
Abstract
Background
Recent reports reveal the presence of Wolbachia in Ae. aegypti. Our study presents additional support for Wolbachia infection in Ae. aegypti by screening field-collected adult mosquitoes using two Wolbachia-specific molecular makers.
Methods
A total of 672 Ae. aegypti adult mosquitoes were collected from May 2014 to January 2015 in Metropolitan Manila. Each individual sample was processed and screened for the presence of Wolbachia by selected markers, Wolbachia-specific 16S rDNA and its surface protein (wsp), under optimized PCR conditions and sequenced.
Results
Totals of 113 (16.8%) and 89 (13.2%) individual mosquito samples were determined to be infected with Wolbachia using the wsp and 16S rDNA markers, respectively. The Ae. aegpyti wsp sample sequences were similar or identical to five known Wolbachia strains belonging to supergroups A and B while the majority of 16S rDNA sample sequences were similar to strains belonging to supergroup B. Overall, 80 (11.90%) individual mosquito samples showed positive amplifications in both markers and 69% showed congruence in supergroup identification (supergroup B).
Conclusions
By utilizing two Wolbachia-specific molecular makers, our study demonstrated the presence of Wolbachia from individual Ae. aegypti samples. Our results showed a low Wolbachia infection rate and inferred the detected strains belong to either supergroups A and B.
Similar content being viewed by others
Background
Wolbachia is a naturally occurring endosymbiont which can be maternally inherited and cause different reproductive alterations in its host to increase their transmission to the next generation [1,2,3]. In insects, it is estimated to be naturally present in 60–65% of known species [4]. Currently, there are 17 identified major clades or supergroups (A–Q), the majority of which are known to infect arthropods such as insects, arachnids and crustaceans [5]. The pathogenic effects of Wolbachia in its host are well-studied and determined to cause sperm-egg incompatibility, parthenogenesis, cytoplasmic incompatibility and feminization [2, 6]. Therefore, utilizing these effects in medically-important mosquito vectors, has resulted in significant progress in the past two decades.
Previous studies claimed that medically important mosquitoes such as Culex spp., Mansonia spp., and Aedes albopictus were naturally infected with Wolbachia, whereas Ae. aegypti was not [7,8,9,10,11,12]. A more recent global survey from 27 countries also established the absence of Wolbachia in Ae. aegypti [13]. However, numerous studies now contradict this claim and present evidence of natural Wolbachia infection in Ae. aegypti, including recent studies from Malaysia [14], India [15] and the USA [16]. The first ever report came from Ae. aegypti larval samples in Malaysia [14]; however, the sample size was too small (n = 16) to affirm such findings. Afterwards, metabarcoding studies by examining bacterial communities in the midgut of Ae. aegypti in the USA and Thailand reported a low presence of Wolbachia sequences [17, 18]. In 2019, evidence of natural Wolbachia infection in Ae. aegypti from India was presented, based on amplification of Wolbachia-specific 16S rRNA, wsp and ftsZ molecular markers [15]. This was followed by a report of Wolbachia presence Ae. aegypti populations in the USA, specifically from the states of New Mexico and Florida, using 16S rDNA, gatB, ftsZ and strain-specific (phosphoesterase and diaminopimelate epimerase) markers [16]. Both demonstrated the persistence of the endosymbiont across the developmental stages of Ae. aegypti through cytological examination and molecular detection. This clearly illustrates that the infection of Wolbachia in Ae. aegypti appears common than previously recognized.
This report provides additional support to the presence of Wolbachia in field-collected Ae. aegypti adult mosquitoes using Wolbachia-specific 16S rDNA and its surface protein (wsp). In comparison to previous studies, we conducted a large sampling of Ae. aegypti mosquitoes (n = 672) in a microgeographical area in order to discern the spatial distribution of Wolbachia-infected mosquitoes at the city scale and also to more accurately understand the infection rate in a natural Ae. aegypti population. Furthermore, our results focus primarily on the global phylogeny of Wolbachia strains within Ae. aegypti. In previous studies [15, 16], the Wolbachia strain isolated belonged to supergroup B or homologous to the strain from Ae. albopictus. Not only do our results conform to these previous findings, but they also reveal other prospective Wolbachia strains (e.g. supergroup A) infecting this mosquito.
