
Abstract
Background
Pain is one of the most important domains affecting health-related quality of life (HRQoL) in patients with psoriatic arthritis (PsA). Secukinumab has demonstrated rapid and sustained improvements in signs and symptoms, including HRQoL, among patients with active PsA. This analysis evaluates the effect of secukinumab on patient-reported pain in PsA through 104 weeks of treatment.
Methods
Pain was assessed through week 104 using clinically relevant measures, including change from baseline in a pain visual analog scale (VAS) and Short Form-36 (SF-36) bodily domain scores; proportion of patients reporting improvements equal to or better than minimum clinically meaningful differences in the pain VAS and SF-36 bodily pain domain scores; and proportion of patients with no, moderate, or extreme pain/discomfort measured by the EuroQoL 5-Dimension 3-Level Questionnaire (EQ-5D-3 L) pain item scores. Correlations of pain measures were analyzed using Pearson’s correlation coefficient. Pre-specified analyses of TNF-naïve patients and patients who stopped TNF-inhibitors (TNFis) due to inadequate responses or safety/tolerability (TNF-IR patients) were performed using “as-observed data.”
Results
Mean improvements from baseline in pain VAS scores were greater with secukinumab versus placebo by week 3 (− 16.9; P < 0.0001 with secukinumab 300 mg and − 12.6; P < 0.05 with secukinumab 150 mg) and sustained through week 104. SF-36 bodily pain domain scores were significantly greater with 300 mg secukinumab and secukinumab 150 mg versus placebo by week 4 (16.2 and 16.3, respectively; P < 0.0001 for both), and these changes were maintained through week 104. With both secukinumab 300 mg and secukinumab 150 mg, improvements equal to or better than the minimum clinically meaningful differences in pain VAS and SF-36 bodily pain were significant versus placebo at week 3 and week 4, respectively. At week 4, 15%, 9%, and 5% of patients receiving secukinumab 300 mg, secukinumab 150 mg, and placebo, respectively, reported “no pain/discomfort” measured by EQ-5D-3 L; these proportions increased to week 104 with both secukinumab doses. Similarly, improvements in pain measures were significant in both TNF-naïve and TNF-IR patients.
Conclusion
Secukinumab provided rapid and sustained pain relief in PsA over 2 years of treatment. Improvements in pain were reported regardless of prior exposure to TNFis.
Trial registration
ClinicalTrials.gov, NCT01752634. Registered on 19 December 2012.
Similar content being viewed by others


Background
Psoriatic arthritis (PsA) is a chronic progressive, clinically heterogeneous inflammatory arthritis that can manifest as peripheral arthritis, axial disease, enthesitis, dactylitis, and psoriasis of the skin/nails [1, 2]. The disease burden of PsA, taking into account patient-reported outcomes and disease activity measures, is similar to the burden of rheumatoid arthritis and axial spondyloarthritis [3]. A recent analysis from the population-based Multinational Assessment of Psoriasis and Psoriatic Arthritis (MAPP) found that 88% of patients questioned had ongoing joint pain or soreness, and 60% reported > 4 affected joints [4]. The most common locations for joint pain were the knee (41%) followed by finger (26%), hip (19%), ankle (19%), back (18%), and wrist (16%) [4].
The combination of ongoing pain, fatigue, physical impairment, and anxiety likely influence the impact of PsA and reduce health-related quality of life (HRQoL) [5]. Specifically, patients with PsA have impaired HRQoL characterized by poor physical functioning and engagement in daily activities, and pain [6]. In a European League Against Rheumatism (EULAR) initiative to develop and validate the Psoriatic Arthritis Impact of Disease questionnaire (PsAID), patients with PsA identified pain as the most important health domain affecting HRQoL [7]. Additionally, arthritis or joint pain causes a significant economic impact that results in US$6773 greater costs compared to individuals without either [8]. Pain poses an important clinical challenge in the treatment of PsA. In a population-based survey, rheumatologists and dermatologists considered joint pain or swelling to be the critical factor contributing to the severity of PsA [9]. Treatment criteria, scoring indices, and clinical trial domains further reflect the importance of pain in PsA. The Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) includes pain as an important factor to examine when treating patients with PsA [10]. A patient pain visual analog scale (VAS) score ≤ 15 is one of seven critical criteria for determining minimal disease activity in PsA [11]. Finally, at the Outcome Measures in Rheumatology (OMERACT) 2016 conference, pain was endorsed as part of a core set of domains recommended for study in randomized controlled trials (RCTs) and longitudinal observational studies (LOS) in PsA [12].
