induced in the OA synovium by transforming growth factor beta (TGF-β1) and its associated signaling pathways [35, 36]. Synovial fibrosis also contributes to joint pain and stiffness observed in advanced OA [31, 37].
In a recent study, we demonstrated that increasing intracellular adenosine 3′, 5′-cyclic monophosphate (cAMP), using forskolin, reduced collagen type-I, PLOD2, and α-SMA expression in TGF-β-stimulated osteoarthritic fibroblast-like synoviocytes (OA FLS) [38]. Associated with this antifibrotic effect was an increase in HA and PRG4 production by OA FLS, whereby native synovial human PRG4 reduced procollagen type-I level in OA FLS [38]. Furthermore, synoviocytes from Prg4 null mice displayed more extensive collagen type-I staining compared to synoviocytes from Prg4 competent mice [38]. In a separate study, we have also shown that PRG4 is a ligand for the HA receptor, CD44 [39]. We have also reported that PRG4-CD44 interaction inhibited interleukin-1 beta (IL-1β) induced OA FLS proliferation and expression of matrix-degrading enzymes [40], via the inhibition of nuclear factor kappa b (NF-κB) nuclear translocation mediated by blocking inhibitory kappa b (IκB) degradation [40].
It remains unknown whether PRG4 has a role in regulating fibroblast to myofibroblast transition and associated cell migration in the fibrotic synovium. Furthermore, it is yet to be determined whether PRG4 regulates synovial fibrosis in vivo and whether this role is due to its interaction with the CD44 receptor. Using recombinant human proteoglycan-4 (rhPRG4), we aimed to study the role of PRG4 in regulating fibroblast to myofibroblast transition and modulating fibroblast migration in response to exogenous TGF-β or co-incubation with lipopolysaccharide (LPS) stimulated macrophages. We also studied the role of PRG4-CD44 interaction, and more specifically CD44-mediated cellular uptake of PRG4, in the regulation of myofibroblast formation in vitro and progression of synovial fibrosis in vivo. We hypothesized that PRG4 regulates fibroblast to myofibroblast transition and prevents synovial fibrosis in a CD44-dependent manner.
Methods
Impact of PRG4 and HA treatments on ACTA2 expression, α-SMA immunostaining, and stress fiber formation in osteoarthritic fibroblast-like synoviocytes (OA FLS) and the role of CD44 in mediating the effect of rhPRG4 in TGF-β-stimulated OA FLS
OA FLS (Cell Applications, USA) were isolated from synovial tissues from de-identified patients undergoing knee replacement surgery (n = 4; 2 males and 2 females; 62–69 years old). Cells were received in their second passage and were cultured as previously described [20]. OA FLS were used between the 3rd and 6th passages to avoid alterations in gene expression pattern and cell proliferation rate [41]. OA FLS (300,000 cells/well) in serum-free DMEM media were treated with recombinant human TGF-β1 (R&D Systems, USA) (1 ng/mL) ± native human synovial PRG4 (apparent mol mass 280 kDa as a monomer; 100 μg/mL) [42] or high-molecular weight hyaluronic acid (HA; ~ 1200 kDa; R&D Systems; 100 μg/mL) for 24 h. RNA isolation, cDNA synthesis, and quantitative PCR were performed as previously described [38]. The cycle threshold (Ct) value of α-SMA (ACTA2) was normalized to the Ct value of GAPDH in the same sample, and the relative expression was calculated using the 2−ΔΔCt method [43]. All primers and probes utilized in our study are commercially available (Thermo Fisher Scientific, USA).
Assessment of α-SMA content in OA FLS was conducted using immunofluorescence and determination of corrected total cell fluorescence (CTCF) using a Nikon E600 fluorescence microscope. OA FLS (200,000 cells/well) were cultured on collagen type-I-coated 22 mm glass coverslips for 48 h in DMEM medium + 10% fetal bovine serum (FBS). Cells were treated with TGF-β1 (1 ng/mL) ± PRG4 or HA (100 μg/mL for both treatments) for 48 h in serum-free DMEM medium. Subsequently, cells were fixed in 10% neutral buffered formalin for 15 min and washed twice with phosphate-buffered saline (PBS). Cells were permeabilized using 0.01% Triton X-100 in PBS and blocked using 2% bovine serum albumin (BSA; Sigma-Aldrich, USA) in PBS for 2 h at room temperature. Probing for α-SMA was performed using FITC-conjugated anti-α-SMA antibody (1:100 dilution; Abcam, USA) and counterstained using Alexa Fluor 594-conjugated anti-α-tubulin antibody (1100 dilution: Abcam) overnight at 4 °C. Following washing with PBS, cells were mounted on DAPI mounting shield (Abcam) and CTCF was quantified using 4 different fields per slide and mean CTCF was calculated. The presence of stress fibers in OA FLS was also evaluated.
Recombinant human PRG4 (rhPRG4; apparent mol mass 240 kDa) is an endotoxin-free full-length product produced by CHO-M cells (Lubris, Framingham, USA) [44]. Rhodamine labeling of rhPRG4 was performed using the Pierce NHS-Rhodamine Antibody Labeling Kit (Thermo Fisher Scientific). OA FLS (200,000 cells/well) were cultured on collagen type-I-coated cover slips and incubated with rhodamine-rhPRG4 (25 μg/mL) ± anti-CD44 or isotype control (IC) antibodies (2 μg/mL for both antibodies; Abcam) for 30 min. Cells were pre-incubated with the antibodies for 1 h prior to rhPRG4 addition. Subsequently, cells were washed twice with PBS and mounted on DAPI mounting media and CTCF was quantified as described above. In another set of experiments, OA FLS (300,000 cells/well) were treated with TGF-β1 (1 ng/mL) ± rhPRG4 (100 μg/mL) ± anti-CD44 or IC antibodies (2 μg/mL for both antibodies) for 24 h followed by determination of ACTA2 expression as described above.
CD44-dependent uptake of rhPRG4, Smad3 phosphorylation, and regulation of TGF-β/Smad pathway activation in TGF-β-stimulated HEK Blue-TGF-β cells
HEK Blue-TGF-β is an engineered reporter cell line produced by transfecting human embryonic kidney (HEK) cells with TGF-β receptor 1 (TGF-β R1), Smad3, and Smad4 genes (Invivogen, USA). TGF-β1-treatment results in phosphorylation of Smad3 (pSmad3) and Smad4 (pSmad4), translocation to the nucleus and expression of secreted alkaline phosphatase (SEAP). The activity of SEAP can be detected colorimetrically in media supernatants using a specific substrate (Quanti Blue) at 620–655 nm (Invivogen). HEK-TGF-β cells (500,000) were plated onto sterile chamber slides and incubated with rhodamine-rhPRG4 (25 μg/mL) for 1 h ± anti CD44 or IC antibodies (2 μg/mL for both antibodies). Cells were pre-incubated with the antibodies for 1 h prior to rhPRG4 addition. Subsequently, cells were washed with PBS and intracellular CTCF was determined as described above. In another set of experiments, rhodamine-labeled rhPRG4 (25 μg/mL) was incubated with HEK-TGF-β cells and CD44 was probed using FITC-conjugated anti-CD44 antibody (Abcam) and CD44 (green) and rhPRG4 (red) co-localization was examined using a confocal microscope.
Immunostaining of pSmad3 was performed using an anti-Smad3 antibody that detects phosphorylated serine residues (1:1000 dilution overnight at 4 °C; Abcam). HEK-TGF-β cells were stimulated with TGF-β1 ± rhPRG4 (150 μg/mL) for 1 h. Subsequently, cells were fixed, washed with PBS, and permeabilized as described above. Following probing with anti-pSmad3 antibody, cells were washed with PBS and incubated with goat anti-rabbit IgG (Alexa Fluor 488) (1:1000 dilution for 1 h at room temperature; Abcam). Cells were subsequently washed with PBS and the green immunofluorescence was quantified across all experimental groups. To investigate the impact of rhPRG4 on TGF-β/Smad pathway activation, HEK-TGF-β cells (100,000 cells/well) were cultured in sterile 96-well plates ± TGF-β1 (1 ng/mL) ± rhPRG4 (50, 100, or 150 μg/mL) for 24 h in Quanti-Blue media and the 655 nm absorbance intensity was determined. Data are presented as fold absorbance intensities across experimental groups normalized to untreated controls.
