
Abstract
Background
Soil microbial fuel cells (MFCs) can remove antibiotics and antibiotic resistance genes (ARGs) simultaneously, but their removal mechanism is unclear. In this study, metagenomic analysis was employed to reveal the functional genes involved in degradation, electron transfer and the nitrogen cycle in the soil MFC.
Results
The results showed that the soil MFC effectively removed tetracycline in the overlapping area of the cathode and anode, which was 64% higher than that of the control. The ARGs abundance increased by 14% after tetracycline was added (54% of the amplified ARGs belonged to efflux pump genes), while the abundance decreased by 17% in the soil MFC. Five potential degraders of tetracycline were identified, especially the species Phenylobacterium zucineum, which could secrete the 4-hydroxyacetophenone monooxygenase encoded by EC 1.14.13.84 to catalyse deacylation or decarboxylation. Bacillus, Geobacter, Anaerolinea, Gemmatirosa kalamazoonesis and Steroidobacter denitrificans since ubiquinone reductase (encoded by EC 1.6.5.3), succinate dehydrogenase (EC 1.3.5.1), Coenzyme Q-cytochrome c reductase (EC 1.10.2.2), cytochrome-c oxidase (EC 1.9.3.1) and electron transfer flavoprotein-ubiquinone oxidoreductase (EC 1.5.5.1) served as complexes I, II, III, IV and ubiquinone, respectively, to accelerate electron transfer. Additionally, nitrogen metabolism-related gene abundance increased by 16% to support the microbial efficacy in the soil MFC, and especially EC 1.7.5.1, and coding the mutual conversion between nitrite and nitrate was obviously improved.
Conclusions
The soil MFC promoted functional bacterial growth, increased functional gene abundance (including nitrogen cycling, electron transfer, and biodegradation), and facilitated antibiotic and ARG removal. Therefore, soil MFCs have expansive prospects in the remediation of antibiotic-contaminated soil. This study provides insight into the biodegradation mechanism at the gene level in soil bioelectrochemical remediation.
Similar content being viewed by others

Background
The widespread use and sustained release of antibiotics pose an enormous threat to the ecological environment, leading to the occurrence of bacterial resistance through antibiotic resistance genes (ARGs) [1, 2]. Global analysis of 1088 soil metagenomic samples found that hot spots of microbial resistance are mainly located in densely populated areas with developed agriculture and animal husbandry, such as the eastern United States, western Europe, South Asia and East Asia [3]. Moreover, antibiotics have been discovered in children and pregnant women [4, 5], 93% of the elderly are harmed by antibiotics [6], and 12% of newborns are diagnosed with invasive bacterial infections [7], which poses a serious threat to human health.
Microbial fuel cells (MFCs) have been confirmed to remove antibiotics and ARGs in wastewater [8,9,10] and soils [11,12,13]. For instance, Xu et al. [14] reported that the removal rate of sulfamethoxazole in wastewater reached 94% using MFC-constructed wetlands. Our previous research showed that 42–50% of tetracycline in soil MFCs could be removed within 7 days, while its degradation rate in control soil was only 6% [13]. Furthermore, the abundance of ARGs in soil MFCs declined by 19–27% compared with the control [13]. However, the heterogeneity of soil leads to difficulties of the MFC in remediation of antibiotic-contaminated soil compared to that of the water environment; therefore, few studies have reported on the removal mechanism of antibiotics and ARGs in soil using MFCs [15].
Microorganisms are important members of the soil system and are crucial in the degradation of pollutants in soils [16]. Soil MFCs can reshape the interspecific relationship of microorganisms and establish a microbial metabolic network with the ability to efficiently degrade antibiotics while also reducing the number of resistant microbes [17]. At present, many potential degraders of antibiotics have been reported, such as Bacillus sp., Shewanella sp., Sphingomonas sp., Phenylobacterium sp., and Paraclostridium sp. [18,19,20,21]. Unfortunately, most microorganisms are uncultivable, which greatly limits the isolation of degrading microbes [22]. It has become a potential pollution remediation strategy to identify functional genes with antibiotic degradation abilities and construct genetically engineered bacteria. However, to our knowledge, few studies have explained the degradation mechanism of antibiotics by soil MFCs based on functional genes, which requires further study.
The shortage of available nitrogen in organic-contaminated soil is the main limiting factor of bioremediation [23]. As is well known, nitrogen availability is closely related to the nitrogen metabolism, whereas it is restrained by antibiotics. For example, sulfadiazine inhibited nitrification functional genes and nitrobacteria in the surface sediment, resulting in NH4+ and NO2− accumulation in overlying water [24]. The presence of oxytetracycline, sulfamethazine, and ciprofloxacin restrained urea decomposition and denitrification by reducing the abundance of functional genes, including ureC, nirK and norB, in soil [25]. Interestingly, soil MFCs promoted cathode-dominated ammoniation and anode-dominated denitrification while degrading petroleum hydrocarbons [26]. However, thus far, it is unclear whether soil MFCs promote nitrogen cycling during antibiotic removal and which key functional genes are involved.