Secukinumab, a fully human monoclonal antibody (mAb) that selectively neutralizes interleukin (IL)-17A, has been shown to have significant efficacy in the treatment of moderate-to-severe psoriasis and active PsA, demonstrating rapid onset of action and sustained responses with a consistent safety profile [13, 14]. Since 2015 in the European Union and 2016 in the USA, secukinumab 300 mg and 150 mg have been approved for the treatment of active PsA. In the phase-3 FUTURE 2 study, secukinumab was effective in treating patients not previously exposed (TNF-naïve patients) to TNF-α inhibitors (TNFis) and patients who stopped using up to three previous TNFis due to inadequate responses or for safety/tolerability reasons (TNF-IR patients) [15]. Over 2 years, secukinumab was shown to exhibit rapid and sustained improvements in the signs and symptoms of active PsA, to inhibit radiographic progression, and to improve HRQoL [16, 17]. This post-hoc analysis of FUTURE 2 examines the effect of secukinumab on patient-reported pain in PsA over 104 weeks.
Methods
Details of FUTURE 2 (NCT01752634), an ongoing, multicenter RCT, have been reported previously [15] and will be briefly summarized here.
Patients
FUTURE 2 was conducted at 76 centers throughout Asia, Australia, Europe, and North America, in accordance with the principles of the Declaration of Helsinki, and all centers received approval from independent ethics committees or institutional review boards. Patients included in this trial were aged ≥ 18 years, met the Classification criteria for Psoriatic Arthritis (CASPAR), had ≥ 3 tender and ≥ 3 swollen joints despite treatment with non-steroidal anti-inflammatory drugs (NSAIDs), disease-modifying anti-rheumatic drugs (DMARDs), or TNFis. Patients who had not responded adequately to up to three treatments with TNFis were eligible for inclusion.
A TNFi wash-out period ≥ 4 weeks prior to randomization was mandatory. Patients on concomitant oral corticosteroids who received a stable dose for ≥ 2 weeks prior to randomization (≤ 10 mg/day) and patients who were on methotrexate and received a stable dose for ≥ 4 weeks prior to randomization (≤ 25 mg/week) were allowed throughout the trial. Patients were excluded from the study for previous use of biologic agents other than TNFis; active inflammatory diseases other than PsA; active infection within 2 weeks prior to randomization or a history of chronic, ongoing, or recurrent infections; pregnancy; or malignancy within the past 5 years, with the exception of basal cell carcinoma or actinic keratosis, in-situ cervical cancer, or non-invasive malignant colon polyps.
Study design and assessments
Patients were randomized 1:1:1:1 to receive subcutaneous secukinumab 300 mg, secukinumab 150 mg, secukinumab 75 mg, or placebo at baseline, weeks 1, 2, and 3, and then every 4 weeks from week 4. At week 16, patients were classified as responders (≥ 20% improvement from baseline in tender and swollen joint counts) or non-responders. Placebo-treated patients were randomly assigned again in a 1:1 ratio to receive subcutaneous secukinumab 300 mg or 150 mg every 4 weeks from week 16 (non-responders) or week 24 (responders). This study was blinded from baseline to week 52 and then open label until week 104.
Pain was assessed through week 104 by the mean change from baseline in the pain VAS and Short Form-36 (SF-36) bodily pain domain scores; proportion of patients reporting improvements equal to or better than the minimum clinically meaningful difference (MCID) in the pain VAS score (mean change from baseline ≥ 20%) [18]; proportion of patients reporting improvements equal to or better than the MCID (5-point improvement from baseline) in SF-36 bodily pain domain score [19]; proportion of patients reporting EuroQoL 5-Dimension 3-Level Questionnaire (EQ-5D-3 L) pain item scores of no, moderate, or extreme pain/discomfort; and correlation coefficients for changes from baseline between pain scores.