Methods
Study area and mosquito collection
The study area was the National Capital Region of the Philippines, also known as Metropolitan Manila. Located on the eastern shore of Manila Bay in southwestern Luzon Island (14°35′58.2432″N, 121°59′3.1992″E), it is considered to be one of the most highly urbanized and densely populated areas in the Philippines. Dengue disease is endemic in this region where it accounted for 15–25% of the total number of reported dengue cases annually in the period 2009–2014 [19].
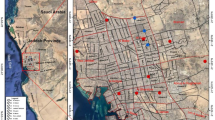
Adult mosquito samples were collected using a commercial branded mosquito UV-light trap (MosquitoTrap®; Jocanima Corporation, Las Piñas City, Philippines) installed in the outdoor premises of 138 residential households (sampling sites) from May 2014 to January 2015 (Fig. 1a). Collected mosquito samples were then sorted and identified as Ae. aegypti using available keys [20]. This was then placed in a tube with 99.5% ethanol for preservation. In total, 672 Ae. aegypti adult mosquito samples were collected, identified, labeled (see Additional file 1: Table S1) and stored at − 20 °C for subsequent processing.

a Spatial distribution of the sampling sites (n = 138) for collecting adult Ae. aegypti. Wolbachia-positive sampling sites (circles) based on b wsp (triangles) and c 16S rDNA (squares). Details of the number of Wolbachia-positive mosquitoes per sampling site are provided in Additional file 1: Table S1
DNA extraction, PCR amplification and sequencing
Total genomic DNA of each mosquito individual was extracted using a Blood and Tissue DNEasy Kit© (Qiagen, Hilden, Germany) following a modified protocol [21]. Our study used two molecular markers for detecting Wolbachia infection, namely wsp [22] and 16S rDNA [23]. The primer sequences were wsp 81F (5′-TGG TCC AAT AAG TGA TGA AGA AAC-3′) and wsp 691R (5′-AAA AAT TAA ACG CTA CTC CA-3′) for the wsp marker and the 16S Wolbachia-specific primers were WolbF (5′-GAA GAT AAT GAC GGT ACT CAC-3′) and Wspecr (5′-AGC TTC GAG TGA AAC CAA TTC-3′).
For the wsp gene amplification, we followed the standard wsp protocol [11] where the suggested annealing temperature and number of cycles were 55 °C and 30 cycles, respectively. To conduct an individual-based detection, we initially performed this protocol using Culex quinquefasciatus as our positive control. Certain modifications were made in the standard protocol based on these results. The annealing temperature was increased to 57 °C and the number of cycles was increased to 35 cycles. This initial modified protocol was performed in individual Ae. aegypti samples where it yielded positive faint bands. As a result, we modified the protocol again, setting the annealing temperature at 59 °C with 40 cycles, and adding 10% DMSO (Sigma-Aldrich, St. Louis, Missouri, USA). This led to desirable results necessary for sequencing. In the end, a 10 μl final reaction volume was used consisting of 10× buffer (TaKaRa, Shiga, Japan), 25 mM MgCl2, 10 mM of each dNTPs, 10 μM forward and reverse primers, 10% DMSO (Sigma-Aldrich) and 5.0 U/μl of Taq DNA polymerase (TaKaRa). The final thermal profile was as follows: initial denaturation at 95 °C for 3 min; 40 cycles of denaturation at 95 °C for 1 min, annealing at 59 °C for 1 min and extension at 72 °C for 1 min; final extension at 72 °C for 3 min.
For the 16S rDNA gene amplification, we used a 10 μl final reaction volume consisting of 10× buffer (TaKaRa), 25 mM MgCl2, 10 mM of each dNTPs, 10 μM forward and reverse primers, 10% DMSO (Sigma-Aldrich) and 5.0U/μl of Taq DNA polymerase (TaKaRa). Thermal profiles followed the protocol of Simões et al. [23]: initial denaturation at 95 °C for 2 min; two cycles of denaturation at 95 °C for 2 min, annealing at 60 °C for 1 min and extension at 72 °C for 1 min; 35 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 1 min and extension at 72 °C for 45 s; final extension at 72 °C for 10 min.
