
Abstract
Background
The apicomplexan haemoparasite Theileria equi, a causative agent of equine piroplasmosis, is an established pathogen of significant welfare and economic concern within the Croatian equine population. A previous large surveillance study of T. equi has identified two distinct parasite populations, one in the north and one in the south, geographically separated by the Dinaric Alps, which traverse the country. This study aimed to further investigate the genetic diversity within these two populations, focussing on allelic variability of the equi merozoite antigen gene, ema-1.
Methods
Following nested PCR of DNA isolates, the generated ema-1 amplicons were subsequently sequenced and compared by phylogenetic analysis to available sequences representing previously described ema-1 genotypes (groups A–C).
Results
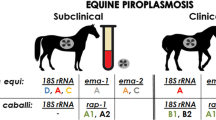
Isolates from the southern T. equi population clustered with the existing ema-1 groups A and B. Strikingly, isolates from the northern population clustered into two novel ema-1 genotypes, named groups D and E.
Conclusions
This detection of hitherto unreported genotypes suggests that historic geographical isolation has led to a degree of divergent evolution in this northern T. equi population. Additionally, current global regulatory testing of equine piroplasmosis relies heavily on EMA-1 based immunodiagnostics, and the discovery of unique ema-1 genotypes may question the efficacy of current diagnostics in international equine movement, with ramifications for the global equine community.
Graphical Abstract

Similar content being viewed by others


Background
Theileria equi has long been established as a pathogen of significant welfare and economic concern within the Croatian equine population [1]. The pathogen is one of the tick-borne apicomplexan parasites responsible for equine piroplasmosis, a disease with ubiquitous global presence which presents in three main clinical forms: acute or sub-acute disease with severe anaemia, pyrexia and dehydration, with death occurring in severe cases; chronic disease with animals displaying fluctuating malaise, weight loss or reduced performance [2]. Importantly, infected animals become carriers for life [3], acting as reservoirs for further infection and demonstrating disease recrudescence in times of illness or stress [4, 5].
Current research has established that Theileria equi demonstrates an unusually high degree of genetic heterogeneity at the 18S rRNA gene compared to other Theileria species [6], and recently a separate species, Theileria haneyi, has been identified within the Theileria equi umbrella [7]. A five-clade (A–E) molecular genotyping system has been established based on this 18S rRNA gene locus [8, 9] and has been used extensively in numerous T. equi surveillance studies in Europe [10, 11], Africa [12], the Americas [13] and the Middle East [14]. Other Theileria species, such as Theileria annulata and Theileria parva, show a high degree of genetic diversity in geographical areas with long-standing parasitic populations [15, 16]. Previous studies have shown this to also be true for T. equi, with historically endemic areas demonstrating not only multiple parasite genotype clades within the equine population [12, 17], but also a multiplicity of infection with individuals [17], whereas areas of more recent parasite introduction can show the presence of only a single clade [10].
As part of a larger surveillance study of piroplasmosis within the Croatian equine population [18], T. equi DNA was identified by PCR in 48 blood samples from equines across Croatia using previously described 18S rRNA gene specific primers [19]. Sequencing of generated amplicons showed the presence of just two T. equi 18S rRNA gene clades, A and E. When the geographical residence of these sampled equines was mapped, a distinct geographical separation of the clades was noted with clade E samples found exclusively in the north and clade A samples restricted to the south, with both populations separated by the Dinaric Alps which traverse Croatia (Fig. 1). This geographic separation is extremely unusual given the heterogenous mixing of clades seen in other historically endemic countries [12, 17]. Given the importance of the disease to the Croatian equine industry, and the endemic presence of the parasite within the country, this finding warranted further, more detailed investigation of the genetic diversity within these two T. equi populations.

The geographical distribution of Theileria equi 18S rRNA clade types within samples collected in northern and southern Croatia. The mountainous area dividing the two regions is marked in grey
The equi merozoite antigen-1 (ema-1) is a surface exposed protein, expressed during the erythrocytic stage of T. equi infection and utilised extensively as a target for commercial ELISA detection [20]. Although initially thought to be well conserved within the species [21, 22], further research has identified variability with three defined ema-1 groups defined [9, 23]. The heterogeneity at this locus makes this an ideal gene for assessing further genetic diversity within T. equi clade populations.
This report describes the methods and findings following investigation of ema-1 diversity within the two geographically separated Croatian T. equi populations.