TGF-β induction of stress fibers, focal adhesions (FAs) and cell migration in murine NIH3T3 fibroblasts, comparison of stress fibers and FAs in Prg4+/+ and Prg−/− synovial fibroblasts and role of rhPRG4 in regulating fibroblast migration
Murine fibroblasts (NIH3T3; ATCC; 20,000 cells/well) were cultured on collagen type-I-coated 22-mm glass coverslips for 24 h in DMEM supplemented with 10% bovine calf serum (BCS). NIH3T3 cells were starved in DMEM containing 1% BCS for 24 h. Murine fibroblasts were treated with murine TGF-β1 (1 ng/mL) ± rhPRG4 (200 μg/mL) for 24 h in serum-free DMEM. Subsequently, fibroblasts were fixed using neutral buffered formalin, washed with PBS, permeabilized using 0.1% triton X-100, and probed using an anti-α-SMA (marker of stress fibers; 1:1,000) or anti-vinculin (marker of FA complex; 1:1,000) overnight at 4 °C. After washing with PBS, cells were incubated with goat anti-rabbit IgG (Alexa Fluor® 488) at 1:1000 dilution for 1 h at 4 °C. Rhodamine-labeled phalloidin (cytoskeleton label; 1:1000) was added with the secondary antibody. All antibodies were obtained from Abcam. Cells were washed and mounted with DAPI medium for 2 h and viewed under a confocal microscope. A blinded investigator determined the number of stress fiber-positive fibroblasts. At least 100 cells over at least 5 different fields were evaluated, and the percentage of stress fiber-positive fibroblasts was calculated. Separately, mean fibroblast cell spread area was determined using the NIS-Elements imaging software (Nikon). The number and mean size of FAs per cell were determined using ImageJ software as previously described [45]. Fibroblast migration was determined using a scratch assay. A 1000-μL pipette tip was used to perform a uniform scratch in the confluent NIH3T3 fibroblast monolayer. TGF-β (1 ng/mL) stimulation was performed for 48 h ± rhPRG4 (200 μg/mL). Subsequently, cells were washed and stained (Cell Biolabs) and imaged using an all-in-one fluorescence microscope (Keyence). A region of interest was defined and scratch widths were determined across multiple locations. Cell migration was expressed as percent wound closure across different experimental groups as previously described [46].
The phenotype of Prg4−/− mice is characterized by synovial membrane hyperplasia, chondrocyte apoptosis, and cartilage surface fibrillation [47]. The Prg4−/− mouse colony is maintained by Dr. Gregory Jay at Rhode Island Hospital (RIH) and is commercially available (stock no. 025737; JAX, USA). The IACUC committee at RIH approved all animal experiments and all experiments were performed according to all applicable guidelines and regulations. Synoviocyte isolation from Prg4−/− and Prg4+/+ synovial tissues was performed as previously described [38, 39]. Prg4−/− and Prg4+/+ synoviocytes were plated onto sterile chamber slides (Thermo Fisher Scientific) and allowed to adhere for 48 h. Subsequently, cells were stained for α-SMA and vinculin as described above. The mean size of FAs per cell in Prg4−/− and Prg4+/+ synoviocytes was determined as described above. Prg4−/− and Prg4+/+ synoviocytes were seeded in 6-well plates and a scratch was performed in the confluent cell monolayer. Basal synovial fibroblast migration of both genotypes was quantified over 48 h ± rhPRG4 (200 μg/mL), anti-CD44 (2 μg/mL), or IC (2 μg/mL) antibodies and expressed as percent wound closure [46].
Generation of active TGF-β in lipopolysaccharide-stimulated murine macrophage J774A and NIH3T3 fibroblast co-culture and impact of rhPRG4 treatment on fibroblast migration
Murine J774A macrophages (ATCC) were cultured in DMEM medium + 10% FBS. Macrophages (300,000 cells in DMEM medium + 10% FBS) were stimulated with lipopolysaccharide (LPS; Invivogen) (5 μg/mL) for 24 h. Subsequently, murine macrophages were washed five times with DMEM medium and transferred to the top chamber of a transwell co-culture system (0.4-μm pore size; Sigma-Aldrich). Murine NIH3T3 cells were seeded in the lower chamber of the transwell system and a scratch was performed as described above. NIH3T3 migration was determined following a 48-h incubation of fibroblasts with macrophages in the co-culture system as described above ± rhPRG4 (200 μg/mL). Active and total TGF-β media levels were determined using an ELISA (R&D Systems).
Age-dependent expression of fibrotic markers in synovial tissues from Prg4GT/GT animals and the role of PRG4/CD44 interaction in modulating synovial fibrosis in vivo
The Prg4 gene-trap (Prg4GT) mouse colony is maintained by Dr. Gregory Jay at RIH and is commercially available (stock no. 025740; JAX) [48]. Prg4GT animal is a genetically engineered PRG4-deficient mouse where the Prg4 expression can be restored via CRE-mediated recombination [48]. The Prg4GT/GT mouse recapitulates the hallmark findings in Prg4−/− mouse, namely synovial tissue hyperplasia and cartilage surface fibrillations and recombination in 3-week-old animals improved but did not completely normalize joint pathological findings [48]. In our studies, recombination (Prg4GTR/GTR) occurred in 3-week-old animals via intraperitoneal injection of tamoxifen (0.1 mg/g in 100 μL corn oil vehicle) daily for 10 days. We compared gene expression and immunostaining of fibrotic markers: α-SMA, COL1A1 (collagen type-I), and PLOD2 in 2-month-old Prg4GT/GT, 2-month-old Prg4GTR/GTR, 9-month-old Prg4GT/GT, and 9-month-old Prg4GTR/GTR animals. ACTA2, COL1A1, and PLOD2 expression levels in murine synovial tissues were performed as previously described [38]. Tissues from each three consecutive mice were pooled and underwent RNA isolation, generating five pooled samples in each experimental group. Separately, animal joints (n = 5 in each group) underwent decalcification and paraffin-embedded sectioning as previously described [48]. Immunostaining of synovial tissues was performed using primary antibodies against α-SMA, COL1A1, or PLOD2 (1:1000 dilutions performed overnight at 4 °C) (All antibodies were purchased from Abcam). Subsequently, sections were washed and incubated with goat anti-rabbit IgG (Alexa Fluor 488) (1:1000 dilution) for 1 h and fluorescence intensities (expressed as lumens per square millimeter) were quantified using a fluorescence microscope.
To appreciate the significance of PRG4 and CD44 interaction in the context of fibrotic markers’ expression in the synovium, Prg4GT/GT animals were crossed with Cd44−/− mice (stock no. 005085; JAX) [49] to generate Cd44−/−&Prg4GT/GT animals. Recombination occurred in 3-week-old animals to generate Cd44−/−&Prg4GTR/GTR animals as described in the last paragraph. Histological analyses of joints harvested from 2-month-old animals (at least 4 animals per group) were performed as described above, using Cd44+/+&Prg4GT/GT animals from the same litters as controls. We probed synovial tissues for the following fibrotic markers: α-SMA and PLOD2 using specific primary antibodies (1:1000 dilutions) followed by goat anti-rabbit IgG (Alexa Fluor 488) and quantitation of fluorescence intensities (expressed as lumens per square millimeter) as described above.
Statistical analyses
Target gene expression was statistically evaluated by comparing ΔCt (Ct value of target gene − Ct value of GAPDH in the same sample) values of different experimental groups. Statistical significance comparing two groups or multiple groups with parametric data was assessed by Student’s t test or ANOVA followed by post hoc multiple comparisons (Tukey’s post hoc test). Statistical significance comparing two groups or multiple groups with nonparametric data was assessed by rank sum test or ANOVA on the ranks. A p value of < 0.05 was considered statistically significant. Data are presented as scatter plots with mean and standard deviations highlighted.
Results
PRG4 reduced ACTA2 expression and stress fiber formation in osteoarthritic fibroblast-like synoviocytes (OA FLS) and CD44 was involved in the uptake of rhPRG4 by OA FLS, whereas rhPRG4-CD44 interaction affected ACTA2 expression in response to TGF-β
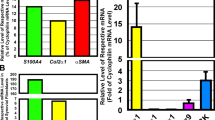
PRG4 and HA treatments demonstrated equivalent efficacies in reducing ACTA2 expression in OA FLS (Fig. 1a). Both treatments reduced ACTA2 expression compared to the positive control TGF-β1 (p < 0.001 for TGF-β1 + PRG4 or TGF-β1 + HA against TGF-β1 alone). In addition to reducing ACTA2 expression, PRG4 reduced α-SMA content in OA FLS (p < 0.001 versus TGF-β1 alone; Fig. 1c). A similar effect was also observed for HA treatment (p < 0.001 versus TGF-β1 alone; Fig. 1c). The effect of PRG4 was biologically significant with approximately 52% reduction in mean α-SMA immunofluorescence compared to TGF-β1 alone. In addition to reducing α-SMA in OA FLS, PRG4, and HA treatments prevented the formation of stress fibers in OA FLS in response to TGF-β1 stimulation (Fig. 1b). rhPRG4 was rapidly internalized by OA FLS and the mechanism of its internalization was related to its interaction with CD44. This was illustrated by a reduction in OA FLS CTCF with an anti-CD44 treatment (Fig. 1d, e). Neutralization of CD44 receptor did not completely block rhPRG4 uptake by OA FLS (Fig. 1e). Alternatively, treatment with an IC antibody did not alter rhPRG4’s uptake by OA FLS (p > 0.05; Fig. 1e). Inhibition of ACTA2 expression by rhPRG4 was attenuated by pre-incubation with an anti-CD44 antibody (p < 0.05 for TGF-β1 + rhPRG4 + anti-CD44 versus TGF-β1 + rhPRG4; Fig. 1f). In contrast, IC antibody treatment did not alter the magnitude of reduction in ACTA2 expression observed with rhPRG4 (Fig. 1f).