All PCR amplification experiments included positive and negative controls. The positive control was a Wolbachia-infected Cx. quinquefasciatus sample while the negative control was water. The product size of each molecular marker was checked through 1.5% agarose gel electrophoresis set at 100 V for 30 min. The size of the amplified wsp gene is approximately 610 bp while the 16S rDNA gene is approximately 850 bp. The PCR amplification process underwent two replicates to validate the results (see Additional file 1: Table S1). A third screening was performed for selected individual samples that had inconsistent results based on the two prior replicates. The criteria set to confirm Wolbachia infection were based on two successful amplifications of the molecular markers. Furthermore, individual samples that met this criterion were subjected for sequencing through Eurofins Genomics, Tokyo.
Identity of Wolbachia strains and their positions in phylogroups
All sequences were subjected to the Nucleotide Basic Local Alignment Search Tool (BLAST) and compared to deposited Wolbachia sequences in GenBank. The selected sequences of Wolbachia strains (Table 1) and those obtained in the study then underwent multiple alignment using Clustal W in MEGA 6 [24]. After editing, the final lengths used for phylogenetic inference analyses were 398 and 721 bp for wsp and 16S rDNA, respectively. The identities and relationships of the Wolbachia strains obtained in our study were determined by performing Bayesian inference analysis using PhyML v.3.0 software with 1000 bootstrap replicates [25]. Smart Model Selection [26] was also utilized to set the parameters for wsp as GTR+G (number of estimated parameters k= 232, Akaike information criterion (AIC)= 4897.31702) and 16S rDNA as GTR+G+1 (number of estimated parameters k= 207, AIC= 5332.88688). All newly generated sequences were submitted to the GenBank database with accession numbers MN046588–MN046789.
Statistical analysis
A Clark-Evans test was performed to determine whether the spatial distribution of Wolbachia-positive mosquito samples from each molecular marker had a pattern of complete spatial randomness. The test uses the aggregation index (R), where a value > 1 suggests an ordered distribution and a value < 1 suggests clustering. This analysis was performed using R v.3.3.5 (package spatstat) [27].
Results
Detection of Wolbachia through wsp and its phylogeny
From a total of 672 adult Ae. aegypti screened using the wsp marker, 113 (16.8%) individual adult mosquito samples were positive for Wolbachia infection (Table 2) based on the study criteria (see Methods). Other than the positive individual adult mosquito samples, there were also 17 individual samples that produced one successful wsp amplification; however, these were excluded in reporting the prevalence and further analysis. The female/male ratio was 0.82 (Table 2). All sequenced amplicons resulted in a high degree of similarity (> 98.0%) with the wsp sequences in GenBank. The spatial distribution showed that 60 (43.0%) sampling sites (Fig. 1b) contained Wolbachia infected mosquitoes. Positive sampling sites had prevalence rates ranging between 7.69–100%. Further analysis showed that the distribution of wsp-positive mosquito samples was significantly clustered (R = 0.003, P < 0.001). The wsp phylogeny indicated that majority of the sequences belong to supergroup B (n = 84) while the remaining were in supergroup A (n = 29) (Fig. 2 and Additional file 2: Figure S1). Based on descending order of sample sizes, sample sequences from supergroup B were identical (> 99.0%) to Wolbachia type strains from selected hosts such as Ae. albopictus (wAlbB), Ae. aegypti (wAegB) (n = 51), Cx. quinquefasciatus, Cx. pipiens (wPip) (n = 23) and Ephestia cautella (wCau) (n = 10). The sample sequences from supergroup A were either similar to (98.0–99.0%) (n = 8) or identical (> 99.0%) (n = 21) with the Wolbachia strain (wAlbA) found in Ae. albopictus.