Statistical methods
Mean changes from baseline in the pain VAS and SF-36 bodily pain domain scores were analyzed using a mixed-effect model for repeated measures (MMRM) through week 24 with treatment regimen, analysis visit, and randomization stratum (TNF-naive or TNF-IR) as factors, weight and baseline score as continuous covariates, and treatment by analysis visit and baseline score by analysis visit as interaction terms, and an unstructured covariance structure. Mean changes from baseline were reported as observed starting from week 28. Frequencies of patient EQ-5D-3 L pain/discomfort domain response were reported using observed data up to week 104. Correlation with pain measures was analyzed using Pearson’s correlation coefficient and P values were calculated using the chi-square likelihood ratio test. The proportion of patients with VAS improvement ≥ 20% were reported using as-observed data and P values were calculated using Fisher’s exact test. The proportion of patients with improvement ≥ 5 in the SF-36 bodily pain domain score were reported using as-observed data and P values were calculated using Fisher’s exact test. Pre-specified subgroup analyses (TNF-naïve and TNF-IR) were performed using MMRM through week 24 and data were reported as observed starting from week 28. Only the results for patients receiving the approved doses – secukinumab 300 mg and secukinumab 150 mg – are reported in this article. Due to the re-randomization of patients in the placebo group at week 16, statistical comparisons of secukinumab versus placebo are presented up to week 16. Analyses were exploratory in nature and conducted without adjustment for multiple comparisons.
Results
Study population
Overall, 397 patients with active PsA were randomized for this trial. Demographics and baseline disease characteristics have been previously reported for patients in this study and are presented in Additional file 1: Table S1. In the overall patient population, mean pain VAS scores were in the range of 55.4–58.9 and mean SF-36 bodily pain scores were in the range of 33.7–37.6. Further, 99% of patients reported moderate-to-extreme pain or discomfort at baseline measured by the EQ-5D-3 L pain/discomfort item. Of 298 patients, 193 (65%) were TNFi naïve at baseline.
Efficacy
Treatment with secukinumab was associated with rapid and sustained relief of pain in patients with PsA. Significant improvements in mean changes from baseline in pain VAS scores were reported beginning at week 3 compared with placebo (− 5.8) with secukinumab 300 mg (−16.9; P < 0.0001) and secukinumab 150 mg (−12.6; P < 0.05). Improvements in mean changes from baseline in pain VAS scores continued to week 16 and were sustained to week 104 with both doses of secukinumab (Fig. 1a). Additionally, most patients treated with secukinumab reported responses equal to or better than the MCID in the pain VAS response. At week 3, a significantly greater number of patients receiving secukinumab 300 mg had ≥ 20% improvement in pain VAS response compared with placebo (60.6% vs 36.6%; P < 0.01). For both doses of secukinumab, the proportion of patients with improvements equal to or better than the MCID in pain VAS scores continued to increase to week 16 and similar levels of attainment were reported at week 104 (Fig. 1b).

Proportion of patients with ≥ 20% improvement in pain visual analog scale (VAS) score through week 104 (a) and proportion of patients with ≥ 20% improvement in pain VAS response (b)
Data are presented as LS mean change using MMRM from weeks 1-24, and observed data are presented from weeks 28-104 (shaded area); P values are calculated from a MMRM analysis (a). Observed data are presented; P values are calculated from Fisher’s exact test (b)
1Number of patients originally randomized to each treatment group. *P < 0.0001; †P < 0.001; ‡P < 0.05 §P < 0.01 versus placebo
LS, least squares; MMRM, mixed-effect model for repeated measures; PBO, placebo; s.c., subcutaneous; SEC, secukinumab
A similar trend for reductions in patient-reported pain with secukinumab was reported for SF-36 bodily pain domain scores. At week 4, mean changes from baseline in SF-36 bodily pain scores were significantly greater with both secukinumab 300 mg (16.2) and secukinumab 150 mg (16.3) compared with placebo (5.9; P < 0.0001 for both). Continued improvement in SF-36 bodily pain domain scores were reported up to week 12 with both secukinumab doses, and initial improvements were sustained to week 104 for both secukinumab doses (Fig. 2a). Most patients treated with secukinumab also reported equal to or better than the MCID in the SF-36 bodily pain domain score. At week 4, a significantly greater number of patients receiving secukinumab 300 mg (72.3%) and secukinumab 150 mg (73.7%) had improvements ≥ 5 points in SF-36 bodily pain domain score compared with placebo (46.9%; P < 0.001 for both). For both doses of secukinumab, the proportion of patients with improvements equal to or better than the MCID in SF-36 bodily pain domain scores were maintained to week 104 (Fig. 2b). The MCID for SF-36 bodily pain domain scores was attained in both anti-TNF-naive and anti-TNF-IR patients receiving secukinumab.