Impact of proteoglycan-4 (PRG4) and high-molecular weight hyaluronic acid (HA) treatments on TGF-β1-induced alpha smooth muscle actin (α-SMA) expression (ACTA2), production and stress fiber formation in fibroblast-like synoviocytes isolated from patients with OA (OA FLS) undergoing knee replacement surgery and CD44-dependent uptake of rhodamine-labeled recombinant human PRG4 (rhPRG4) into OA FLS and associated regulation of ACTA2 expression. OA FLS were treated with TGF-β1 (1 ng/mL) ± rhPRG4 (~ 240 kDa) or HA (~ 1200 kDa) (100 μg/mL for both treatments) for 24 h followed by RNA isolation and qPCR using GAPDH as an internal reference gene. OA FLS were stained with anti-α-SMA (green) and counterstained for α-tubulin (red) and DAPI (blue). Corrected total cell fluorescence (CTCF) of α-SMA was determined following a 48-h treatment, and normalized to controls. Rhodamine-labeled rhPRG4 was incubated with OA FLS ± anti-CD44 or isotype control (IC) antibodies for 30 min, and CTFC was quantified. OA FLS were treated with TGF-β1 ± rhPRG4 ± anti-CD44 or IC for 24 h and ACTA2 expression was determined. Data are presented as a scatterplot with means and standard deviations highlighted utilizing OA FLS from 3 to 4 different patients. *p < 0.001; ***p < 0.05. Scale = 50 μm. a PRG4 and HA reduced ACTA2 expression in OA FLS. b Representative images showing TGF-β1-induced stress fiber formation in OA FLS (white arrows) and PRG4 or HA treatments prevented their formation. c PRG4 and HA reduced α-SMA CTCF in OA FLS. d Representative images showing rhodamine-rhPRG4 intracellular localization (white arrows), following incubation with OA FLS. e Co-incubation of rhPRG4 and anti-CD44 reduced cellular uptake of rhPRG4. f Anti-CD44 antibody co-treatment reduced rhPRG4’s antifibrotic effect in OA FLS
CD44 receptor facilitated rhPRG4 uptake by HEK-TGFβ cells and rhPRG4 inhibited TGF-β1/Smad pathway activation
Representative images demonstrate intracellular red fluorescence following the incubation of rhodamine-labeled rhPRG4 with HEK-TGFβ cells (Fig. 2a). Antibody-mediated CD44 receptor neutralization reduced the intensity of red fluorescence, indicative of reduced rhPRG4 uptake into HEK-TGFβ cells (p < 0.001 vs. rhPRG4 alone; Fig. 2a, b). This effect was specific to CD44 receptor, as a non-specific IC antibody pre-incubation did not alter the extent of rhPRG4 uptake into HEK-TGFβ cells (p > 0.05; Fig. 2a, b). Further evidence of CD44 involvement in rhPRG4 uptake by HEK-TGFβ cells is provided by the intracellular co-localization of rhPRG4 (red) and CD44 (green) as shown by arrows (yellow color) (Fig. 2a). TGF-β1 stimulation resulted in Smad3 phosphorylation as shown in representative images (Fig. 2c). Intense green fluorescence in TGF-β1-stimulated HEK-TGF-β cells indicated the formation of pSmad3. rhPRG4 treatment reduced mean pSmad3 staining intensity subsequent to TGF-β1 stimulation (p < 0.001; Fig. 2d). In addition, rhPRG4 treatment dose-dependently reduced TGF-β1/Smad pathway activation as shown by a reduction in the formation of SEAP colored product (p < 0.05 for 100 and 150 μg/mL concentrations versus TGF-β1 alone; Fig. 2e).

CD44-dependent interaction of recombinant human proteoglycan-4 (rhPRG4) with HEK Blue-TGF-β cells and associated modulation of phosphorylated Smad3 (pSmad3) and TGF-β/Smad signaling pathway. HEK Blue-TGF-β is an engineered cell line produced by transfecting human embryonic kidney (HEK) cells with TGF-β receptor 1 (TGF-β R1), Smad3, and Smad4 genes. Activation of TGF-β R1 results in expression of secreted alkaline phosphatase (SEAP) whose activity can be determined colorimetrically. Rhodamine-labeled rhPRG4 was incubated with HEK Blue-TGF-β cells ± anti-CD44 or isotype control (IC) antibodies and corrected total cell fluorescence (CTCF) was determined. pSmad3 immunocytostaining was performed using an antibody against pSmad3 and pSmad3 fluorescence intensity was determined. Activity of TGF-β/Smad pathway was determined colorimetrically. *p < 0.001; **p < 0.01; n.s., non-significant. Scale in a = 40 μm; scale in c = 50 μm. a Representative images showing rhPRG4 internalization (white arrows) by HEK-TGF-β cells and co-localization with CD44 receptor (white arrows in merged image). b rhPRG4 uptake by HEK-TGF-β cells was reduced by CD44 receptor neutralization. c Representative images showing pSmad3 staining in HEK-TGF-β cells following TGF-β stimulation (white arrows) ± rhPRG4 treatment. d pSmad3 immunocytostaining was reduced following rhPRG4 treatment. e rhPRG4 treatment dose-dependently reduced TGF-β/Smad signaling pathway in TGF-β-stimulated HEK-TGF-β cells
TGF-β induced the formation of stress fibers and FAs in murine fibroblast NIH3T3 cells resulting in enhanced cell migration, and these effects were reduced with rhPRG4 treatment
Representative images highlight that murine TGF-β induced the formation of stress fibers and vinculin expression and thus FAs in NIH3T3 cells (Fig. 3a). TGF-β increased the percentage of stress fiber-positive NIH3T3 cells by ~ 5-fold (p < 0.001 against control cells; Fig. 3b) and increased mean cell spread area by ~ 2-fold (p < 0.001 against control cells; Fig. 3c). rhPRG4 treatment reduced the percentage of stress fiber-positive NIH3T3 cells (p < 0.001; Fig. 3b) and mean cell spread area (p < 0.001; Fig. 3c) to control levels. TGF-β increased the number of FAs in NIH3T3 cells by ~ 4-fold (p < 0.001; Fig. 3d), and the mean size of FAs per cell by ~ 2-fold (p < 0.05; Fig. 3e). Likewise, rhPRG4 treatment reduced the number of FAs (p < 0.001; Fig. 3d) and mean FA size (p < 0.05; Fig. 3e) to control levels. A functional outcome of TGF-β induced stress fiber and FAs formation was the enhanced migration of NIH3T3 fibroblasts (Fig. 3f). rhPRG4 reduced TGF-β-stimulated NIH3T3 fibroblast migration (p < 0.001; Fig. 3g) while rhPRG4 alone did not change basal NIH3T3 fibroblast migration (p > 0.05 against control; Fig. 3g).

Impact of recombinant human proteoglycan-4 (rhPRG4) treatment on TGF-β induced stress fiber formation, vinculin expression, and formation of focal adhesions (FAs) in murine fibroblasts (NIH3T3) and its relationship to cell migration. Stress fiber formation was probed using an anti-alpha smooth muscle actin (α-SMA) antibody (green) and a blinded investigator evaluated % stress fiber-positive fibroblasts. Vinculin (a marker of FAs) expression was probed using an anti-vinculin antibody (green) and cells were counterstained using rhodamine-labeled phalloidin (cytoskeleton label; red). NIH3T3 Fibroblast migration was performed using a scratch assay and the wound closure percentage was determined. *p < 0.001; **p < 0.01; ***p < 0.05; n.s., non-significant. Scale in a and f = 20 μm; a Representative images showing TGF-β induced stress fiber formation and vinculin staining (white arrows) in NIH3T3 fibroblasts. b rhPRG4 reduced stress fiber formation in NIH3T3 fibroblasts. c rhPRG4 reduced mean NIH3T3 fibroblast spread area. d rhPRG4 reduced the number of FAs in NIH3T3 fibroblasts. e rhPRG4 reduced the mean size of FAs in NIH3T3 fibroblasts. f Representative images showing NIH3T3 migration in response to TGF-β ± rhPRG4. g rhPRG4 reduced NIH3T3 migration
Lack of Prg4 expression in murine synovial fibroblasts was associated with increased formation of stress fibers, mean FA size, and enhanced basal cell migration, which was reduced by rhPRG4 or anti-CD44 antibody treatments
Representative images demonstrate enhanced α-SMA and vinculin staining in Prg4−/− synoviocytes compared to Prg4+/+ synoviocytes (Fig. 4a). The mean size of FAs was ~ 2-folds higher in Prg4−/− synoviocytes compared to Prg4+/+ synoviocytes (p < 0.001; Fig. 4b). Prg4−/− synoviocytes displayed enhanced basal migration compared to Prg4+/+ synoviocytes (p < 0.01; Fig. 4c, d). rhPRG4 and anti-CD44 antibody treatments reduced Prg4−/− synoviocyte migration (p < 0.01 for both treatments; Fig. 4d) with no significant difference between both treatments (p > 0.05).