Previous studies showed that the bioelectricity generated by soil MFCs could stimulate the growth of functional microbes, thus promoting the degradation of pollutants [13, 27, 28]. This is mainly because the metabolism of organic matter is accelerated through electron transfer, hence it is necessary to study the electron transfer process in soil MFCs. Bidirectional extracellular electron transfer (EET), namely, outwards EET and inwards EET, is regarded as the key for the electrochemical activity of electrically active bacteria [29]. In soil MFCs, outwards EET normally occurs at the anode, and the cathode serves as a sustained electron donor for electrotrophic bacteria to conduct inwards EET [13]. Zhang et al. [30] found that conjugated polymers improved the bidirectional EET efficiency by the close biointerface interactions of conjugated polymer-microbe biohybrid systems. Riboflavin is also conducive to enhancing bidirectional EET, and its mechanism is determined by the EET direction: for outwards EET, free riboflavin serves as a redox mediator; for inwards EET, bound riboflavin is involved in electricity consumption [31]. Recently, the impact of adding exogenous substances to electron transfer in soil MFCs has received widespread attention. Chen et al. [32] reported that the addition of insulative ferrihydrite in a soil MFC generated more bioelectricity than conductive magnetite, possibly because ferrihydrite was turned into small particles of semiconductive lepidocrocite/goethite, which was likely to promote long-distance electron transfer. Zhang et al. [33] showed that both dissolved (Fe2+) and solid-state (Fe2O3) electron media acted as electron transporters in soil MFCs. Moreover, the possible electron transfer pathways in soil MFCs also need to be further studied.
In this study, tetracycline was selected to study soil MFCs with the following aims: first, to identify potential degrading bacteria and functional genes to reveal the biodegradation mechanism of tetracycline by soil MFCs; second, to explore the electrically active bacteria and functional genes involved in electron transfer in soil MFCs to speculate on probable electron transfer pathways; and third, to reveal functional genes involved in the nitrogen cycle of soil MFCs and their contribution to tetracycline removal.
Results and discussion
Reciprocal action of the anode and cathode was more conducive to tetracycline removal
The anode (area A) of the soil MFC treatment (MT) showed superior removal capacity for tetracycline, with a degradation rate of 76%, which increased by 34% (p < 0.05) compared with that in the corresponding area of the anaerobic controls spiked with tetracycline (AT) (Fig. 1). To our knowledge, most studies have mainly focused on the degradation mechanism of tetracycline at the MFC anode, while fewer studies have studied the removal mechanism of tetracycline at the cathode. In this study, a similar degradation rate was found between the cathode and anode of the soil MFC, suggesting great potential for the cathode removal of tetracycline [13, 17]. Wang et al. [34] found that electroactive bacteria (Rhodopseudomonas sp. and Acetobacter sp.) enriched on biocathodes accelerated electron transfer, thereby enhancing the metabolic activity of degraders and ultimately promoting the removal of methyl, hydroxyl, dimethyl, and amide groups on tetracycline. Our previous study also showed that flavoprotein 2,3-oxidoreductase, quinol oxidase and fumarate reductase encoded by EC 1.3.8.7, EC 1.10.3.14 and EC 1.3.5.4, respectively, might promote the electron transfer efficiency from the cathode to the cell, thus strengthening tetracycline removal [13]. Interestingly, in the soil MFC, the highest degradation rate of tetracycline was found in area C (the overlap area of the cathode and anode, 87%), which was 14–72% higher (p < 0.05) than other regions of the MFC (Fig. 1). The reason might be that the biodegradation of tetracycline in the C region of the MFC was influenced by both the cathode and anode. In contrast, the tetracycline removal in area B was the lowest but 36% higher (p < 0.05) than the corresponding area of the AT treatment.

Contents (column graph) and degradation rates (scatter graph) of tetracycline in different treatments. Different lowercase letters represent significant differences at the 0.05 level. The soil MFC and its anaerobic control spiked with tetracycline were labelled MT and AT, respectively
Soil bacterial community
Anodes dominated the bacterial diversity of the soil MFC
A total of 78,552–78,879 effective tags with average lengths of 254 bp were obtained (Additional file 1: Table S1). The coverage indices were all above 98%, suggesting that the sequencing depths could reflect the real situation of the bacterial community (Additional file 1: Table S1). Compared with the anaerobic controls without antibiotic (AN), the richness declined more obviously (12%) than diversity (4%) in terms of Shannon and Chao1 indices in the AT treatment (Additional file 1: Table S1). In contrast, these two indices in the MT treatment were comparable to those in the AT treatment. In the soil MFC, the highest Shannon index was observed in area A, followed by area C (Additional file 1: Table S2). Furthermore, the Chao1 index of area A was also the highest in the soil MFC, and these among the other three areas were similar. Principal coordinate analysis (PCoA) was conducted for the bacterial community of different treatments based on Bray‒Curtis distances. Axis 1 explained 45.78% of the variance, and Axis 2 explained 14.68% (Fig. 2a). Samples from the AN, AT and MT treatments were clustered together. Furthermore, the distance between MT and AT or AN was farther than that between AT and AN, which indicated that the bacterial community was greatly shifted by the soil MFC.