Change from baseline in the Short Form-36 (SF-36) bodily pain score through week 104 (a) and proportion of patients with improvement ≥ 5 in the SF-36 bodily pain domain score (b)
Data are presented as LS mean change using MMRM from weeks 1-24, and observed data are presented from weeks 52-104 (shaded area); P values are calculated from a MMRM analysis (a). Observed data are presented; P values are calculated from Fisher’s exact test (b)
1Number of patients originally randomized to each treatment group. *P < 0.0001; †P < 0.001; ‡P < 0.05 versus placebo. LS, least squares; MMRM, mixed-effect model for repeated measures; PBO, placebo; s.c., subcutaneous; SEC, secukinumab
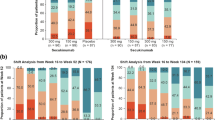
At baseline, only 1% of patients reported no pain or discomfort in the EQ-5D-3 L pain/discomfort item. Following 4 weeks of secukinumab treatment, no pain or discomfort was reported by 14.7% of patients receiving secukinumab 300 mg, 9.1% of patients receiving secukinumab 150 mg, and 5.2% of patients receiving placebo (Fig. 3). With secukinumab treatment, the proportion of patients reporting no pain or discomfort continued to increase over time, especially with secukinumab 300 mg and after 2 years of treatment with secukinumab 300 mg almost 30% of patients experienced no pain or discomfort by EQ-5D-3 L.

Proportion of patients with no pain or discomfort in the EuroQoL 5-Dimension (EQ-5D-3 L) domain through week 104
Observed data are presented. Data are presented only for evaluable patients at each time point
s.c., subcutaneous
At weeks 16, 24, 52, and 104, improvements from baseline in mean pain VAS scores were significantly correlated with improvements from baseline in mean SF-36 bodily pain scores (correlation coefficients of − 0.56 to − 0.62; P < 0.01 for all) and EQ-5D pain/discomfort domain scores (correlation coefficients of 0.36 to 0.47; P < 0.01 for all). Similarly, improvements from baseline in mean SF-36 bodily pain scores were significantly correlated with improvements in EQ-5D pain/discomfort domain scores at weeks 16, 24, 52, and 104 (correlation coefficients of − 0.45 to − 0.52; P < 0.01 for all).
Response by prior TNF inadequate response
TNF-naive and TNF-IR patients both demonstrated significant improvements in mean changes from baseline in pain VAS scores and SF-36 bodily pain scores. TNF-naïve patients receiving secukinumab 300 mg reported significant improvements in mean change from baseline in pain VAS score compared with placebo beginning at week 3 (− 19.7 vs − 8.5; P < 0.01). With both doses of secukinumab, improvements continued to week 104 (Table 1). Although a similar trend was observed in TNF-IR patients, the magnitude of improvements was generally lower. In TNF-IR patients, significant mean improvements from baseline in pain VAS scores were reported at week 3 with secukinumab 300 mg (− 13.1; P < 0.05 vs placebo) and secukinumab 150 mg (− 14.8; P < 0.01 vs placebo). Due to the smaller number of TNF-IR patients there was more volatility in responses over time. Nonetheless, improvements were maintained over time.
Significant mean improvements from baseline in SF-36 bodily pain scores were also reported by both TNF-naive and TNF-IR patients receiving secukinumab (Table 1). At week 4, mean changes from baseline in SF-36 bodily pain scores in TNF-naïve patients improved by 18.4 with secukinumab 300 mg, by 18.9 with secukinumab 150 mg compared with 5.6 for placebo (P < 0.0001 for both versus placebo). Over time, the degrees of improvement in SF-36 bodily pain scores were similar with both doses of secukinumab in TNF-naïve patients. In TNF-IR patients, there were significant mean improvements from baseline in SF-36 bodily pain scores at week 8 with both secukinumab 300 mg (18.7; P < 0.01) and 150 mg (21.0; P < 0.001) compared to 5.7 with placebo. Mean changes from baseline in SF-36 bodily pain scores were similar over time between TNF-naive and TNF-IR patients with secukinumab 300 mg.