Stress fibers and focal adhesions (FAs) in murine synovial fibroblasts isolated from Prg4−/− and Prg4+/+ mice and their relationship to cell migration. Stress fiber formation was probed using an anti-alpha smooth muscle actin (α-SMA) antibody (green). Vinculin (a marker of FAs) expression was probed using an anti-vinculin antibody (green) and cells were counterstained using rhodamine-labeled phalloidin (cytoskeleton label; red). The impact of rhPRG4 treatment (200 μg/mL) on basal Prg4−/− synovial fibroblast migration was assessed using the scratch assay and the wound closure percentage was determined. To highlight the CD44-dependency of the enhanced basal migration of Prg4−/− synovial fibroblasts, cells were also treated with either an anti-CD44 or isotype control (IC) antibodies (2 μg/mL for both antibodies). *p < 0.001; **p < 0.01; n.s., non-significant. Scale in a = 50 μm (α-SMA) and 100 μm (vinculin); scale in c = 50 μm. a Representative images showing increased stress fibers and FAs in Prg4−/− synoviocytes. b Prg4−/− synoviocytes demonstrated a higher mean FA size than Prg4+/+ synoviocytes. c Representative images showing enhanced basal migration of Prg4−/− synoviocytes compared to Prg4+/+ synoviocytes. d rhPRG4 and anti-CD44 treatments were equally effective in reducing basal migration of Prg4−/− synoviocytes
LPS stimulation of macrophages increased active and total TGF-β levels in a macrophage and fibroblast co-culture model, induced fibroblast migration and rhPRG4 treatment reduced fibroblast migration without altering active or total TGF-β levels
In response to LPS stimulation and co-culture of macrophages (M) and fibroblasts (F), active TGF-β levels in the macrophage and fibroblast co-culture were higher compared to fibroblasts alone (p < 0.001; Fig. 5a). Correspondingly, total TGF-β levels in the macrophage and fibroblast co-culture were higher compared to fibroblasts alone (p < 0.001; Fig. 5a). The increase in active and total TGF-β levels resulted in enhanced fibroblast migration in the macrophage and fibroblast co-culture compared to fibroblasts alone (p < 0.001; Fig. 5b, c). rhPRG4 treatment reduced fibroblast migration in the co-culture model (p < 0.001; Fig. 5c). The effect of rhPRG4 was not mediated by an alteration in active or total TGF-β levels, shown by the lack of differences in active or total TGF-β concentrations between rhPRG4-treated and untreated cells (p > 0.05; Fig. 5d).

Impact of co-culturing murine fibroblasts (F) and murine macrophages (M) on active and total supernatant TGF-β levels, fibroblast migration, and efficacy of rhPRG4 in regulating fibroblast migration in the co-culture model. Murine macrophages (J774A) were stimulated with lipopolysaccharide (LPS; 5 μg/mL) for 24 h prior to seeding in the top chamber of a co-culture system. Murine fibroblasts (NIH3T3) were cultured in the lower chamber of the same system. Active and total TGF-β media levels were determined using an ELISA. A scratch was performed in the fibroblast monolayer and fibroblast migration in the lower chamber was determined at 48 h ± rhPRG4 (200 μg/mL). *p < 0.001; **p < 0.01; n.s., non-significant. Scale = 40 μm. a Active and total TGF-β concentrations were higher in the fibroblast and macrophage co-culture compared to fibroblasts alone. b Representative images of fibroblast migration across different experimental groups. Fibroblast migration was highest in co-cultured fibroblasts and macrophages and rhPRG4 treatment reduced fibroblast migration. c rhPRG4 reduced fibroblast migration in a fibroblast and macrophage co-culture model. d rhPRG4 did not alter active or total TGF-β media levels in the fibroblast and macrophage co-culture model
Lack of Prg4 expression resulted in progressive synovial tissue fibrosis, shown by enhanced α-SMA, collagen type-I, and PLOD2 production and Prg4 re-expression reduced synovial tissue fibrosis and this effect was evident in CD44-deficient mice
Synovial tissues from Prg4GT/GT animals displayed higher ACTA2, COL1A1, and PLOD2 expression levels compared to Prg4 wildtype synovia (p < 0.001 for all comparisons; Fig. 6a, d, g). Interestingly, lack of Prg4 expression resulted in a greater increase in ACTA2 expression compared to COL1A1 and PLOD2 (p < 0.001 for all comparisons). The expression of fibrotic markers in synovial tissues from Prg4GT/GT animals progressed with age. ACTA2, COL1A1, and PLOD2 expression levels in 9-month-old Prg4GT/GT animals were higher than corresponding expression levels in 2-month-old Prg4GT/GT animals (p < 0.001 for all comparisons). The regulation of fibrotic gene expression in the synovium by PRG4 was further confirmed by the reduction in ACTA2, COL1A1, and PLOD2 expression in 9-month-old Prg4GTR/GTR animals compared to age-matched Prg4GT/GT animals (p < 0.001 for all comparisons). Representative images of synovial tissues stained for α-SMA, collagen type-I, and PLOD2 proteins are shown in Fig. 6b, e, and h. There was no significant difference in mean collagen type-I staining intensity between 2-month-old Prg4GT/GT and age-matched Prg4GTR/GTR animals (p > 0.05; Fig. 6f). Alternatively, mean α-SMA, collagen type-I, and PLOD2 staining intensities were lower in 9-month-old Prg4GTR/GTR animals compared to age-matched Prg4GT/GT animals (p < 0.05 for all comparisons; Fig. 6c, f, i).

Gene expression and immunohistological analysis of fibrotic markers: alpha smooth muscle actin (α-SMA) (ACTA2), collagen type-1 (COL1A1) and procollagen-lysine, 2-oxoglutarate 5-dioxygenase (PLOD2) in synovial tissues isolated from 2- and 9-month-old gene-trap (Prg4GT/GT) and 2- and 9-month-old recombined gene-trap (Prg4GTR/GTR) mice. The Prg4 gene-trap allele is a loss of function allele (Prg4GT/GT) whose function is restored with Cre-excision (Prg4GTR/GTR). Recombination occurred at 3 weeks of age using intraperitoneal tamoxifen (0.1 mg/g in 100 μL corn oil vehicle) daily for 10 days. Fibrotic markers were probed using specific antibodies and staining intensities (lumens per square millimeter) were quantified. *p < 0.001; **p < 0.01; ***p < 0.05. Scale = 100 μm. a ACTA2 expression was higher in 9-month Prg4GT/GT compared to 2-month littermates and was reduced by Prg4 re-expression. b Representative images showing enhanced α-SMA immunostaining in 2- and 9-month Prg4GT/GT synovia. c Prg4 re-expression reduced α-SMA content in 2- and 9-month-old animals. d COL1A1 expression was higher in 9-month-old Prg4GT/GT animals compared to 2-month and was reduced by Prg4 re-expression. e Representative images showing enhanced collagen type-I immunostaining in 9-month-old Prg4GT/GT synovia. f Collagen type-I content was higher in 9-month-old Prg4GT/GT animals and was reduced with Prg4 re-expression. g PLOD2 expression was higher in 9-month-old Prg4GT/GT animals compared to 2-month and was reduced by Prg4 re-expression. h Representative images showing enhanced PLOD2 immunostaining in Prg4GT/GT synovia. i Prg4 re-expression reduced PLOD2 content in Prg4GT/GT synovia in 2- and 9-month-old animals
The contribution of CD44 to PRG4’s effect on α-SMA and PLOD2 expression was further studied in vivo by comparing histological staining intensities in synovial tissues from Cd44+/+&Prg4GT/GT, Cd44−/−&Prg4GT/GT, and Cd44−/−&Prg4GTR/GTR (Fig. 7). While mean α-SMA staining intensity trended higher in Cd44−/−&Prg4GT/GT synovia compared to Cd44+/+&Prg4GT/GT synovia, this increase did not reach statistical significance (p = 0.16). Mean α-SMA staining intensity in Cd44−/−&Prg4GTR/GTR synovia was lower than the corresponding mean intensity in Cd44−/−&Prg4GT/GT synovia (p < 0.05). The absence of CD44 receptor increased PLOD2 staining in synovial tissues as evidenced by a higher mean staining intensity in Cd44−/−&Prg4GT/GT animals compared to Cd44+/+&Prg4GT/GT animals (p < 0.001). In addition, Prg4 re-expression reduced mean PLOD2 staining in otherwise Cd44 null animals (p < 0.001).