Phylogenic analysis based on wsp gene. The alignment was analyzed in PhyML. Sample sequences of Ae.aegypti collected in Metropolitan Manila are in red, labeled as AAML (Ae. aegypti Metropolitan ManiLa) and alphanumeric values indicate the unique code assigned to each Ae. aegypti individual sample. Merging (gray triangles) of sample and representative Wolbachia sequences was done to show degree of similarity (98–100%). Supergroups are indicated as A–C depending on the representative sequences used. The phylogenetic trees are re-drawn for better visualization; the expanded version is provided in Additional file 2: Figure S1. Please refer to Table 1 for the Wolbachia type sequences (ingroup and outgroup) for both markers
Detection of Wolbachia through 16S rDNA and its phylogeny
For the 16S rDNA, 89 (13.2%) individual adult mosquito samples were infected with Wolbachia (Table 2) based on the study criteria. In addition to these, 20 individual mosquito samples generated one successful 16S rDNA amplification, but were excluded in reporting the prevalence and further analysis. The female/male ratio was 0.85 (Table 2). Fifty (36.0%) sampling sites (Fig. 1c) contained Wolbachia-infected mosquitoes. Positive sampling sites had prevalence rates ranging from 3.9 to 100%. The distribution of 16S rDNA-positive individuals was revealed to be clustered or aggregated (R = 0.001, P < 0.001). All sequenced amplicons resulted in a high degree of similarity (> 98%) with 16S rDNA Wolbachia sequences in GenBank. Nearly all 16S rDNA sample sequences (n = 84) (Fig. 3, Additional file 3: Figure S2) belonged to supergroup B. Only one sample sequence was identical to the endosymbiont found in Nasonia vitripennis while 27 sample sequences were identical to Wolbachia isolated from Ae. aegypti. The remaining sample sequences from supergroup B were 99% similar from selected hosts of the supergroup. Five sample sequences were grouped together with Wolbachia hosts in supergroups C, D and J. Only one sample sequence was highly similar (> 99%) to Dirofilaria immitis while the remaining were 98–99% similar to the selected hosts of the supergroup.

Phylogenic analysis based on 16S rDNA. The alignment was analyzed in PhyML. Sample sequences of Ae.aegypti collected in Metropolitan Manila are in red, labeled as AAML (Ae. aegypti Metropolitan ManiLa) and alphanumeric values indicate the unique code assigned to each Ae. aegypti individual sample. Merging (gray triangles) of sample and representative Wolbachia sequences was done to show degree of similarity (98–100%). Supergroups are indicated as A to J depending on the representative sequences used. The phylogenetic trees are re-drawn for better visualization; the expanded version is provided in Additional file 3: Figure S2. Please refer to Table 1 for the Wolbachia type sequences (ingroup and outgroup) for both markers
Comparison of 16S rDNA and wsp for Wolbachia detection and phylogeny
A total of 80 (11.90%) individual samples yielded positive amplification in both markers (Table 2). In the wsp positive samples (n = 113), 80 had two successful amplifications of the 16S rDNA amplification while 27 had only one successful 16S rDNA amplification and the remaining 6 had no successful 16S rDNA amplification. For the 16S rDNA positive samples (n = 89), there were 80 individuals with two successful wsp amplification, while 9 had only one successful wsp amplification. We then focused on the supergroup classification of the 80 individual samples based on the wsp and 16S rDNA phylogeny. It was found that 55 samples (69%) belonged to supergroup B while the remaining 25 samples (31%) showed disparity. In certain instances, wsp identified an individual sample as supergroup A, but 16S rDNA revealed it as either supergroup B, C or J.
Discussion
In our study, we found a low infection rate (11%) of Wolbachia in the Ae. aegypti population studied. This finding coincides with the low infection rate reported in Florida [16]; however, a higher infection rate (> 50%) was observed in Ae. aegypti populations in Malaysia [14] and New Mexico [16]. It has been established that it is common to see varying Wolbachia infection rates of the same insect host from different geographical locations such as that observed in Cx. quienquefasciatus [28, 29] and Cx. pipiens [30,31,32]. The variation of infection rates could be driven by either genetic or environmental factors [16]. Wolbachia density in Ae. albopictus tends to decrease if exposed to increasing temperatures [33]. Removal of the endosymbiont from its host could be achieved by exposure to heat treatment or even antibiotics [34, 35]. The observed low infection rate could be attributed to the low density of the endosymbiont in Ae. aegypti. This is further supported by metabarcoding studies which yielded a low number (2–10) [16,17,18] of sequence reads in the midgut of Ae. aegpyti which indicate a low probable density of the endosymbiont. Although our study did not measure the actual density, a 40-cycle PCR amplification procedure or long PCR run [36] was needed to amplify and confirm a positive infection of the endosymbiont in our Ae. aegypti samples.