Both TNF-naive and TNF-IR patients also reported improvements in the EQ-5D-3 L pain item scores. At week 16, 22% of TNF-naive patients receiving either dose of secukinumab reported no pain or discomfort compared with 6.9% of patients receiving placebo (Table 1). The proportion of TNF-naive patients receiving secukinumab 300 mg that reported no pain or discomfort continued to increase to week 104 when 32.8% TNF-naive patients had no pain or discomfort. A similar trend was observed in TNF-IR patients but fewer of these patients reported no pain or discomfort compared with TNF-naïve patients. There was a small numerical increase in the number of TNF-IR patients reporting no pain or discomfort at week 104.
Discussion
Pain is one of the most important domains affecting HRQoL in patients with PsA [7], and patients with PsA report role limitations caused by both emotional problems and bodily pain [5, 20]. Pain has also been linked to fatigue in other inflammatory arthritides [21]. Additionally, joint pain has a quantifiable burden to society as demonstrated by work limitations in patients with PsA [22]. Taken together, these findings exemplify the importance of managing pain when treating patients with PsA.
In this post-hoc analysis of the phase-3, FUTURE 2 study, secukinumab 300 mg and secukinumab 150 mg both provided rapid and sustained pain relief to patients with PsA over 2 years’ treatment. Improvements in pain with secukinumab were consistent across three different patient-reported measures (pain VAS, SF-36 bodily pain, and EQ-5D-3 L pain/discomfort) and were observed regardless of prior exposure to TNFis. Improvements in pain reported in this trial are consistent with findings of improved pain and HRQoL in other secukinumab trials. In FUTURE 1, improvement was observed in both mean pain VAS and SF-36 bodily pain domain scores [19]. Further, secukinumab 150 mg demonstrated sustained improvement in HRQoL with an SF-36 physical component summary score of 4.89 at week 104 [17]. In FUTURE 2, improved physical function was reported at week 24 with a change from baseline in the Health Assessment Questionnaire Disability Index (HAQ-DI) of − 0.56 with secukinumab 300 mg (P = 0.004 vs placebo) and − 0.48 with secukinumab 150 mg (P = 0.0555 vs placebo) [15].
In this trial, improvements in pain with secukinumab treatment were sufficient to meet or exceed established definitions for MCID in pain for PsA and were observed irrespective of previous exposure to TNFis. Due to the level of improvement in pain with secukinumab treatment, it is interesting to consider whether pain alleviation is due to overall improvement in inflammation or through a direct role of IL-17 inhibition. Plasma levels of IL-17A are elevated in patients with fibromyalgia and in mice, IL-17 contributes to neuroinflammatory responses and pain hypersensitivity following neuropathic injury [23, 24]. These findings suggest that additional investigation is warranted on the role of IL-17 in other conditions associated with pain such as PsA.
There was significant correlation between the improvement from baseline in mean pain VAS scores and mean SF-36 bodily pain scores and EQ-5D pain/discomfort domain scores and significant correlation between improvement from baseline in mean SF-36 bodily pain scores and EQ-5D pain/discomfort scores. Together these correlations indicate that secukinumab consistently improves pain regardless of the specific measurement used for assessment.
Secukinumab 300 mg and secukinumab 150 mg also improved pain in both TNF-naive and TNF-IR patients measured by the pain VAS, SF-36 bodily pain scores, and EQ-5D-3 L pain/discomfort domain. Not surprisingly, TNF-naïve patients reported numerically greater changes from baseline in all three pain outcomes than those reported by TNF-IR patients.
Limitations of this study include that it was a post-hoc analysis, the lack of an active comparator, and lack of placebo control after week 24.
Conclusions
Secukinumab has previously demonstrated efficacy in treatment of the signs and symptoms of PsA, inhibition of radiographic progression in patients with PsA, and a favorable safety profile [15,16,17]. In this study, secukinumab provided rapid pain relief for patients with PsA as assessed by multiple clinically relevant patient-reported pain measures. Pain relief began as early as week 3 and improvements were sustained through 104 weeks. Further, improvements in pain were reported by patients regardless of prior exposure to TNFis.