Modulation of expression of fibrotic markers: alpha smooth muscle actin (α-SMA) and procollagen-lysine, 2-oxoglutarate 5-dioxygenase (PLOD2) in synovial tissues from Prg4 gene-trap animals by CD44. The Prg4 gene-trap allele is a loss of function allele (Prg4GT/GT) whose function is restored with Cre-excision (Prg4GTR/GTR). Prg4GT/GT animals were crossed with Cd44−/− mice to generate PrgGT/GT&Cd44−/− animals. Recombination occurred at 3 weeks of age using intraperitoneal tamoxifen (0.1 mg/g in 100 μL corn oil vehicle) daily for 10 days. Fibrotic markers were probed using specific antibodies and staining intensities (lumens per square millimeter) were quantified. Prg4 recombination reduced α-SMA and PLOD2 staining in synovial tissues of 2-month-old Cd44+/+ and Cd44−/− animals. *p < 0.001; ***p < 0.05; n.s., non-significant
Discussion
PRG4, also called lubricin and superficial zone protein, is a mucinous glycoprotein with a protein core of 1404 amino acids and a central mucin domain that is extensively glycosylated with O-linked β (1-3) Gal-GalNAc oligosaccharides [29, 30, 50, 51]. PRG4 is secreted from synovial fibroblasts and superficial zone chondrocytes and is found adsorbed on articular surfaces and the synovial membrane in addition to its presence in SF [52, 53]. PRG4 has a multifaceted role in the joint and one aspect of its role is to provide boundary lubrication, to prevent friction-induced mitochondrial dysregulation and chondrocyte apoptosis [54,55,56]. Synthesis of PRG4 by cartilage and synovium was reduced in posttraumatic OA (PTOA) animal models and inflammatory cytokines, e.g., IL-1β reduced PRG4 secretion by synovial fibroblasts while TGF-β produced an opposite effect [57,58,59]. Furthermore, SF PRG4 levels decreased following acute joint trauma and in patients with advanced OA and rheumatoid arthritis (RA) [60,61,62]. Therapeutically, native and rhPRG4 demonstrated disease-modifying activities in pre-clinical PTOA mediated by their ability to reduce cartilage degeneration, enhance cartilage repair, and reduce chondrocyte apoptosis [42, 63,64,65].
In this investigation, we studied rhPRG4’s antifibrotic biological effects on fibroblasts in response to exogenously introduced recombinant TGF-β or TGF-β produced by co-cultured macrophages in response to TLR4 receptor stimulation. We identified that PRG4 and HA provide a similar outcome in TGF-β-stimulated OA FLS in the form of preventing transition of fibroblasts to myofibroblasts with inhibition of α-SMA expression and thus stress fiber formation. PRG4 and HA produced an antifibrotic effect in OA FLS using concentrations that are well below what is normally found in SF [60,61,62]. As HA is the prototypical CD44 ligand, we used HA as a positive control and our findings support that CD44 modulates fibroblast to myofibroblast transition in OA synoviocytes. PRG4 was rapidly internalized by OA FLS, with a prominent role for CD44 in this process. However, CD44 was not the only mechanism by which OA synoviocytes internalized rhPRG4 as approximately 60% of rhPRG4 entry was blocked subsequent to CD44 receptor neutralization. CD44-mediated rhPRG4’s uptake partially contributed to the latter’s antifibrotic role as rhPRG4’s effect on α-SMA expression was significantly weakened, but not completely abolished, when CD44-mediated uptake was blocked. CD44 also played a role in facilitating the enhanced migration of murine synovial fibroblasts that lack Prg4 expression as antibody-mediated neutralization of the receptor reduced cell migration. This finding is consistent with our prior observation that Prg4 null synoviocytes had higher CD44 receptor expression levels compared to Prg4 competent synoviocytes and is supported by an independent finding that CD44 plays a role in facilitating cell migration [39, 66, 67]. Synovial fibrosis developed in Prg4 null mice and the expression of SMA, PLOD2, and collagen type-I progressively increased as animals aged. Interestingly, the in vivo antifibrotic effect due to Prg4 re-expression was not dependent on CD44 as endogenously produced PRG4 reduced SMA and PLOD2 expression in Cd44 null animals.
CD44 is a single-pass transmembrane glycoprotein that is widely expressed in immune and connective tissues, and different isoforms, generated by alternate splicing, have been characterized [66, 67]. The extracellular domain of CD44 binds different ligands and thus can sense a broad array of microenvironmental signals resulting in ligand-specific effects in inflammation, cell migration, and growth [67,68,69]. In addition to HA and PRG4, other CD44 ligands include osteopontin, chondroitin, fibronectin, and matrix metalloproteinases (MMPs) [69, 70]. The intracellular domain of CD44 lacks intrinsic kinase activity, but it transduces extracellular ligand-specific signals via a combination of proteolytic fragmentation or interactions with different signaling pathways including members of Src and Ras family of GTPases and protein phosphatase-2A [69, 71, 72]. HA is cellularly uptaken by CD44, and this interaction mediates HA’s ability to suppress MMP-13 and aggrecanase-1 expression in OA chondrocytes and synoviocytes [73,74,75]. In addition to inhibiting synovitis, HA treatment inhibited synovial fibrosis in a murine model of TGF-β and treadmill running [75,76,77]. The effect of HA was CD44-dependent as its antifibrotic effect was abolished in CD44-deficient mice [77]. Contrary to HA, PRG4 had an in vivo antifibrotic effect that was not dependent on CD44. This might be related to PRG4’s ability to regulate other signaling pathways apart from CD44. PRG4 binds to and inhibits the activation of TLR2 and TLR4 receptors by their ligands and by SF aspirates from patients with OA [78, 79]. In addition, PRG4 was internalized by murine macrophages and co-localized with both CD44 and TLR2 receptors in the cytosol of these cells [80]. As such, it is expected that the enhanced TLR2 and TLR4 receptor signaling in the absence of PRG4 would propagate synovitis in Prg4 null mice. The innate immune-mediated synovitis would in turn contribute to the development of synovial fibrosis. Upon Prg4 re-expression in 3-week-old animals, PRG4 would attenuate TLR2- and TLR4-mediated synovitis, thereby potentially reducing TLR-regulated fibrotic tissue remodeling [81].
Myofibroblasts are characterized by α-SMA expression, formation of stress fibers, and FAs and are thought to play an effector role in the pathophysiology of tissue fibrosis [81, 82]. Compared to fibroblasts, myofibroblasts produce more collagen and respond to mitogenic signals resulting in cell proliferation, migration, and secretion of matrix-degrading enzymes [81, 83]. While stress fibers and FAs are unique structures within the cell, they are highly interconnected, where they coordinate cell responses to mechanical signals in their microenvironment [84]. Interestingly, the mean size of FAs, and not their molecular composition or density, can accurately predict cell migration [85]. Normal synovial fibroblasts do not appear to express α-SMA, even with TGF-β stimulation [86]. In our study, OA FLS had a basal level of α-SMA expression and in response to TGF-β, α-SMA expression was enhanced and stress fibers appeared. This was also evident in murine NIH3T3 fibroblasts as the transition from a fibroblast to a myofibroblast phenotype was accompanied by stress fiber and FAs formation, with subcellular and cellular changes in the form of increased number and mean size of FAs and cell spread area and thus enhanced cell migration. rhPRG4 acted biologically to reduce the formation of stress fibers and to reduce the mean number and size of FAs in NIH3T3 fibroblasts and this reduction resulted in arresting fibroblast migration. Thus, it is unlikely that the observed antimigratory effect of rhPRG4 was a physical effect consequent to PRG4’s amphipathic nature and its adsorption on hydrophilic and hydrophobic surfaces [87]. The higher mean size of FAs in synoviocytes from Prg4 null mice, and its resultant enhanced migration argues that PRG4 plays a role in regulating the expression of the cellular machinery mediating migration in vivo. This may have biological relevance in the realm of synovial fibrosis where PRG4 may function to reduce the migration of fibrocytes to the inflamed joint and therefore the likelihood of adding to the pool of existing myofibroblasts in the synovium [88, 89].