PCoA of the bacterial community (a). The anaerobic control without antibiotics was labelled AN. Chord charts of the top 10 bacterial communities at the phylum level in different treatments (b) and different areas (c). The last capital letter of the sample name represents the sampling area; for example, ANA represents area A of the AN treatment. Heatmaps of the bacterial community at the genus level in different treatments (d) and different areas (e). Networks of the potential functional bacteria at the genus level (f). Red and blue edges represent significant positive and negative relationships, respectively (p < 0.05, Spearman test), and the size of each node is proportional to the abundance of the corresponding bacteria
Soil bacterial community composition
At the phylum level, Proteobacteria, Bacteroidetes and Acidobacteria were the top three phyla, with their abundances accounting for 25–37%, 16–19% and 11–13% of the total bacteria, respectively (Fig. 2b). The abundances of Proteobacteria and Acidobacteria in the AT treatment were 18% and 13% lower than those in the AN treatment, respectively. However, the amounts of Proteobacteria and Acidobacteria in the MT treatment increased by 19% and 6% compared with those in the AN treatment, respectively. For Proteobacteria, the highest increase was found in the area B of MT (40%), followed by the area A of MT (34%, Fig. 2c). For Acidobacteria, however, the highest increase was observed in the area C of MT (24%). The abundance of Gemmatimonadetes exhibited a similar trend to that of Acidobacteria.
The abundances of the top fifty genera, accounting for 24–34% of the total composition, were chosen to construct the heatmap (Fig. 2d). The top five genera were Pontibacter (2.3–11%), unidentified Acidobacteria (1.4–4.8%), Anaerolinea (0.4–5.9%), Sphingomonas (1.2–3.7%) and Bacillus (0.7–2.2%). The abundance of Pontibacter in the AT treatment increased by 49% compared with that in the AN, and it further increased by 64% in the MT treatment (dominant in area C) relative to that in the AT treatment (Fig. 2d, e). Compared with the AN treatment, the abundance of unidentified Acidobacteria and Sphingomonas declined by 9% and 22%, respectively, in the AT treatment, whereas they were enhanced by 15% and 14%, respectively, in the MT treatment (Fig. 2d). There were sixteen bacteria showing the same trend as unidentified Acidobacteria and Sphingomonas, nine of which belonged to Proteobacteria (Steroidobacter, Phenylobacterium, Massilia, Azoarcus, Microvirga, Woeseia, unidentified Burkholderiaceae, Skermanella and Lysobacter), one belonged to Acidobacteria (Stenotrophobacter), one belonged to Bacteroidetes (Adhaeribacter), two belonged to Actinobacteria (Gaiella and Marmoricola), one belonged to Gemmatimonadetes (Gemmatirosa), one belonged to unidentified Bacteria (Candidatus Entotheonella) and one belonged to Firmicutes (unidentified Clostridiales). Unidentified Acidobacteria, Stenotrophobacter, Phenylobacterium and Adhaeribacter were dominant in area C of the MT treatment; Gemmatirosa and Lysobacter were dominant in areas C and D; Sphingomonas, Massilia, Woeseia, and Candidatus Entotheonella were dominant in area D; Steroidobacter, Azoarcus, unidentified Burkholderiaceae and Skermanella were dominant in areas A and D; Microvirga, Gaiella, unidentified Clostridiales and Marmoricola were dominant in area A (Fig. 2e). In addition, although the amount of Bacillus in the AT treatment declined by 21% compared with that in the AN treatment, it rose by 17% in the MT treatment (dominant in area A) relative to that in the AT treatment, as did Acidibacter.
Potential degrading bacteria and electroactive bacteria
The correlation results showed that the degradation rate was significantly positively correlated with Sphingomonas, Stenotrophobacter, Phenylobacterium, Massilia, Adhaeribacter, Acidibacter, Woeseia and Gemmatirosa (p < 0.05, Additional file 1: Table S3). Previous studies indicated that Sphingomonas sp. can use tetracycline as a nutrient source for growth and reproduction [18, 19]. Phenylobacterium sp. was reported to be capable of degrading sulfonamide antibiotics [35]. Massilia sp. and Gemmatirosa sp. are potential degrading bacteria of polycyclic aromatic hydrocarbons [36, 37]. Woeseia sp. may utilize plastics and hydrocarbons as energy substances [38, 39]. Therefore, the species of these genera might be crucial in tetracycline removal, especially Sphingomonas and Phenylobacterium.
Bacillus sp. and Geobacter sp. are potential electroactive bacteria in MFCs [40, 41]. For example, Bacillus cereus is capable of promoting electron transfer by aligning the cytochrome complex and excreting flavin molecules [42]. Geobacter sulfurreducens can obtain electrons from fumarate and solid donors and conduct extracellular electron transfer through type IV conductive pili and c-type cytochromes [43, 44]. In the current research, the abundance of Bacillus in the soil MFC was 17% higher than that in the AT treatment and dominant at the anode. Furthermore, Geobacter showed enrichment in the MFC anode. Therefore, Bacillus and Geobacter were likely to contribute to electricity production. Moreover, the abundance of Anaerolinea showed a similar tendency to that of Geobacter. Anaerolinea sp. was reported to be enriched at the MFC anode and could transfer electrons [45], which might also be a potential exoelectrogen.
The twelve major genera were selected to explore the evolution of interspecific relationships through network analysis (Fig. 2f). Compared with the AN treatment, the numbers of nodes and edges in the network decreased by 29% and 20% in the AT treatment, respectively. This result indicated that the interspecific relationship was weakened after tetracycline was added, which might be attributed to the inhibition of bacteria by tetracycline. In the soil MFC, most of the selected bacteria had a close relationship with each other, and the numbers of nodes and edges were 29–80% and 100–150% higher than those in the other two treatments, respectively (Fig. 2f). This indicated that the interspecific relationship between microorganisms in the soil MFC was strengthened by biocurrent stimulation [17].