Methods
DNA was extracted using a commercial extraction kit (DNeasy blood & tissue kit, Qiagen, Germany) from equine blood samples previously collected between 2012 and 2014 as part of a larger national equine haemoparasite surveillance study in Croatia. A total of 32 samples were available from the original 48 samples previously identified as containing T. equi DNA following screening with a Theileria/Babesia spp. 18S rRNA gene targeting PCR protocol [19]. Sequencing of the generated amplicons allowed clade genotyping based on this locus.
These 32 DNA samples were then further screened using novel primers targeting a hyper variable region of the ema-1 gene in a nested PCR protocol (Table 1). Reaction conditions for the first round of the PCR were a final volume of 20 μl with 5 × GoTaq Colorless Reaction Buffer (Promega, USA) (final concentrations of 1.5 mM MgCl2), Deoxynucleotide (dNTP) Solution Mix (New England Biolabs, USA) (final concentrations of 0.2 mM dATP, 0.2 mM dCTP, 0.2 mM dGTP and 0.2 mM dTTP), 0.5 μM of each outer primer (RC_EMA_F1 and RC_EMA_R1), 0.025 units/μl DNA polymerase (GoTaq G2, Promega, USA) and 2 μl of template genomic DNA solution.
The cycling conditions for the primary reaction were an initial denaturation of 94 °C for 5 min, then 30 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C for 30 s, and extension at 74 °C for 60 s, with a final extension at 72 °C for 5 min. Reactions for the secondary PCR were performed using identical reagent concentrations and cycling conditions except using the inner primer pair (RC_EMA_F2 and RC_EMA_R2). A 1:10 dilution of the primary reaction product was used as the DNA template in the secondary PCR reaction. The final product was visualised using gel electrophoresis with a 1% agarose gel. The PCR product was purified (QIAquick PCR purification kit, Qiagen, Germany) and submitted for Sanger sequencing (Eurofins Genomics, Germany). Amplicons were sequenced in both directions with a consensus sequence generated. Known positive and negative samples from a previous study were used as controls [10].
Species identification of sequences obtained in this study was achieved using the basic local alignment search tool (BLAST) and comparison with sequences deposited in the non-redundant National Center for Biotechnology Information (NCBI) GenBank database (https://blastncbi.nlm.nih.gov/). The MUSCLE function [24], within the AliView alignment viewer and editor [25], was used to compare the study sequences with those previously determined and deposited in the NCBI GenBank database. Nucleotide diversity statistics were generated using DnaSP [26].
A maximum likelihood phylogenetic tree was constructed using MEGA11 software [27] to compare the genetic diversity of the ema-1 gene sequences generated from the study samples with previously defined ema-1 groups [9, 23], based on 1000 replications. Piroplasm surface protein gene sequences of Theileria buffeli (D78015) and Theileria sergenti (D11046) and a Plasmodium falciparum surface antigen (AY861651) were included in the trees as outgroups [23]. All sequences generated in this report were submitted to the NCBI GenBank database (https//www.ncbi.nlm.nih.gov/genbank/) accession numbers ON502853—ON502883 inclusve.
Results
Amplicons were generated and successfully sequenced from 31 of the 32 available equine DNA samples, and BLAST analysis demonstrated all amplicons had an identity of between 92 and 100% with existing T. equi ema-1 sequences in the NCBI GenBank database.
The maximum-likelihood phylogenetic tree constructed to compare the Croatia ema-1 sequences with the previously defined ema-1 groups is shown in Fig. 2.

A maximum-likelihood tree inferring the evolutionary relationship of the ema-1 sequences from the Theileria equi samples identified in this survey. Included are all detected T. equi sequences from northern Croatia (blue) and southern Croatia (red). Other representative sequences of T. equi, Theileria buffeli, Theileria sergenti and Plasmodium falciparum (to which the tree is rooted) are included with GenBank accession numbers. Bootstrap values are shown as a percentage, based on 1000 replications. The previously described ema-1 groups, A–C, and the novel ema-1 groups, D, E, have been annotated, and external branches have been collapsed to the clade level
All samples from southern Croatia, previously identified as belonging to the 18S rRNA gene clade A, demonstrated ema-1 homology to the previously described ema-1 genotype groups A and B. All samples from northern Croatia, previously identified as belonging to the 18S rRNA gene clade E, were assembled distinct from the previously described ema-1 groups and clustered in to two novel genotype groups, tentatively named ema-1 groups D and E.