Based on the results of our phylogenetic analysis, the Wolbachia strains found in our sampled Ae. aegypti belong to supergroups A and B. Both wsp and 16S rDNA phylogeny showed that the majority of the individual samples belong to supergroup B while a small number of individual samples belong to supergroup A (based on wsp). The same observation has been reported in previous studies [14,15,16,17,18]. Detecting different Wolbachia strains in a single mosquito species is relatively common, especially in medically-important mosquitoes such as Ae. albopictus [22, 37], An. gambiae [38] and other insect host species (e.g. Drosophila species [37]). Dipterans, especially mosquitoes, are commonly infected by Wolbachia strains from supergroups A and B. They have been shown to cause parasitism towards the insect host by producing phenotypic effects such as cytoplasmic incompatibility, male killing and feminization [39]. However, it remains unclear whether the identified Wolbachia strains in Ae. aegypti induce these phenotypic effects. Further studies are needed to confirm the pathogenic impact of this endosymbiont to the mosquito vector. It is also important to determine whether these identified Wolbachia strains could inhibit the replication of arboviruses such as dengue, rendering Ae. aegypti a less effective vector. A few individual samples (n = 5) were shown to be similar to Wolbachia strains found in supergroups C, D and J based on 16S rDNA. It is likely that our 16S rDNA amplified the Wolbachia strain found in the roundworm, Dirofilaria immitis, a parasitic nematode that Ae. aegpyti mosquitoes also carry and transmit to certain mammals, such as dogs [40]. This observation was also reported in one of the metabarcoding studies that showed sequences of Wolbachia from Dirofilaria immitis. However, when these 16S rDNA results were compared to the wsp results in our study, it showed the Wolbachia wsp sample sequence of the same mosquito individuals belonged to supergroup B. We assume that this discordance may stem from the different mutation rates of the markers used. 16S rDNA is known to be a conserved gene; however, in some instances the typing system of this marker has been shown to be insufficient in establishing correct supergroup classification due to its low evolutionary rate [41]. This indicates a potential drawback of 16S rDNA as a less robust marker in estimating intraspecific phylogenetic relationship among Wolbachia supergroup members.
Previous studies reported the non-detection of Wolbachia in Ae. aegypti which is in contrast with both our results and with recent Wolbachia detection reports in this mosquito vector from India, Malaysia and the USA [14,15,16]. The reasons for these contrasting observations could be attributed to the following: (i) individual vis-à-vis pooled detection assays; (ii) procedural modifications; and (iii) sample size. Kulkarni et al. [16] emphasized that individual screening is more suitable in detecting Wolbachia in Ae. aegypti due to the low density load of the endosymbiont in the mosquito vector. They tested the sensitivity of a PCR assay containing a pool of 19 Wolbachia-negative individuals and one positive individual each from Ae. albopictus and Ae. aegypti. The results showed that Wolbachia could be detected in a pool containing DNA from a single positive Ae. albopictus but not in a pool containing DNA from a single Ae. aegypti specimen. These results strengthen the notion that Wolbachia prevalence studies in Ae. aegypti could be inaccurate due to pooled detection assays. Non-optimal DNA amplification and extraction methods could also compromise the results of detection assays. This was demonstrated and emphasized in studies detecting Wolbachia from An. gambiae [38, 42, 43]. Our study conducted a longer PCR run (i.e. 40 PCR cycles) as compared to the general protocol just to produce satisfactory positive bands in gel electrophoresis. Due to the varying and potentially low infection rate of Ae. aegypti observed in different studies including ours, a larger sample size would provide a more accurate estimate of its prevalence. Assessing several reports of the prevalence of Wolbachia published before 2017 showed that the greatest number of individuals screened was 119 [11], resulting in the non-detection of the endosymbiont. This sample size is very small compared to our study which screened 672 individual mosquitoes, while similar studies screened 288–554 mosquito individuals. In a more recent report in 2018, 2663 Ae. aegypti mosquitoes were screened from 27 countries, yet the results showed no presence of Wolbachia in these samples [13] However, the screening of Wolbachia in this global survey was done in pools consisting up to 20 individuals, which may have compromised the detection of this endosymbiont in Ae. aegypti as previously discussed by Kulkarni et al. [16].