Potential functions of microbiomes in the soil MFC
To identify the potential functions of the soil microbiomes in the MFC, samples from area C (the highest degradation rate in the MT) were chosen for metagenomic sequencing. The clean data were between 6732 and 7183, and the clean Q30 was over 93%, which suggested that the quantity and quality of sequencing data were sufficient (Additional file 1: Table S4). After metagenomic assembly, 186,023–247389 gene fragments were obtained, with N50 lengths reaching 673–958 bp, indicating that the splicing quality was satisfactory for gene prediction.
Pathway and enzyme-encoding genes of tetracycline biodegradation
Biological metabolic pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database mostly included energy metabolism, carbohydrate metabolism, amino acid metabolism, metabolism of other amino acids, metabolism of cofactors and vitamins, nucleotide metabolism, xenobiotic biodegradation and metabolism, metabolism of terpenoids and polyketides, and lipid metabolism. In this study, the relative abundance of metabolic pathways in the area C of AT (ATC) declined by 5.1% compared with that in the area C of AN (ANC), whereas it was enhanced by 7.6% in the area C of MT (MTC) compared with ATC, suggesting that soil MFC could restore the metabolism inhibited by tetracycline (Fig. 3a). Especially for xenobiotic biodegradation and metabolism, its abundance in MTC was 25% higher than that in ATC, which reflected the superior degradation performance of MFC. In addition, DNA polymerase (encoded by EC 2.7.7.7, EC 2.7.7.6) and RNA helicase (EC 3.6.4.13) abundances in the MTC treatment were enhanced by 16–27% and 42%, respectively, compared with those in the ATC treatment (Fig. 3e), suggesting that MFC could promote microorganism growth, which confirmed a previous conjecture [13, 26, 46].

The abundance of metabolism in different treatments (a). Proposed nitrogen cycle pathway (b), degradation pathway of tetracycline (c) and electron transfer pathway (d) in the soil MFC. The abundances of enzyme-encoding genes (e) and ARGs (f) in different treatments
Previous studies have shown that the biodegradation pathway of tetracycline mostly included oxidation, demethylation, decarbonylation, dehydrogenation, deamination, dehydroxylation, loss of acyl-amino groups (deacylation) and ring opening, and the degraders included Sphingobacterium changzhouense TC931, Sphingobacterium mizutaii S121, Klebsiella sp. SQY5 and Alcaligenes sp. R3 [47,48,49,50,51]. The biodegradation of tetracycline is achieved by complicated metabolic reactions catalysed by enzymes. For example, Yin et al. [52] found that isomerase-, oxidoreductase-, and transferase-encoding genes were possibly involved in tetracycline degradation. In the present study, amidase (EC 3.5.1.4) and 4-hydroxyacetophenone monooxygenase (EC 1.14.13.84) were found to have potential degradation functions. Although the abundance of EC 3.5.1.4 was similar in the ANC and ATC, its abundance in the MTC was 31% higher than that in the ATC (Fig. 3e). Compared with that of the ANC, the abundance of EC 1.14.13.84 decreased by 67% in the ATC, whereas it increased by 59% in the MTC. According to the KEGG database, EC 3.5.1.4 is involved in the degradation of aminobenzoate (reaction: R05590, pathway: map00627, Additional file 1: Figure S1), and it mainly promotes the removal of amino groups [53, 54]. Therefore, the enzyme-encoding gene EC 3.5.1.4 was likely to catalyse the deamination reaction of tetracycline biodegradation in this study (Fig. 3c). Zhou et al. [55] reported that the –NH2 in sulfamethoxazole easily underwent a substitution reaction with –OH. Moreover, EC 1.14.13.84 can insert oxygen atoms between the aromatic ring and a ketone side chain to degrade bisphenol (reaction: R06892, pathway: map00363, Additional file 1: Figure S2) [56]. Thus, the EC 1.14.13.84 gene might be involved in the deacylation of tetracycline degradation or act on the bond breaking of the –COOH group (Fig. 3c).
Nitrogen cycle processes were enhanced by the soil MFC
Nitrogen limitation, as the bottleneck of soil bioremediation, needs attention [57]. Our results showed that the abundance of nitrogen metabolism (ko00910) in the ANC decreased by 11% compared with that in the ATC (Additional file 1: Figure S3). Previous studies showed that tetracyclines might inhibit nitrification, denitrification and anammox processes, and the rates of nitrification, denitrification and anammox activity declined by 50%, 44% and 81% relative to nonantibiotic treatment, respectively [58,59,60]. However, whether tetracyclines inhibit the nitrogen cycle process is related to the concentration of tetracyclines and the duration of the experiment [61]. Interestingly, the gene abundance of nitrogen metabolism increased by 16% in the soil MFC compared with ATC (Additional file 1: Figure S3). The possible reasons were as follows: first, the electrochemically active microbes enriched in the MFC promoted the geochemical cycle (including the carbon cycle, nitrogen cycle, sulfur cycle, iron cycle, etc.) [62]; second, the growth and metabolism of nitrogen cycle microorganisms might be activated in MFCs [26]; third, the MFC enhanced the shedding of nitrogen-containing groups (amino and amide groups) of tetracycline, thus providing a more abundant nitrogen source for nitrogen cycle microbes.