Synovial tissue macrophages are critical regulators of the initiation, maintenance, and resolution phases of synovitis [90]. The complex functions of macrophages are enabled by the presence of different macrophage subpopulations within the joint, which fulfill distinct inflammatory and antiinflammatory roles [90]. In synovial tissue fibrosis, bone marrow-derived monocytes and macrophages are found in increased numbers, which may signal their involvement in the development of synovial fibrosis [91, 92]. In response to an inflammatory signal, macrophages produce TGF-β as a regulatory feedback signal to aid in the resolution of inflammation [93, 94]. TGF-β in turn triggers myofibroblast differentiation and production of matrix proteins [38]. In our study, LPS stimulation resulted in increased secretion of TGF-β by macrophages. Cells secrete a latent form of TGF-β, which in turn is proteolytically converted to active TGF-β by a variety of proteases, including MMPs [95]. The generation of active TGF-β in our co-culture system was associated with fibroblast migration. Interestingly, the introduction of rhPRG4 after macrophage activation did not alter the total TGF-β quantity produced by macrophages. Furthermore, rhPRG4 did not alter the rate of activation of latent TGF-β, indicating that the antimigratory effect of rhPRG4 was at the level of fibroblasts’ response to TGF-β receptor activation. This is supported by our observation that rhPRG4 reduced Smad3 phosphorylation in TGF-β reporter cells and that reduction in pSmad3 intracellular levels attenuated the TGF-β/Smad pathway activation. In TGF-β reporter cells, the attenuation of Smad signaling pathway was downstream to rhPRG4 uptake by these cells in a mechanism that involved CD44 receptor, similar to what was observed in OA FLS. However, the precise mechanism that is activated by CD44 engagement that inhibits TGF-β/Smad signaling pathway remains unclear. Another limitation of our study was that we did not study the antifibrotic effect of rhPRG4 in vivo.
Conclusion
In summary, we demonstrated that PRG4 is an important regulator of synovial tissue fibrosis using a combination of in vitro and in vivo models. PRG4 prevented the transition of human and murine fibroblasts to a myofibroblast phenotype and in OA FLS; the mechanism of PRG4’s antifibrotic effect was partially linked to CD44-mediated cell uptake. rhPRG4 acted biologically to reduce Smad3 phosphorylation, the activation of TGF-β/Smad signaling pathway, and FA mean size which translated to inhibiting fibroblast migration. The lack of Prg4 expression in murine synovial tissues resulted in enhanced expression of SMA, collagen type-I, and PLOD2, the enzyme responsible for collagen cross-linking. Synovial fibrosis in Prg4GT/GT animals progressed with age and re-establishing Prg4 expression was antifibrotic. The role of PRG4 in regulating synovial fibrosis in vivo extends beyond its interaction with CD44 receptor, as Prg4 re-expression was antifibrotic in Cd44 null mice. Therapeutically, overexpressing PRG4 in the articular joint, using a viral mediated gene delivery approach, may prove beneficial in reducing synovial fibrosis in addition to reducing cartilage degeneration [96, 97]. The study of PRG4 as an antifibrotic modulator of the joint’s soft tissues is further warranted especially with the known contribution of synovial fibrosis in advanced OA.
Availability of data and materials
Not applicable.
Abbreviations
- α-SMA:
-
Alpha smooth muscle actin
- ACTA2 :
-
Gene symbol for alpha smooth muscle actin
- ANOVA:
-
Analysis of variance
- BCS:
-
Bovine calf serum
- BSA:
-
Bovine serum albumin
- CD44:
-
Cluster determinant 44
- COL1A1 :
-
Gene symbol for collagen type-I
- Ct:
-
Cycle threshold
- DMEM:
-
Dulbecco’s Modified Eagle Media
- ELISA:
-
Enzyme-linked immunosorbent assay
- FBS:
-
Fetal bovine serum
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- OA:
-
Osteoarthritis
- OA FLS:
-
Osteoarthritic fibroblast-like synoviocytes
- PBS:
-
Phosphate-buffered saline
- PCR:
-
Polymerase chain reaction
- PLOD2:
-
Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2
- PRG4:
-
Proteoglycan-4
- RA:
-
Rheumatoid arthritis
- rhPRG4:
-
Recombinant human PRG4
- TLR2:
-
Toll-like receptor 2
- TLR4:
-
Toll-like receptor 4
References
Kloppenburg M, Berenbaum F. Osteoarthritis year in review 2019: epidemiology and therapy. Osteoarthritis Cartilage. 2020 doi: https://doi.org/10.1016/j.joca.2020.01.002. [Epub ahead of print].
Cross M, Smith E, Hoy D, Nolte S, Ackerman I, et al. The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann rheum Dis. 2014;73(7):1323–30.
Fu K, Robbins SR, JJ MD. Osteoarthritis: the genesis of pain. Rheumatology (Oxford). 2018;57(suppl_4):iv43–50.
Wang Y, UDT N, Lane NE, Lu N, Wei J, et al. Knee osteoarthritis, potential mediators, and risk of all-cause mortality: data from the osteoarthritis initiative. Arthritis Care Res. 2020. https://doi.org/10.1002/acr.24151 [Epub ahead of print].
Cleveland RJ, Alvarez C, Schwartz TA, Losina E, Renner JB, et al. The impact of painful knee osteoarthritis on mortality: a community-based cohort study with over 24 years of follow-up. Osteoarthritis Cartilage. 2019;27(4):593–602.
Veronnese N, Cereda E, Maggi S, Luchini C, Solmi M, et al. Osteoarthritis and mortality: a prospective cohort study and systematic review with meta-analysis. Semin Arthritis Rheum. 2016;46(2):160–7.
O’Neill TW, PS MC, McBeth J. Update on the epidemiology, risk factors and disease outcomes of osteoarthritis. Best Pract Res Clin Rheumatol. 2018;32(2):312–26.
Silverwood V, Blagojevic-Bucknall M, Jinks C, Jordan JL, Protheroe J, et al. Current evidence on risk factors for knee osteoarthritis in older adults: a systematic review and meta-analysis. Osteoarthritis Cartilage. 2015;23(4):507–15.
Carbone A, Rodeo S. Review of current understanding of post-traumatic osteoarthritis resulting from sports injuries. J Orthop Res. 2017;35(3):397–405.
Poulsen E, Goncalves GH, Bicca A, Roos EM, Thorlund JB, et al. Knee osteoarthritis risk is increased 4-6 fold after knee injury-a systematic review and meta-analysis. Br J Sports Med. 2019;53(23):1454–63.
Poole AR. Osteoarthritis as a whole joint disease. HSS J. 2012;8(1):4–6.
Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, et al. Osteoarthritis. Nat Rev Dis Primers. 2016;2:16072.
Wenham CY, Congahan PG. The role of synovitis in osteoarthritis. Ther Adv Musculoskelet Dis. 2010;2:349–59.
Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19:18.
Guermazi A, Hayashi D, Roemer FW, Zhu Y, Niu J, et al. Synovitis in knee osteoarthritis assessed by contrast-enhanced magnetic resonance imaging (MRI) is associated with radiographic tibiofemoral osteoarthritis and MRI-detected widespread cartilage damage: the MOST study. J Rheumatol. 2014;41(3):501–8.
Baker K, Grainger A, Niu J, Clancy M, Guermazi A, et al. Relation of synovitis to knee pain using contrast-enhanced MRIs. Ann Rheum Dis. 2010;69(1):1779–83.
Gomez R, Villavilla A, Largo R, Gualillo O, Herrero-Beaumont G. TLR4 signaling in osteoarthritis-finding targets for candidate DMOADs. Nat Rev Rheumatol. 2015;11:159–70.
Scanzello CR, Plaas A, Crow MK. Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound? Curr Opin Rheumatol. 2008;20:565–72.
Sharma N, Drobinski P, Kayed A, Chen Z, Kjelgaard-Petersen CF, et al. Inflammation and joint destruction may be linked to the generation of cartilage metabolites of ADAMTS-5 through activation of toll-like receptors. Osteoarthritis Cartilage. 2019. https://doi.org/10.1016/j.joca.2019.11.002 [Epub ahead of print].
Herrero-Beaumont G, Perez-Baos S, Sanchez-Pernaute O, Roman-Blas JA, Lamuedra A, et al. Targeting chronic innate inflammatory pathways, the main road to prevention of osteoarthritis progression. Biochem Pharmacol. 2019;165:24–32.
Kalaitzoglou E, Griffin TM, Humphrey MB. Innate immune responses and osteoarthritis. Curr Rheumatol Rep. 2017;19(8):45.
Conaghan PG, D’Agostino MA, Le Bars M, Schmidely N, Wakefield R, et al. Clinical and ultrasonographic predictors of joint replacement for knee osteoarthritis: results from a large, 3-year, prospective EULAR study. Ann Rheum Dis. 2010;69(4):644–7.
Wang X, Hunter DJ, Ding C. The importance of synovial inflammation in osteoarthritis: current evidence from imaging assessments and clinical trials. Osteoarthritis Cartilage. 2018;26(2):165–74.
Collins JE, Losina E, Nevitt MC, Roermer FW, Guermazi A, et al. Semiquantitative imaging biomarkers of knee osteoarthritis progression: data from the foundation for the national institutes of health osteoarthritis biomarkers consortium. Arthritis Rheumatol. 2016;68(10):2422–31.
Atukorala I, Kwoh CK, Guermazi A, Roemer FW, Boudreau RM, et al. Synovitis in knee osteoarthritis: a precursor of disease? Ann Rheum Dis. 2016;75(2):390–5.