Our study acknowledges the uncertainties associated with conventional PCR detection such as high false positive detection rates. With this in mind, we were cautious in affirming a positive infection in each Ae. aegypti adult sample. First, the selection of markers is based on the study of Simoes et al. [23] which produced low false positive and false negative rates. Secondly, our study performed replications with strict criteria for a successful Wolbachia infection in each mosquito sample. Additionally, a similar study conducted in the USA [16] identified individual mosquito samples with Wolbachia by conducting two rounds of PCR detection. Although there are several genetic markers (e.g. MLST genes) and techniques (e.g. IFA, FISH or whole-genome sequencing) available, this short report is limited in presenting the possible detection of Wolbachia using a conventional PCR-based approach. We are conducting similar experiments (see recent studies [15, 16]) to substantiate the infection status of Wolbachia in this mosquito vector. Mosquito colonies are now being reared in order to establish the maternal inheritance and persistence of Wolbachia infection through different mosquito developmental stages and generations.
Conclusions
The study demonstrated the detection of Wolbachia from field-collected Ae. aegypti in Metropolitan Manila, Philippines. Totals of 113 (16.8%) and 89 (13.2%) individual mosquito samples were determined to be infected with Wolbachia using the wsp and 16S rDNA markers, respectively. Overall, 80 (11.90%) individual mosquito samples showed positive amplifications in both markers, indicating a low infection rate. Our study supports previous studies that the potential Wolbachia strain in Ae. aegypti belongs to supergroup B. In addition, other Wolbachia strains (e.g. supergroup A) could potentially infect this mosquito vector.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files. All newly generated sequences are available in the GenBank database under the Accession Numbers MN046588–MN046789.
References
Serbus LR, Casper-Lindley C, Landmann F, Sullivan W. The genetics and cell biology of Wolbachia-host interactions. Annu Rev Genet. 2008;42:683–707.
Werren JH, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741.
Saridaki A, Bourtzis K. Wolbachia: more than just a bug in insects genitals. Curr Opin Microbiol. 2010;13:67–72.
Rasgon JL. Using predictive models to optimize Wolbachia-based strategies for vector-borne disease control. In: Aksoy S, editor. Transgenesis and the management of vector-borne disease. New York: Springer; 2008. p. 114–25.
Glowska E, Dragun-Damian A, Dabert M, Gerth M. New Wolbachia supergroups detected in quill mites (Acari: Syringophilidae). Infect Genet Evol. 2015;30:140–6.
Ahmed MZ, Breinholt JW, Kawahara AY. Evidence for common horizontal transmission of Wolbachia among butterflies and moths. BMC Evol Biol. 2016;16:118.
Kittayapong P, Baisley KJ, Baimai V, O’Neill SL. Distribution and diversity of Wolbachia infections in Southeast Asian mosquitoes (Diptera: Culicidae). J Med Entomol. 2000;37:340–5.
Ricci I, Cancrini G, Gabrielli S, D’amelio S, Favia G. Searching for Wolbachia (Rickettsiales: Rickettsiaceae) in mosquitoes (Diptera: Culicidae): large polymerase chain reaction survey and new identifications. J Med Entomol. 2002;39:562–7.
Rasgon JL, Scott TW. Impact of population age structure on Wolbachia transgene driver efficacy: ecologically complex factors and release of genetically modified mosquitoes. Insect Biochem Mol Biol. 2004;34:707–13.
Hilgenboecker K, Hammerstein P, Schlattmann P, Telschow A, Werren JH. How many species are infected with Wolbachia?–a statistical analysis of current data. FEMS Microbiol Lett. 2008;281:215–20.
Wiwatanaratanabutr I. Geographic distribution of wolbachial infections in mosquitoes from Thailand. J Invertebr Pathol. 2013;114:337–40.
Nugapola NWNP, De Silva WAPP, Karunaratne SHPP. Distribution and phylogeny of Wolbachia strains in wild mosquito populations in Sri Lanka. Parasit Vectors. 2017;10:230.