The nitrogen cycle mostly includes dissimilatory nitrate reduction, assimilatory nitrate reduction, denitrification, nitrification and anammox [63, 64], and enzymes are vital driving factors in nitrogen transformation processes [65]. Nine enzyme-encoding genes related to the nitrogen cycle were identified (Fig. 3b), of which EC 1.7.5.1 showed the highest abundance, followed by EC 1.7.2.5, EC 1.7.99.-, EC 1.7.2.4, EC 1.7.2.1, EC 1.7.1.15, EC 1.7.99.1, EC 1.7.2.6 and EC 1.7.7.1 (Additional file 1: Figure S4). For dissimilatory nitrate reduction, the nitrate reductase encoding EC 1.7.5.1 catalyses the conversion of nitrate to nitrite [66], and the presence of nitrite reductase encoding EC 1.7.1.15 is beneficial for the formation of ammonia [67]. The enzyme-encoding gene EC 1.7.5.1 simultaneously participates in nitrification (nitrite → nitrate) [68]. In addition, the reaction from hydroxylamine to nitrite in nitrification was equally important and was catalysed by hydroxylamine dehydrogenase (EC 1.7.2.6) [69]. The assimilatory nitrate reduction process mostly involves assimilatory nitrate reductase encoded by EC 1.7.99.- and ferredoxin-nitrite reductase encoded by EC 1.7.7.1 [70]. Denitrification was divided into four steps, namely, nitrate → nitrite → nitric oxide → nitrous oxide → nitrogen. Nitrite reductase (EC 1.7.2.1) and hydroxylamine reductase (EC 1.7.99.1) jointly promote the reaction from nitrite to nitric oxide [23, 71]. Nitric oxide reductase (EC 1.7.2.5) and nitrous oxide reductase (EC 1.7.2.4) catalyse the conversion of nitric oxide to nitrous oxide and nitrous oxide to nitrogen, respectively [72]. As expected, the nine enzyme-encoding genes mentioned above were inhibited by tetracycline; however, their abundances were upregulated in the soil MFC, with EC 1.7.5.1 showing the largest increase in abundance, an increase of 79% in the MTC treatment compared to the ATC treatment (Fig. 3e). This result indicated that MFC promoted the nitrogen cycle in soil, especially the mutual conversion between nitrite and nitrate (NO2−NO3−) driven by the enzyme-encoding gene EC 1.7.5.1 (Fig. 3b). This meant that there were sufficient electron receptors (NO3−) and donors (NO2−), as electron shuttles in the overlapping area of the cathode and anode, which might serve tetracycline degradation.
Genes of electron transfer were upregulated by the soil MFC
Complexes (I, II, III, IV) containing electronic carriers are crucial in the respiratory chain, which participates in inwards and outwards electron transfer [73]. For instance, our previous research found that the succinate dehydrogenase complex encoded by EC 1.3.5.4 (EC 1.3.5.1) catalysed the conversion of succinate to fumarate at the cathode [13]. A total of four related enzyme-encoding genes (including EC 1.6.5.3, EC 1.3.5.1, EC 1.10.2.2 and EC 1.9.3.1) were discovered in this study. Although their abundances in the ATC declined by 11–34% relative to the ANC, they increased by 20–36% in the MTC compared with the ATC (Fig. 3e). EC 1.6.5.3 encodes ubiquinone reductase (complex I), which is a very large complex that participates in the electron transfer chains of mitochondria and aerobic bacteria, transferring electrons from NADH to the ubiquinone pool [74]. Coenzyme Q-cytochrome c reductase encoded by EC 1.10.2.2 could act as complex III for electron transfer [75]. EC 1.9.3.1 encoded cytochrome-c oxidase that acted on a haem group of donors and could serve as complex IV [75]. In addition, as a liposoluble coenzyme, ubiquinone could accept electrons transferred from complex I or complex II in the respiratory chain and then transfer the electrons to complex III [76]. In the current study, the abundance of electron transfer flavoprotein-ubiquinone oxidoreductase encoded by EC 1.5.5.1 in the ATC treatment declined by 21% compared with that in the ANC treatment, whereas it was enhanced by 44% in the MTC treatment compared with that in the ATC treatment (Fig. 3e). Based on the above analysis, a schematic diagram of intracellular electron transfer is presented in soil MFC (Fig. 3d). Ultimately, the electrons transmitted within the cell are transferred to the electrode through direct contact or electron mediators, thereby accelerating the electron transfer of the soil MFC, which was beneficial for tetracycline degradation.