To investigate the variation in diversity between the samples from the different regions at the sequence level, within-region expected heterozygosity (He) was calculated using the allelic sequences derived from the northern Croatian, southern Croatian and ema-1 reference samples (Table 2). A relatively lower heterozygosity (He 0.24) was evident in the northern sample set compared to those in the south (He 0.64) and the reference samples (He 0.93). In addition, the nucleotide diversity (π) and average number of differences between samples (k) were calculated for each group (Table 2). This analysis further highlighted the reduced ema-1 diversity among the northern Croatian samples, which contrasted to that of the southern Croatian samples and those from other countries which showed substantial variation at the nucleotide level.
Discussion
The geographic distribution of T. equi phylogeny in Croatia is unique compared to that of other endemic countries [11], with a northern population clearly derived from the 18S rRNA clade E and a southern population derived from 18S rRNA clade A, separated by the robust geographic boundary of the Dinaric Alps.
The findings of the ema-1 phylogeny in this report show a homology between the southern T. equi population and previously defined ema-1 groups from global samples. This suggests the southern population may be a result of introduction from different geographic areas, most likely from historical international movement of horses into Croatia.
In contrast, the northern T. equi samples revealed the identification of two novel ema-1 genotypes. The detection of multiple novel gene groups implies that geographic isolation of this northern population has been maintained for a substantial chronological period. The northern region has a continental climate and geography, where the tick species of Dermacentor reticulatus, Dermacentor marginatus and Rhipicephalus sanguineus predominate, contrasting with the southern region which possesses a coastal climate and geography where Hyalomma marginatum, Rhipicephalus bursa and Rhipicephalus turanicus populations exist as [28]. The unique selection pressures of the northern region, likely a combination of the environment, presence of local tick species and equine host availability, has led to a degree of divergent evolution and development of unique genotypes within this isolated population.
As well as providing a distinctive example of divergent evolution in a T. equi population, these results further expand on the complexity of the species’ phylogeny being depicted in recent literature [9]. The relative homogeneity of the northern T. equi population and unique ema-1 genotypes may even be suggestive of a separate subspecies or species, such as the recently described T. haneyi, which clusters with the T. equi 18S rRNA clade C but has been demonstrated to lack the ema-1 gene completely [7]. However, this conclusion is beyond the scope of the results presented in the current report.
The results of this report also have relevance to the global equine community. Current regulatory testing of equine piroplasmos is heavily on EMA-1 based cELISA diagnostics [20], and despite reduced efficacy of commercial EMA-1 diagnostics having already been suggested within the previously known ema-1 genotypes [23], this method is still recommended by the World Organisation for Animal Health (OIE) for use in international equine movement [29]. The presence of the additional and substantial ema-1 heterogeneity described in this report further questions this existing dependence on EMA-1 as a target for immunodiagnostic screening, especially when the national biosecurity of countries without endemic equine piroplasmosis is at risk and introduction of the disease can lead to prolonged and costly eradication [30]. In light of this increasing evidence of diversity at this locus, newer diagnostics now additionally combine EMA-2 detection as part of immunodiagnostic screening methods [9, 31], and the international equine community would be wise to continually evaluate the robustness of current protocols and ideally seek more conserved, but specific targets for diagnostic testing.
The relatively small sample size, especially the available samples from southern regions, is a limitation of this study, and consequently the findings may not be representative of the country as a whole. In addition, the role of iatrogenic transmission events, which can be significant in the spread of infection [30], could not be assessed from the available data. However, given the infrequent occurrence of these events, and the wide-geographical area over which the isolates and novel genotypes have been detected, any iatrogenic transmission is unlikely to have had a significant impact on the reported findings.
Conclusion
This study has identified two hitherto unreported ema-1 genotypes suggesting that historic geographical isolation in Croatia has led to a degree of divergent evolution between the northern versus southern T. equi population. In addition to expanding current scientific understanding of T. equi species diversity, the discovery of unique ema-1 genotypes adds to current equine community concerns over the efficacy of current diagnostics in international equine movement.
Availability of data and materials
The datasets generated and analysed during the current study are available in the NCBI GenBank repository (accession numbers ON502853—ON502883 openly accessible at https://www.ncbi.nlm.nih.gov/genbank/).