Abbreviations
- DMARD:
-
Disease-modifying anti-rheumatic drug
- EQ-5D-3 L:
-
EuroQoL 5-Dimension 3-Level Questionnaire
- EULAR:
-
European League Against Rheumatism
- GRAPPA:
-
Group for Research and Assessment of Psoriasis and Psoriatic Arthritis
- HRQoL:
-
Health-related quality of life
- IL:
-
Interleukin
- LOS:
-
Longitudinal observational studies
- MAPP:
-
Multinational Assessment of Psoriasis and Psoriatic Arthritis
- MCID:
-
Minimum clinically meaningful difference
- MMRM:
-
Mixed-effect model for repeated measures
- NSAID:
-
Non-steroidal anti-inflammatory drug
- OMERACT:
-
Outcome Measures in Rheumatology
- PsA:
-
Psoriatic arthritis
- PsAID:
-
Psoriatic Arthritis Impact of Disease
- RCT:
-
Randomized controlled trial
- SF-36:
-
Short Form-36
- TNF:
-
Tumor necrosis factor
- TNFi:
-
Tumor necrosis factor-α inhibitor
- TNF-IR:
-
TNF inadequate response
- VAS:
-
Visual analog scale
References
Gladman DD. Psoriatic arthritis. Dermatol Ther. 2009;22:40–55.
Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med. 2017;376:957–70.
Michelsen B, Fiane R, Diamantopoulos AP, Soldal DM, Hansen IJ, Sokka T, et al. A comparison of disease burden in rheumatoid arthritis, psoriatic arthritis and axial spondyloarthritis. PLoS One. 2015;10:e0123582.
Kavanaugh A, Helliwell P, Ritchlin CT. Psoriatic arthritis and burden of disease: patient perspectives from the population-based Multinational Assessment of Psoriasis and Psoriatic Arthritis (MAPP) Survey. Rheumatol Ther. 2016;3:91–102.
Lee S, Mendelsohn A, Sarnes E. The burden of psoriatic arthritis: a literature review from a global health systems perspective. P T. 2010;35:680–9.
Gratacós J, Daudén E, Gómez-Reino J, Moreno JC, Casado MA, Rodriguez-Valverde V. Health-related quality of life in psoriatic arthritis patients in Spain. Reumatol Clin. 2014;10:25–31.
Gossec L, de Wit M, Kiltz U, Braun J, Kalyoncu U, Scrivo R, et al. A patient-derived and patient-reported outcome measure for assessing psoriatic arthritis: elaboration and preliminary validation of the Psoriatic Arthritis Impact of Disease (PsAID) questionnaire, a 13-country EULAR initiative. Ann Rheum Dis. 2014;73:1012–9.
Williams EM, Walker RJ, Faith T, Egede LE. The impact of arthritis and joint pain on individual healthcare expenditures: findings from the Medical Expenditure Panel Survey (MEPS), 2011. Arthritis Res Ther. 2017;19:38.
Lebwohl MG, Kavanaugh A, Armstrong AW, Van Voorhees AS. US perspectives in the management of psoriasis and psoriatic arthritis: patient and physician results from the population-based Multinational Assessment of Psoriasis and Psoriatic Arthritis (MAPP) Survey. Am J Clin Dermatol. 2016;17:87–97.
Coates LC, Kavanaugh A, Mease PJ, Soriano ER, Laura Acosta-Felquer M, Armstrong AW, et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis Rheumatol. 2016;68:1060–71.
Coates LC, Fransen J, Helliwell PS. Defining minimal disease activity in psoriatic arthritis: a proposed objective target for treatment. Ann Rheum Dis. 2010;69:48–53.
Orbai AM, de Wit M, Mease PJ, Callis Duffin K, Elmamoun M, Tillett W, et al. Updating the Psoriatic Arthritis (PsA) Core Domain Set: a report from the PsA Workshop at OMERACT 2016. J Rheumatol. 2017;44:1522–8.
Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371:326–38.
Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N Engl J Med. 2015;373:1329–39.
McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2015;386:1137–46.
McInnes IB, Mease PJ, Ritchlin CT, Rahman P, Gottlieb AB, Kirkham B, et al. Secukinumab sustains improvement in signs and symptoms of psoriatic arthritis: 2 year results from the phase 3 FUTURE 2 study. Rheumatology (Oxford). 2017;56:1993–2003.
Kavanaugh A, Mease PJ, Reimold AM, Tahir H, Rech J, Hall S, et al. Secukinumab for long-term treatment of psoriatic arthritis: a two-year followup from a phase III, randomized, double-blind placebo-controlled study. Arthritis Care Res (Hoboken). 2017;69:347–55.