ARGs were reduced efficiently by the soil MFC
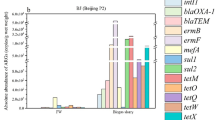
The thirty most abundant ARGs were selected to draw the bubble chart (Fig. 3f). The total abundance in the ATC increased by 14% compared with that in the ANC; however, it decreased by 17% in the MTC relative to that in the ATC (Additional file 1: Figure S5), indicating that MFC could effectively remove ARGs [13, 17]. Among these thirty ARGs, a total of twenty-five ARGs (Excluding Streptomyces_cinnamoneus_EF-Tu, aminocoumarin_resistant_alaS, Mycobacterium_tuberculosis_katG, lrfA and farB) increased in the ATC compared with the ANC, with an average increase of 60% (Fig. 3f). In the twenty-five ARGs, 54% belonged to efflux pump genes, 33% belonged to target alteration genes, and 13% belonged to inactivation genes (Additional file 1: Figure S6a). Therefore, efflux pump resistance was the main resistance mechanism of soil microorganisms to tetracycline. The efflux pump, as a transport protein present on the cell membrane, can pump antibiotics out of the cells, thereby reducing the concentration of intracellular antibiotics and leading to bacterial resistance [77, 78]. Furthermore, the efflux pump is the vital cross-resistance mechanism; that is, different pollutants (such as tetracycline or heavy metals) attack the same target and activate the bacterial efflux pump system, causing it to be resistant to multiple pollutants [79]. In this study, the addition of tetracycline not only increased the abundance of tetracycline resistance genes but also amplified other ARGs after tetracycline exposure. For example, among the efflux pump genes, only adeC, adeG, mexW, mexI and tetV belonged to tetracycline resistance genes. The other efflux pump genes were as follows: abcA, peptide antibiotic; penam, cephalosporin resistance gene; TaeA, pleuromutilin resistance gene; mdtB and mdtC, aminocoumarin resistance genes; patA, fluoroquinolone resistance gene; and msbA, nitroimidazole resistance gene. Zhang et al. [80] also found that the abundances of sulII and blaTEM-1 were elevated by tetracycline, indicating that tetracycline might function as a coselection pressure for ARGs corresponding to other antibiotics. In addition, nineteen of the thirty ARGs (Streptomyces_cinnamoneus_EF-Tu, Bifidobacteria_intrinsic_ileS, abcA, TaeA, desR, TriC, adeG, Mycobacterium_tuberculosis_katG, Mycobacterium_tuberculosis_gyrA, lrfA, OXA-368, mexW, aminocoumarin_resistant_cysB, mdtC, PmrF, mdtB, patA, msbA and tetV) in the MTC treatment were reduced compared with those in the ANC treatment, and the abundance of the nineteen ARGs mentioned above in the MTC treatment was 33% lower on average than that in the ANC (Fig. 3f). Similarly, the efflux pump gene accounted for most ARGs (61%), followed by the target alteration gene (33%) (Additional file 1: Figure S6b).
Functional microbes at the species level were discovered in the soil MFC
The abundances of the top thirty species were chosen to cluster the split heatmap (Fig. 4). Phenylobacterium zucineum was the top species. Compared with ANC, the amount of P. zucineum in the ATC treatment decreased by 52%, whereas it was 637% higher in MTC than in ATC (Fig. 4). A total of fourteen species exhibited the same trend as P. zucineum, such as Gemmatirosa kalamazoonesis, Nitrospira moscoviensis, Ramlibacter tataouinensis, Steroidobacter denitrificans, Nitrospira sp. SG-bin1, Candidatus Nitrospira nitrificans and Nitrospira japonica. Furthermore, the abundances of Pontibacter roseus and Luteitalea pratensis in the ATC, respectively, increased by 18% and 1.2% compared with the ANC treatment, and they rose by 10% and 51% in the MTC treatment relative to the ATC treatment, respectively (Fig. 4).

Split heatmap of the microbial community at the species level in different treatments
Chen et al. [35] found that Phenylobacterium sp. was a degrader of sulfadiazine and sulfamethoxazole. Interestingly, EC 1.14.13.84 was found in the species P. zucineum, which suggested that P. zucineum was likely to secrete 4-hydroxyacetophenone monooxygenase to degrade tetracycline (Fig. 3c). Our previous study indicated that the species G. kalamazoonesis and S. denitrificans were potential electrotrophic microbes; G. kalamazoonesis and S. denitrificans secreted quinoloxidase encoded by EC 1.10.3.14 and flavoprotein 2,3-oxidoreductase encoded by EC 1.3.8.7 to accelerate electron transfer, respectively [13]. In the current study, EC 1.6.5.3 and EC 1.9.3.1 were found in both G. kalamazoonesis and S. denitrificans. In addition, EC 1.3.5.1 and EC 1.10.2.2 existed in G. kalamazoonesis and S. denitrificans, respectively. G. kalamazoonosis and S. denitrificans were likely to be electrotrophic bacteria and contributed to electricity generation in this experiment. Previous studies showed that the species N. moscoviensis, Nitrospira sp. SG-bin1, Candidatus Nitrospira nitrificans and N. japonica are involved in the nitrogen cycle [81,82,83,84]. For instance, Candidatus Nitrospira nitrificans could fully oxidize ammonia via nitrite to nitrate [83]. However, unfortunately, no association was found between these species and the nitrogen cycle genes mentioned above.
Conclusions
Tetracycline and ARGs were effectively removed simultaneously by the soil MFC, which was due to the enrichment of degrading bacteria and electroactive bacteria and their close interactions. Based on the metagenomic analysis, G. kalamazoonesis and S. denitrificans are likely to be electrotrophic bacteria, while P. zucineum can secrete the 4-hydroxyacetophenone monooxygenase encoded by EC 1.14.13.84 to catalyse the deacylation or decarboxylation of tetracycline. Substantially, the soil MFC enhanced microbial metabolism, especially xenobiotic biodegradation and nitrogen metabolism, and corresponding functional genes, including degradation, electron transfer and the nitrogen cycle. In addition, efflux pump resistance, as the main resistance of microbes to tetracycline, was reduced by the soil MFC. Overall, the key functional genes in soil bioelectrochemical remediation were revealed in this study.