Abbreviations
- 18S rRNA:
-
18S (Svedberg units) small subunit ribosomal ribonucleic acid
- BLAST:
-
Basic local alignment search tool
- cELISA:
-
Competitive enzyme-linked immunosorbent assay
- EMA-1:
-
Equi merozoite antigen one protein
- ema-1 :
-
Equi merozoite antigen one gene
- OIE:
-
World Organisation for Animal Health
References
Richter S. Vrste haemosporidia u konja i goveda NR Hrvatskoj. Thesis: Veterinary faculty university of zagreb; 1954.
Rothschild CM. Equine piroplasmosis. J Equine Vet Sci. 2013;33:497–508.
Ueti MW, Palmer GH, Scoles GA, Kappmeyer LS, Knowles DP. Persistently infected horses are reservoirs for intrastadial tick-borne transmission of the apicomplexan parasite Babesia equi. Infect Immun. 2008;76:3525–9.
Hailat NQ, Lafi SQ, AlDarraji AM, AlAni FK. Equine babesiosis associated with strenuous exercise: clinical and pathological studies in Jordan. Vet Parasitol. 1997;69:1–8.
Piantedosi D, D’Alessio N, Di Loria A, Di Prisco F, Mariani U, Neola B, et al. Seroprevalence and risk factors associated with Babesia caballi and Theileria equi infections in donkeys from Southern Italy. Vet J. 2014;202:578–82.
Bhoora R, Franssen L, Oosthuizen MC, Guthrie AJ, Zweygarth E, Penzhorn BL, et al. Sequence heterogeneity in the 18S rRNA gene within Theileria equi and Babesia caballi from horses in South Africa. Vet Parasitol. 2009;159:112–20.
Knowles DP, Kappmeyer LS, Haney D, Herndon DR, Fry LM, Munro JB, et al. Discovery of a novel species, Theileria haneyi n sp, infective to equids, highlights exceptional genomic diversity within the genus Theileria: implications for apicomplexan parasite surveillance. Int J Parasitol. 2018;48:679–90.
Bishop RP, Kappmeyer LS, Onzere CK, Odongo DO, Githaka N, Sears KP, et al. Equid infective Theileria cluster in distinct 18S rRNA gene clades comprising multiple taxa with unusually broad mammalian host ranges. Parasit Vectors. 2020;13:261.
Tirosh-Levy S, Gottlieb Y, Fry LM, Knowles DP, Steinman A. 20 years of equine piroplasmosis research: global distribution, molecular diagnosis, and phylogeny. Pathogens. 2020;9:926.
Coultous RM, Leadon DP, Shiels BR, Sutton D, Weir W. Investigating the presence of equine piroplasmosis in Ireland. Vet Rec. 2020;187:e97.
Camino E, Cruz-Lopez F, de Juan L, Dominguez L, Shiels B, Coultous RM. Phylogenetic analysis and geographical distribution of Theileria equi and Babesia caballi sequences from horses residing in Spain. Ticks Tick Borne Dis. 2020;11:101521.
Bhoora RV, Collins NE, Schnittger L, Troskie C, Marumo R, Labuschagne K, et al. Molecular genotyping and epidemiology of equine piroplasmids in South Africa. Ticks Tick Borne Dis. 2019;11:101358.
Peckle M, Pires MS, Silva CBD, Costa RLD, Vitari GLV, Senra MVX, et al. Molecular characterization of Theileria equi in horses from the state of Rio de Janeiro Brazil. Ticks Tick Borne Dis. 2018;9:349–53.
Qablan MA, Obornik M, Petrželková KJ, Sloboda M, Shudiefat MF, Horin P, et al. Infections by Babesia caballi and Theileria equi in Jordanian equids: epidemiology and genetic diversity. Parasitology. 2013;140:1096–103.
Weir W, Karagenc T, Gharbi M, Simuunza M, Aypak S, Aysul N, et al. Population diversity and multiplicity of infection in Theileria annulata. Int J Parasitol. 2011;41:193–203.
Hemmink JD, Sitt T, Pelle R, de Klerk-Lorist LM, Shiels B, Toye PG, et al. Ancient diversity and geographical sub-structuring in African buffalo Theileria parva populations revealed through metagenetic analysis of antigen-encoding loci. Int J Parasitol. 2018;48:287–96.