Henrotin Y, Pesesse L, Lambert C. Targeting the synovial angiogenesis as a novel treatment approach to osteoarthritis. Ther Adv Musculoskelet Dis. 2014;6(1):20–34.
Benito M, Veale D, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64(9):1263–7.
Smith MD. The normal synovium. Open Rheumatol J. 2015;5:100–6.
Jay GD, Waller KA. The biology of lubricin: near frictionless joint motion. Matrix Biol. 2014;39:17–24.
Jay GD, Britt DE, Cha CJ. Lubricin is a product of megakaryocyte stimulating factor gene expression by human synovial fibroblasts. J Rheumatol. 2000;27:594–600.
Loeuille D, Chary-Valckenaere I, Champigneulle J, Rat AC, Toussaint F, et al. Macroscopic and microscopic features of synovial membrane inflammation in the osteoarthritic knee: correlating magnetic resonance imaging findings with disease severity. Arthritis Rheum. 2005;52(11):3492–501.
Abdul N, Dixon D, Walker A, Horabin J, Smith N, et al. Fibrosis is a common outcome following total knee arthroplasty. Sci Rep. 2015;5:16469.
Remst DF, Blaney Davidson EN, Vitters EL, Blom AB, Stoop R, et al. Osteoarthritis-related fibrosis is associated with both elevated pyrdinoline cross-link formation and lysyl hydroxylase 2b expression. Osteoarthritis Cartilage. 2013;21:157–64.
Remst DF, Blom AB, Vitters EL, Bank RA, van den Berg WB, et al. Gene expression analysis of murine and human osteoarthritis synovium reveals elevation of transforming growth factor β-responsive genes in osteoarthritis-related fibrosis. Arthritis Rheumatol. 2014;66:647–56.
van der Kraan PM. The changing role of TGFβ in healthy, aging and osteoarthritic joints. Nat Rev Rheumatol. 2017;13:155–63.
Remst DF, Blaney Davidson EN, Vitters EL, Bank RA, van den Berg WB, et al. TGF-β induces lysyl hydroxylase 2b in human synovial osteoarthritic fibroblasts through ALK5 signaling. Cell Tissue Res. 2014;355:163–71.
Bong MR, Di Cesare PE. Stiffness after total knee arthroplasty. J Am Acad Orthop Surg. 2004;12(3):f–71.
Qadri MM, Jay GD, Ostrom RS, Zhang LX, Elsaid KA. cAMP attenuates TGF-β’s profibrotic responses in osteoarthritic synoviocytes: involvement of hyaluronan and PRG4. Am J Physiol Cell Physiol. 2018;315:C432–43.
Al-Sharif A, Jamal M, Zhang LX, Larson K, Schmidt TA, et al. Lubricin/proteoglycan 4 binding to CD44 receptor: a mechanism of the suppression of proinflammatory cytokine-induced synoviocyte proliferation by lubricin. Arthritis Rheumatol. 2015;67:1503–13.
Alquraini A, Jamal M, Zhang L, Schmidt T, Jay GD, et al. The autocrine role of proteoglycan-4 (PRG4) in modulating osteoarthritic synoviocyte proliferation and expression of matrix degrading enzymes. Arthritis res Ther. 2017;19:89.
Neumann E, Riepl B, Knedla A, Lefevre S, Tarner IH, et al. Cell culture and passaging alters gene expression pattern and proliferation rate in rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther. 2010;12:R83.
Jay GD, Fleming BC, Watkins BA, KA MH, Anderson SC, et al. Prevention of cartilage degeneration and restoration of chondroprotection by lubricin tribosupplementation in the rat following anterior cruciate ligament transection. Arthritis Rheum. 2010;62:2382–91.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8.
Samson ML, Morrison S, Masala N, Sullivan BD, Sullivan DA, et al. Characterization of full-length recombinant human proteoglycan 4 as an ocular surface boundary lubricant. Exp Eye Res. 2014;127C:14–9.
Acharya PS, Majumdar S, Jacob M, Hayden J, Mrass P, et al. Fibroblast migration is mediated by CD44-dependent TGF beta activation. J Cell Sci. 2008;121:1393–402.
Ascione F, Vasaturo A, Caserta S, D’Esposito V, Formisano P, et al. Comparison between fibroblast wound healing and cell random migration assays in vitro. Exp Cell Res. 2016;347:123–32.
Rhee DK, Marcelino J, Baker M, Gong Y, Smits P, Lefebvre V, et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J Clin Invest. 2005;115:622–31.
Hill A, Walker KA, Cui Y, Allen JM, Smits P, et al. Lubricin restoration in a mouse model of congenital deficiency. Arthritis Rheumatol. 205;67:3070–81.
Protin U, Schweighoffer T, Jochum W, Hilberg F. CD44-deficient mice develop normally with changes in subpopulations and recirculation of lymphocyte subsets. J Immunol. 1999;163:4917–23.
Swann DA, Silver FH, Slayter HS, Stafford W, Shore E. The molecular structure and lubricating activity of lubricin isolated from bovine and human synovial fluids. Biochem J. 1985;225(1):195–201.
Jay GD, Habertstroh K, Cha CJ. Comparison of the boundary lubricating ability of bovine synovial fluid, lubricin, and Healon. J Biomed Mater Res. 1998;49(3):414–8.
Jay GD, Tantravahi U, Britt DE, Barrach HJ, Cha CJ. Homology of lubricin and superficial zone protein (SZP): products of megakaryocyte stimulating factor (MSF) gene expression by human synovial fibroblasts and articular chondrocytes localized to chromosome 1q25. J Orthop Res. 2001;19(4):677–87.
Flannery CR, Hughes CE, Schumacher BL, Tudor D, Aydelotte MB, et al. Articular cartilage superficial zone protein (SZP) is homologous to megakaryocyte stimulating factor precursor and is a multifunctional proteoglycan with potential growth-promoting cytoprotective, and lubricating properties in cartilage metabolism. Biochem Biophys Res Commun. 1999;254(3):535–41.
Schmidt TA, Gastelum NS, Nguyen QT, Schumacher BL, Sah RL. Boundary lubrication of articular cartilage: role of synovial fluid constituents. Arthritis Rheum. 2007;56(3):882–91.
Waller KA, Zhang LX, Jay GD. Friction-induced mitochondrial dysregulation contributes to joint deterioration in Prg4 knockout mice. Int J Mol Sci. 2017;18(6):E1252.
Larson KM, Zhang L, Badger GJ, Jay GD. Early genetic restoration of lubricin expression in transgenic mice mitigates chondrocyte peroxynitrite release and caspase-3 activation. Osteoarthritis Cartilage. 2017;25(9):1488–95.
Elsaid KA, Machan JT, Waller K, Fleming BC, Jay GD. The impact of anterior cruciate ligament injury on lubricin metabolism and the effect of inhibiting tumor necrosis factor alpha on chondroprotection in an animal model. Arthritis Rheum. 2009;60(10):2997–3006.
Schmidt TA, Gastelum NS, Han EH, Nugent-Derfus GE, Schumacher BL, et al. Differential regulation of proteoglycan 4 metabolism in cartilage by IL-1 alpha, IGF-1, and TGF-beta 1. Osteoarthritis Cartilage. 2008;16(1):90–7.
Blewis ME, Lao BJ, Schumacher BL, Bugbee WD, Sah RL, et al. Interactive cytokine regulation of synoviocyte lubricant secretion. Tissue Eng Part A. 2010;16(4):1329–37.
Elsaid KA, Fleming BC, Oskendahl HL, Machan JT, Fadale PD, et al. Decreased lubricin concentrations and markers of joint inflammation in the synovial fluid of patients with anterior cruciate ligament injury. Arthritis Rheum. 2008;58(6):1707–15.
Catterall JB, Stabler TV, Flannery CR, Kraus VB. Changes in serum and synovial fluid biomarkers after acute injury (NCT00332254). Arthritis Res Ther. 2010;12(6):R229.
Kosinska MK, Ludwig TE, Liebisch G, Zhang R, Siebert HC, et al. Articular joint lubricants during osteoarthritis and rheumatoid arthritis display altered levels and molecular species. PLoS One. 2015;10:e0125192.
Elsaid KA, Zhang L, Waller K, Tofte J, Teeple E, et al. The impact of forced joint exercise on lubricin biosynthesis from articular cartilage following ACL transection and intra-articular lubricin’s effect in exercised joints following ACL transection. Osteoarthritis Cartilage. 2012;20:940–8.
Cui Z, Xu C, Li X, Song J, Yu B. Treatment with recombinant lubricin attenuates osteoarthritis by positive feedback loop between articular cartilage and subchondral bone in ovariectomized rats. Bone. 2015;74:37–47.