Material and methods
Tested soils and chemicals
The tested soil sample was collected from Wuqing farmland in Tianjin (coordinate: N39°27′20.59″, E117°09′26.18″) and then air-dried, ground, and passed through a 2-mm sieve. The soil properties are shown in Additional file 1: Table S5. Tetracycline was purchased from Dr. Ehrenstorfer LGC (Augsburg, Germany). Chemicals such as methanol, acetonitrile and acetone were of chromatography grade.
Soil MFC configuration and operation
The configuration and operation of the soil MFC were performed according to our previous method [85]. Briefly, the cylindrical reactor was composed of a graphite rod anode and an activated carbon air–cathode (Fig. 5). Each reactor was filled with 1000 g of soil (400 mL deionized water, 10 mg·kg−1 tetracycline) and connected to an external resistance of 100 Ω (labelled MT). Carbon fibre was mixed into the soil at a 1% mass fraction to reduce soil internal resistance [28]. Furthermore, nonelectrode controls (spiked with 10 mg·kg−1 tetracycline, AT; no antibiotic added, AN) were set. The experimental details are shown in the Supplementary Information (Additional file 1: Table S6). All reactors were placed at a constant temperature of 30 °C for 53 days without light.

Picture (a), schematic drawing of the soil MFC (b), open-circuit group (c) and nonelectrode group (d). The yellow area is the sampling area
Electrochemical and chemical analysis
Voltage (U) was recorded using a data acquisition system (PISO-813, ICP DAS Co., Ltd) [13]. The soil samples were collected at the anode (area A), away from the cathode and anode (area B), overlapping area of the cathode and anode (area C), cathode (area D) of the soil MFC, and the corresponding control areas (Fig. 5). The partial samples were stored at − 80 °C for biological analysis. Others were freeze-dried for soil property analysis and tetracycline quantification. The soil pH and electrical conductivity were measured by a metre at a 1:5 soil:water ratio [86]. The total nitrogen, phosphorus and organic matter were analysed by common methods [87]. The tetracycline content was determined by a published protocol [88].
Analysis of 16S rRNA gene amplicons
Total genomic DNA was extracted from soil samples by a Power Soil DNA isolation kit (Mo Bio, America). The concentration and purity of DNA were determined in 1% agarose gels and then diluted to 1 ng·μL−1 with sterile water. The universal primers 515F (GTGCCAGCMGCCGCGGTAA) and 806R (GGACTACHVGGGTWTCTAAT) were used to amplify the V3-V4 regions of the 16S rRNA gene by PCR. The PCR conditions referred to our previous method [13]: 3 min at 95 °C (initial denaturation); 30 cycles consisting of denaturation at 95 °C for 30 s, renaturation at 55 °C for 30 s, and extension at 72 °C for 45 s; and a final extension at 72 °C for 10 min. The samples were assessed in 2% agarose gels by electrophoresis. After purification, a high-quality sequencing library was constructed and sequenced by Novogene Company (Beijing, China).
Metagenomic analysis
The extracted DNA samples were sequenced on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) according to our previous report [13]. Briefly, the genomic DNA was randomly sheared into fragments using a Covaris S2 System (Massachusetts, USA) for library construction. Libraries were quantified using real-time PCR and then sequenced by Novogene Company. Subsequently, the quality of the raw data was filtered to acquire reliable data for assembly. Open reading frame prediction was performed based on each sample and the mixed assembled scaffolds (≥ 500 bp), and reads with a length less than 100 nt were filtered out. CD-HIT software was used to remove redundancy to obtain a nonredundant initial gene catalogue. The clean data of each sample were compared to the initial gene catalogue to obtain the number of gene reads. DIAMOND software was employed to compare the gene catalogue with the sequences of bacteria, fungi, archaea and viruses extracted from the National Center for Biotechnology Information (NCBI) database to obtain species information. Enzyme genes were annotated by the KEGG database, and ARGs were annotated by the Comprehensive Antibiotic Research Database (CARD) database.
Statistical analysis
Microsoft Excel 2010 (Redmond, USA) was employed to acquire averages and standard deviations of the data. The significant differences and Spearman correlation between samples were determined by IBM SPSS Statistics 22 software (New York, USA). Networks were constructed to reveal the interspecific relationships between the potential functional bacteria at the genus level using Cytoscape 3.9.1 software (California, USA). To reduce the network complexity, Spearman correlation analysis was conducted between functional bacteria, and a correlation between two bacteria was regarded as statistically robust if p < 0.05. Nodes represented functional bacteria, and edges represented the interaction between these bacteria.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- MFCs:
-
Microbial fuel cells
- ARGs:
-
Antibiotic resistance genes
- MT:
-
Soil MFC spiked with tetracycline
- MTC:
-
The area C of MT
- AT:
-
Anaerobic controls spiked with tetracycline
- ATC:
-
The area C of AT
- AN:
-
Anaerobic controls without antibiotic
- ANC:
-
The area C of AN
- NCBI:
-
National Center for Biotechnology Information
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- CARD:
-
Comprehensive Antibiotic Research Database
References
Roemhild R, Bollenbach T, Andersson DI. The physiology and genetics of bacterial responses to antibiotic combinations. Nat Rev Microbiol. 2022;20:478–90. https://doi.org/10.1038/s41579-022-00700-5.