Coultous RM, McDonald M, Raftery AG, Shiels BR, Sutton DGM, Weir W. Analysis of Theileria equi diversity in The Gambia using a novel genotyping method. Transbound Emerg Dis. 2019;53:385.
Gotić J. Klinička i serološka dijagnostika te molekularna tipizacija uzročnika piroplazmoze konja na području republike hrvatske. Thesis. Faculty of veterinary medicine university of zagreb; 2015. https://www.bib.irb.hr/802430 Accessed 17 May 2022.
Beck R, Vojta L, Mrljak V, Marinculic A, Beck A, Zivicnjak T, et al. Diversity of Babesia and Theileria species in symptomatic and asymptomatic dogs in Croatia. Int J Parasitol. 2009;39:843–8.
Wise LN, Kappmeyer LS, Knowles DP, White SN. Evolution and diversity of the EMA families of the divergent equid parasites, Theileria equi and T. haneyi. Infect Genet Evol. 2019;68:153–60.
Kappmeyer LS, Perryman LE, Knowles DP. A Babesia equi gene encodes a surface protein with homology to Theileria species. Mol Biochem Parasitol. 1993;62:121–4.
Xuan X, Larsen A, Ikadai H, Tanaka T, Igarashi I, Nagasawa H, et al. Expression of Babesia equi merozoite antigen 1 in insect cells by recombinant baculovirus and evaluation of its diagnostic potential in an enzyme-linked immunosorbent assay. J Clin Microbiol. 2001;39:705–9.
Bhoora R, Quan M, Matjila PT, Zweygarth E, Guthrie AJ, Collins NE. Sequence heterogeneity in the equi merozoite antigen gene (ema-1) of Theileria equi and development of an ema-1-specific TaqMan MGB assay for the detection of T equi. Vet Parasitol. 2010;172:33–45.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res. 2004;32:1792–7.
Larsson A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics. 2014;30:3276–8.
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299–302.
Tamura K, Stecher G, Kumar S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021;38:3022–7.
Krčmar S, Klobučar A, Vucelja M, Boljfetić M, Kučinić M, Madić J, et al. DNA barcoding of hard ticks (Ixodidae), notes on distribution of vector species and new faunal record for Croatia. Ticks Tick Borne Dis. 2022;13:101920.
OIE (World Organisation for Animal Health). Chapter 3.6.8. Equine piroplasmosis. In: manual of diagnostic tests and vaccines for terrestrial animals. 2021. https://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/3.06.08_EQUINE_PIROPLASMOSIS.pdf Accessed 17 May 2022.
Short MA, Clark CK, Harvey JW, Wenzlow N, Hawkins IK, Allred DR, et al. Outbreak of equine piroplasmosis in Florida. J Am Vet Med Assoc. 2012;240:588–95.
Kumar S, Kumar R, Gupta AK, Yadav SC, Goyal SK, Khurana SK, et al. Development of EMA-2 recombinant antigen based enzyme-linked immunosorbent assay for seroprevalence studies of Theileria equi infection in Indian equine population. Vet Parasitol. 2013;198:10–7.
Acknowledgements
The authors thank all owners and veterinarians who provided equine samples for the study.
Funding
A bursary supporting MM and the costs of the ema-1 PCR and sequencing were funded by a summer scholarship from Beaufort Cottage Educational Trust. RC is currently supported by a Horserace Betting Levy Board research fellowship (VET/EPDF/2019–1).
Author information
Authors and Affiliations
Contributions
RC was involved in conceptualisation, data acquisition (laboratory), methodology, data analysis, visualisation, writing of original draft and manuscript review and editing. JG was involved in conceptualisation, data acquisition (fieldwork), writing of original draft and manuscript review and editing. MM was involved in methodology and data acquisition (laboratory). DS was involved in conceptualisation, manuscript review and editing. RB was involved in data acquisition (laboratory), methodology, manuscript review and editing. BS was involved in conceptualisation, methodology and manuscript review and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Committee of Ethics at the University of Zagreb, Faculty of Veterinary Medicine (Permit Number 640–01/14–17/60, 251–61-01/139–14-3).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Coultous, R., Gotić, J., McCann, M. et al. Novel equi merozoite antigen (ema-1) gene heterogeneity in a geographically isolated Theileria equi population in Croatia. Parasites Vectors 15, 401 (2022). https://doi.org/10.1186/s13071-022-05484-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05484-4