Waller KA, Chin KE, Jay GD, Zhang LX, Teeple E, et al. Intra-articular recombinant human proteoglycan-4 mitigates cartilage damage after destabilization of the medial meniscus in the Yucatan minipig. Am J Sports Med. 2017;45(7):1512–21.
Cutly M, Nguyen HA, Underhill CB. The hyaluronan receptor (CD44) participates in the uptake and degradation of hyaluronan. J Cell Biol. 1992;116(4):1055–62.
Underhill C. CD44: the hyaluronan receptor. J Cell Sci. 1992;103(Pt 2):293–8.
Weber GF, Ashkar S, Glimcher MJ, Cantor H. Receptor-ligand interaction between CD44 and osteopontin (Eta-1). Science. 1996;271:509–12.
Senbanjo LT, Chellaiah M. CD44: a multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front Cell Dev Biol. 2017;5:18.
Chen C, Zhao S, Karnad A, Freeman JW. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol. 2018;11(1):64.
Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 2015;6:201.
Mellor L, Knudson CB, Hida D, Askew EB, Knudson W. Intracellular domain fragment of CD44 alters CD44 function in chondrocytes. J Biol Chem. 2013;288(36):25838–50.
Julovi SM, Yasuda T, Shimizu M, Hiramitsu T, Nakamura T. Inhibition of interleukin-1 beta-stimulated production of matrix metalloproteinases by hyaluronan via CD44 in human articular cartilage. Arthritis Rheum. 2004;50(2):516–25.
Yatabe T, Mochizuki S, Takizawa M, Chijiwa M, Okada A, et al. Hyaluronan inhibits expression of ADAMTS4 (aggrecanases-1) in human osteoarthritic chondrocytes. Ann Rheum Dis. 2009;68(6):1051–8.
Kataoka Y, Ariyoshi W, Okinaga T, Kaneuki T, Mitsugi S, et al. Mechanisms involved in suppression of ADAMTS4 expression in synoviocytes by high molecular weight hyaluronic acid. Biochem Biophys Res Commun. 2013;432(4):580–5.
Waddell DD, Kolomytkin OV, Dunn S, Marino AA. Hyaluronan suppresses IL-1 beta-induced metalloproteinase activity from synovial tissue. Clin Orthop Relat Res. 2007;465:241–8.
Plaas A, Li J, Riesco J, Das R, Sandy JD, et al. Intrarticular injection of hyaluronan prevents cartilage erosion, periarticular fibrosis and mechanical allodynia and normalizes stance time in murine knee osteoarthritis. Arthritis Res Ther. 2011;13:R46.
Alquraini A, Garguilo S, D’Souza G, Zhang LX, Schmidt TA, et al. The interaction of lubricin/proteoglycan 4 (PRG4) with toll-like receptors 2 and 4: an anti-inflammatory role of PRG4 in synovial fluid. Arthritis Res Ther. 2015;17:353.
Iqbal SM, Leonard C, Regmi SC, De Rantere D, Tailor P, et al. Lubricin/proteoglycan 4 binds to and regulates the activity of toll-like receptors in vitro. Sci Rep. 2016;6:18910.
Qadri M, Jay GD, Zhang LX, Wong W, Reginato M, et al. Recombinant human proteoglycan-4 reduces phagocytosis of urate crystals and downstream nuclear factor kappa B and inflammasome activation and production of cytokines and chemokines in human and murine macrophages. Arthritis Res Ther. 2018;20(1):192.
Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–40.
Sarini G, Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res. 1999;250:273–83.
Ehrlich HP, Allison GM, Leggett M. The myofibroblast, cadherin, alpha smooth muscle actin and the collagen effect. Cell Biochem Funct. 2006;24:63–70.
Livne A, Geiger B. The inner workings of stress fibers-from contractile machinery to focal adhesions and back. J Cell Sci. 2016;129(7):1293–304.
Kim DH, Wirtz D. Focal adhesion size uniquely predicts cell migration. FASEB J. 2013;27(4):1351–61.
Steenvoorden MM, Tolboom TC, van der Pluijm G, Löwik C, Visser CP, et al. Transition of healthy to diseased synovial tissue in rheumatoid arthritis is associated with gain of mesenchymal/fibrotic characteristics. Arthritis Res Ther. 2006;8:R165.
Zappone B, Ruths M, Greene GW, Jay GD, Israelachvili JN. Adsorption, lubrication, and wear of lubricin on model surfaces: polymer brush-like behavior of a glycoprotein. Biophys J. 2007;92(5):1693–708.
Keeley EC, Mehrad B, Strieter RM. Fibrocytes: bringing new insights into mechanisms of inflammation and fibrosis. Int J Biochem Cell Biol. 2010;42(4):535–42.
Reikoff RA, Bucala R, Herzog EL. Fibrocytes: emerging effector cells in chronic inflammation. Nat Rev Immunol. 2011;11(6):427–35.
Kurowska-Stolarska M, Alivernini S. Synovial tissue macrophages: friend or foe? RMD Open. 2017;3(2):e000527.
Bosch U, Zeichen J, Skutek M, Haeder L, van Griensven M. Arthrofibrosis is the result of a T cell mediated immune response. Knee Surg Sports Traumatol Arthrosc. 2001;9(5):282–9.
Freeman TA, Parvizi J, Della Valle CJ, Steinbeck MJ. Reactive oxygen and nitrogen species induce protein and DNA modifications driving arthrofibrosis following total knee arthroplasty. Fibrogenesis Tissue Repair. 2009;2(1):5.
Wermuth PJ, Jiminez SA. The significance of macrophage polarization subtypes for animal models of tissue fibrosis and human fibrotic diseases. Clin Transl Med. 2015;4:2.
Donlin LT, Jayatilleke A, Giannopoulou EG, Kalliolias GD, Ivashkiv LB. Modulation of TNF-induced macrophage polarization by synovial fibroblasts. J Immunol. 2014;193(5):23773–2383.
Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-β signaling in fibrosis. Growth Factor. 2011;29(5):196–202.
Stone A, Grol MW, Ruan MZC, Dawson B, Chen Y, et al. Combinatorial Prg4 and IL-1ra gene therapy protects against hyperalgesia and cartilage degeneration in post-traumatic osteoarthritis. Human Gene Ther. 2019;30(2):225–35.
Ruan MZ, Erez A, Guse K, Dawson B, Bertin T, et al. Proteoglycan 4 expression protects against the development of osteoarthritis. Sci Transl Med. 2013;5(176):176ra34.
Acknowledgements
Not applicable.
Funding
This work is supported by R01AR067748 to KE and GJ. 5P30GM122732 which supported GDJ per the NIH.
Author information
Authors and Affiliations
Contributions
Authors MQ, GJ, LZ, and KE carried out experiments and participated in analysis of data. Authors TS and HR participated in study design and critical interpretation of results. Authors GJ and KE conceived the study and participated in data analysis and interpretation. All authors have participated in drafting and critical evaluation of the manuscript. All authors have read and approved the final version of the manuscript.
Authors’ information
Marwa Qadri, MSc: Ph.D. student, Chapman University School of Pharmacy, Irvine, CA, USA.
Gregory D. Jay, MD, Ph.D.: Professor, Emergency Medicine and Engineering, Brown University, Providence, RI, USA.
Ling Zhang, MD: Senior Research Assistant, Rhode Island Hospital, Providence, RI, USA.
Holly Richendrfer: Research Associate, Rhode Island Hospital, Providence, RI, USA.
Tannin A. Schmidt, Ph.D.: Associate Professor of Biomedical Engineering, University of Connecticut Health Center, Farmington, CT, USA.
Khaled A. Elsaid, Pharm.D, Ph.D.: Associate Professor of Biomedical and Pharmaceutical Sciences, Chapman University, Irvine, CA, USA.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
OA synoviocytes were obtained from a commercial source (Cell Applications Inc.). Harvest of synoviocytes was performed following appropriate IRB approvals from partner sites with informed written consent from the donor. Patients were de-identified. Authors did not seek patients’ consent to study these biological samples. This study was approved by the IRB at Chapman University, Orange, CA, USA.
Harvest of synovial tissues from mice and histological evaluations of mice joints were approved by the IACUC Committee at Rhode Island Hospital, Providence, RI, USA. All experiments were performed in accordance with established guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
Authors MQ, HR, and LZ have nothing to disclose.
Author GJ authored patents on rhPRG4 and holds equity in Lubris LLC, MA, USA.
Author TS authored patents on rhPRG4; is a paid consultant for Lubris LLC, MA, USA; and holds equity in Lubris LLC, MA, USA.
Author KE authored patents on rhPRG4.
All authors have no non-financial competing interests related to this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qadri, M., Jay, G.D., Zhang, L.X. et al. Proteoglycan-4 regulates fibroblast to myofibroblast transition and expression of fibrotic genes in the synovium. Arthritis Res Ther 22, 113 (2020). https://doi.org/10.1186/s13075-020-02207-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-020-02207-x