Gloria-Soria A, Chiodo TG, Powell JR. Lack of evidence for natural Wolbachia infections in Aedes aegypti (Diptera: Culicidae). J Med Entomol. 2018;55:1354–6.
Teo CH, Lim PK, Voon K, Mak JW. Detection of dengue viruses and Wolbachia in Aedes aegypti and Aedes albopictus larvae from four urban localities in Kuala Lumpur, Malaysia. Trop Biomed. 2017;34:583–97.
Balaji S, Jayachandran S, Prabagaran SR. Evidence for the natural occurrence of Wolbachia in Aedes aegypti mosquitoes. FEMS Microbiol Lett. 2019;366:fnz055.
Kulkarni A, Yu W, Jiang J, et al. Wolbachia pipientis occurs in Aedes aegypti populations in New Mexico and Florida, USA. Ecol Evol. 2019;9:6148–56.
Coon KL, Brown MR, Strand MR. Mosquitoes host communities of bacteria that are essential for development but vary greatly between local habitats. Mol Ecol. 2016;25:5806–26.
Thongsripong P, Chandler JA, Green AB, Kittayapong P, Wilcox BA, Kapan DD, Bennett SN. Mosquito vector-associated microbiota: Metabarcoding bacteria and eukaryotic symbionts across habitat types in Thailand endemic for dengue and other arthropod-borne diseases. Ecol Evol. 2018;8:1352–68.
Carvajal TM, Viacrusis KM, Hernandez LF, Ho HT, Amalin DM, Watanabe K. Machine learning methods reveal the temporal pattern of dengue incidence using meteorological factors in metropolitan Manila, Philippines. BMC Infect Dis. 2018;18:183.
Rueda LM. Pictorial keys for the identification of mosquitoes (Diptera: Culicidae) associated with dengue virus transmission. Zootaxa. 2004;589:1–60.
Crane S. DNA extraction from archival museum insect specimens. 2011. https://s3-eu-west-1.amazonaws.com/pfigshare-u-files/1114092/extractionmuseum.pdf. Accessed 10 Mar 2015
Zhou W, Rousset F, OʼNeill S. Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc Biol Sci. 1998;265:509–15.
Simoes PM, Mialdea G, Reiss D, Sagot MF, Charlat S. Wolbachia detection: an assessment of standard PCR protocols. Mol Ecol Resour. 2011;11:567–72.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Guindon S, Dufayard J, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Lefort V, Longueville JE, Gascuel O. SMS: smart model selection in PhyML. Mol Biol Evol. 2017;34:2422–4.
R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2016.
Sunish IP, Rajendran R, Paramasivan R, Dhananjeyan KJ, Tyagi BK. Wolbachia endobacteria in a natural population of Culex quinquefasciatus from filariasis endemic villages of south India and its phylogenetic implication. Trop Biomed. 2011;28:569–76.
Carvajal TM, Capistrano JD, Hashimoto K, Go KJ, Cruz MA, Martinez MJ, et al. Detection and distribution of Wolbachia endobacteria in Culex quinquefasciatus populations (Diptera: Culicidae) from Metropolitan Manila, Philippines. J Vector Borne Dis. 2018;55:265.
Vinogradova EB, Shaikevich EV, Ivanitsky AV. A study of the distribution of the Culex pipiens complex (Insecta: Diptera: Culicidae) mosquitoes in the European part of Russia by molecular methods of identification. Comp Cytogenet. 2007;1:129–38.
Rasgon JL, Scott TW. Wolbachia and cytoplasmic incompatibility in the California Culex pipiens mosquito species complex: parameter estimates and infection dynamics in natural populations. Genetics. 2003;165:2029–38.
Chen L, Zhu C, Zhang D. Naturally occurring incompatibilities between different Culex pipiens pallens populations as the basis of potential mosquito control measures. PLoS Negl Trop Dis. 2013;7:e2030.
Wiwatanaratanabutr I, Kittayapong P. Effects of temephos and temperature on Wolbachia load and life history traits of Aedes albopictus. Med Vet Entomol. 2006;20:300–7.
Hermans PG, Hart CA, Trees AJ. In vitro activity of antimicrobial agents against the endosymbiont Wolbachia pipientis. J Antimicrob Chemother. 2001;47:659–63.