Busse JW, Bartlett SJ, Dougados M, Johnston BC, Guyatt GH, Kirwan JR, et al. Optimal strategies for reporting pain in clinical trials and systematic reviews: recommendations from an OMERACT 12 workshop. J Rheumatol. 2015;42:1962–70.
Strand V, Mease P, Gossec L, Elkayam O, van den Bosch F, Zuazo J, et al. Secukinumab improves patient-reported outcomes in subjects with active psoriatic arthritis: results from a randomised phase III trial (FUTURE 1). Ann Rheum Dis. 2017;76:203–7.
Husted JA, Gladman DD, Farewell VT, Cook RJ. Health-related quality of life of patients with psoriatic arthritis: a comparison with patients with rheumatoid arthritis. Arthritis Rheum. 2001;45:151–8.
Pollard LC, Choy EH, Gonzalez J, Khoshaba B, Scott DL. Fatigue in rheumatoid arthritis reflects pain, not disease activity. Rheumatology (Oxford). 2006;45:885–9.
Tillett W, de-Vries C, NJ MH. Work disability in psoriatic arthritis: a systematic review. Rheumatology (Oxford). 2012;51:275–83.
Kim CF, Moalem-Taylor G. Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J Pain. 2011;12:370–83.
Pernambuco AP, Schetino LP, Alvim CC, Murad CM, Viana RS, Carvalho LS, et al. Increased levels of IL-17A in patients with fibromyalgia. Clin Exp Rheumatol. 2013;31:S60–3.
Acknowledgements
The authors thank the patients and investigators who participated in the study.
Funding
Technical assistance with editing and styling of the manuscript for submission was provided by Oxford PharmaGenesis Inc. and was funded by Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA. The FUTURE 2 study was funded by Novartis Pharma AG, Basel, Switzerland.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Consortia
Contributions
The authors were fully responsible for all content and editorial decisions and received no financial support or other form of compensation related to the development of this manuscript. The opinions expressed in the manuscript are those of the authors and Novartis Pharmaceuticals had no influence on the contents. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the principles of the Declaration of Helsinki, and all centers received approval from independent ethics committees or institutional review boards.
Competing interests
IB McInnes received research grants, consultation fees, or speaker honoraria from AbbVie, Amgen, BMS, Celgene, Janssen, Lilly, Novartis, Pfizer, and UCB. PJ Mease received research grants, consultation fees, or speaker honoraria from AbbVie, Amgen, BMS, Celgene, Crescendo Bioscience, Genentech, Janssen, Eli Lilly, Merck, Novartis, Pfizer, and UCB. G Schett received speaker fees from Abbvie, BMS, Celgene, Chugai, Eli Lilly, Janssen, Novartis, Roche, and UCB. B Kirkham received research support from AbbVie, Novartis, Roche; served as a consultant and speaker for Abbott, BMS, Chugai, MSD, Novartis, Pfizer, Roche, UCB; and participated in speakers bureaus for Abbott, BMS, Chugai, MSD, Novartis, Pfizer, Roche, UCB. V Strand is a founding member of the executive of Outcome Measures in Rheumatoid Arthritis Clinical Trials (OMERACT), an organization that develops outcome measures in rheumatology and receives arms-length funding from 36 companies; a member of the American College of Rheumatology’s Clinical Trials Task Force and Chair of the American College of Rheumatology/OMERACT Imaging Subcommittee. Dr. Strand also has served as a consultant and member of advisory boards for AbbVie, Alder, Amgen Corporation, AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Celltrion, Genentech/Roche, GlaxoSmithKline, Janssen, Eli Lily, Novartis, Pfizer, Regeneron, Sandoz, Sanofi, and UCB. N Williams is a consultant for Novartis through employment at RTI Health Solutions and is an employee of RTI Health Solutions. T Fox, L Pricop, SM Jugl, and KK Gandhi are employees of Novartis and own Novartis stock.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Baseline demographics and disease severity characteristics. (DOCX 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
McInnes, I.B., Mease, P.J., Schett, G. et al. Secukinumab provides rapid and sustained pain relief in psoriatic arthritis over 2 years: results from the FUTURE 2 study. Arthritis Res Ther 20, 113 (2018). https://doi.org/10.1186/s13075-018-1610-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-018-1610-3