Van Opijnen T, Breeuwer JA. High temperatures eliminate Wolbachia, a cytoplasmic incompatibility inducing endosymbiont, from the two-spotted spider mite. Exp Appl Acarol. 1999;23:871–81.
Jeyaprakash A, Hoy MA. Long PCR improves Wolbachia DNA amplification: wsp sequences found in 76% of sixty-three arthropod species. Insect Mol Biol. 2000;9:393–405.
Kitrayapong P, Baimai V, O’Neill SL. Field prevalence of Wolbachia in the mosquito vector Aedes albopictus. Am J Trop Med Hyg. 2002;66:108–11.
Baldini F, Segata N, Pompon J, Marcenac P, Shaw WR, Dabiré RK, et al. Evidence of natural Wolbachia infections in field populations of Anopheles gambiae. Nat Commun. 2014;5:3985.
Ellegaard KM, Klasson L, Näslund K, Bourtzis K, Andersson SG. Comparative genomics of Wolbachia and the bacterial species concept. PLoS Genet. 2013;9:e1003381.
Kramer L, Grandi G, Leoni M, Passeri B, McCall J, Genchi C, et al. Wolbachia and its influence on the pathology and immunology of Dirofilaria immitis infection. Vet Parasitol. 2008;158:191–5.
Ma Y, Chen WJ, Li ZH, Zhang F, Gao Y, Luan YX. Revisiting the phylogeny of Wolbachia in Collembola. Ecol Evol. 2017;7:2009–17.
Shaw WR, Marcenac P, Childs LM, Buckee CO, Baldini F, Sawadogo SP, et al. Wolbachia infections in natural Anopheles populations affect egg laying and negatively correlate with Plasmodium development. Nat Commun. 2016;7:11772.
Gomes FM, Hixson BL, Tyner MDW, Ramirez JL, Canepa GE, Silva TL, et al. Effect of naturally occurring Wolbachia in Anopheles gambiae sl mosquitoes from Mali on Plasmodium falciparum malaria transmission. Proc Natl Acad Sci USA. 2017;114:12566–71.
Acknowledgements
We would like to thank M. J. L. B. Martinez, J. D. R. Capistrano, V. S. P. Tiopianco, B. M. C Orantia, C. R. Estrada, M. G. Cuenca, K. M. Viacrusis and L. F. T. Hernandez for their valuable help in the collection of mosquitoes.
Funding
This work was funded by the Japanese Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research (16H05750, 17H01624, 17K18906), JSPS Bilateral Joint Research Projects, JSPS Core-to-Core programme (Asia-Africa Science Platforms), Leading Academia in Marine, Environmental Pollution Research (LaMER), Ehime University (Y29-1-8), The Sumitomo Electric Industries Group Corporate Social Responsibility Foundation and the Research Unit Programme of Ehime University.
Author information
Authors and Affiliations
Contributions
TMC, DMA and KW conceptualized and designed the experiment. TMC and DMA collected and identified the adult mosquito samples for the study. TMC, KH and RKH conducted the detection process using a molecular approach. TMC, DMA and KW performed the analysis. TMC and KW wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Demographic profile (sex, sampling site code, location), detection status (wsp and 16S rDNA) of all individual adult Aedes aegypti mosquitoes used in the study and their supergroup classification.
Additional file 2: Figure S1.
Complete wsp phylogeny of Wolbachia from Ae. aegypti (n = 113). The alignment was analyzed in the program PHYML and Wolbachia host Dirofilaria immitis and Brugia malayi were selected as outgroups. All sample sequences are indicated as red dots. The condensed version of this tree is presented in Fig. 2.
Additional file 3: Figure S2.
Complete 16S rDNA phylogeny of Wolbachia from Ae. aegypti (n = 85). The alignment was analyzed in the program PHYML and Rickettsia sp. was selected as an outgroup. All sample sequences are indicated as red dots. The condensed version of this tree is presented in Fig. 3.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Carvajal, T.M., Hashimoto, K., Harnandika, R.K. et al. Detection of Wolbachia in field-collected Aedes aegypti mosquitoes in metropolitan Manila, Philippines. Parasites Vectors 12, 361 (2019). https://doi.org/10.1186/s13071-019-3629-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3629-y