Zhang R, Yang S, An YW, Wang YQ, Lei Y, Song LY. Antibiotics and antibiotic resistance genes in landfills: a review. Sci Total Environ. 2022;806:150647. https://doi.org/10.1016/j.scitotenv.2021.150647.
Zheng DS, Yin GY, Liu M, Hou LJ, Yang Y, Van Boeckel TP, Zheng YL, Li Y. Global biogeography and projection of soil antibiotic resistance genes. Sci Adv. 2022;8:eabq8015. https://doi.org/10.1126/sciadv.abq8015.
Zhou YJ, Zhu F, Zheng DY, Mm G, Guo BF, Zhang N, Meng Y, Gl Wu, Yl Z, Huo X. Detection of antibiotics in the urine of children and pregnant women in Jiangsu China. Environ Res. 2021;196:110945. https://doi.org/10.1016/j.envres.2021.110945.
Wang HX, Wang B, Zhao Q, Zhao YP, Fu CW, Feng X, Wang N, Su MF, Tang CX, Jiang F, Zhou Y, Chen Y, Jiang QW. Antibiotic body burden of Chinese school children: A multisite biomonitoring-based study. Environ Sci Technol. 2015;49:5070–9. https://doi.org/10.1021/es5059428.
Zhu YT, Liu KY, Zhang JJ, Liu XJ, Yang LS, Wei R, Wang SF, Zhang DM, Xie SY, Tao FB. Antibiotic body burden of elderly Chinese population and health risk assessment: a human biomonitoring-based study. Environ Pollut. 2020;256:113311. https://doi.org/10.1016/j.envpol.2019.113311.
Tan JT, Wang YW, Gong XH, Li J, Zhong WH, Shan LQ, Lei XP, Zhang Q, Zhou Q, Zhao YY, Chen C, Zhang YJ. Antibiotic resistance in neonates in China 2012–2019: a multicenter study. J Microbiol Immunol Infect. 2022;55:454–62. https://doi.org/10.1016/j.jmii.2021.05.004.
Yan WF, Xiao Y, Yan WD, Ding R, Wang SH, Zhao F. The effect of bioelectrochemical systems on antibiotics removal and antibiotic resistance genes: a review. Chem Eng J. 2019;358:1421–37. https://doi.org/10.1016/j.cej.2018.10.128.
Miran W, Jang J, Nawaz M, Shahzad A, Lee DS. Biodegradation of the sulfonamide antibiotic sulfamethoxazole by sulfamethoxazole acclimatized cultures in microbial fuel cells. Sci Total Environ. 2018;627:1058–65. https://doi.org/10.1016/j.scitotenv.2018.01.326.
Chen P, Guo XY, Li SN, Li FX. A review of the bioelectrochemical system as an emerging versatile technology for reduction of antibiotic resistance genes. Environ Int. 2021;156:106689. https://doi.org/10.1016/j.envint.2021.106689.
Song HL, Zhang C, Lu YX, Li H, Shao Y, Yang YL. Enhanced removal of antibiotics and antibiotic resistance genes in a soil microbial fuel cell via in situ remediation of agricultural soils with multiple antibiotics. Sci Total Environ. 2022;829:154406. https://doi.org/10.1016/j.scitotenv.2022.154406.
Li T, Song HL, Xu H, Yang XL, Chen QL. Biological detoxification and decolorization enhancement of azo dye by introducing natural electron mediators in MFCs. J Hazard Mater. 2021;416:125864. https://doi.org/10.1016/j.jhazmat.2021.125864.
Zhao XD, Li XJ, Li Y, Zhang XL, Zhai FH, Ren TZ, Li YT. Metagenomic analysis reveals functional genes in soil microbial electrochemical removal of tetracycline. J Hazard Mater. 2021;408:124880. https://doi.org/10.1016/j.jhazmat.2020.124880.
Xu WX, Yang BS, Wang H, Zhang LD, Dong JH, Liu CC. Simultaneous removal of antibiotics and nitrogen by microbial fuel cell-constructed wetlands: microbial response and carbon–nitrogen metabolism pathways. Sci Total Environ. 2023;893:164855. https://doi.org/10.1016/j.scitotenv.2023.164855.
Li SN, Ondon BS, Ho S-H, Li FX. Emerging soil contamination of antibiotics resistance bacteria (ARB) carrying genes (ARGs): new challenges for soil remediation and conservation. Environ Res. 2023;219:115132. https://doi.org/10.1016/j.envres.2022.115132.
Lu L, Huggins T, Jin S, Zuo Y, Ren ZJ. Microbial metabolism and community structure in response to bioelectrochemically enhanced remediation of petroleum hydrocarbon-contaminated soil. Environ Sci Technol. 2014;48:4021–9. https://doi.org/10.1021/es4057906.
Zhao XD, Li XJ, Li Y, Sun Y, Zhang XL, Weng LP, Ren TZ, Li YT. Shifting interactions among bacteria, fungi and archaea enhance removal of antibiotics and antibiotic resistance genes in the soil bioelectrochemical remediation. Biotechnol Biofuels. 2019;12:160. https://doi.org/10.1186/s13068-019-1500-1.
Sun YF, Lyu HH, Cheng Z, Wang YZ, Tang JC. Insight into the mechanisms of ball-milled biochar addition on soil tetracycline degradation enhancement: physicochemical properties and microbial community structure. Chemosphere. 2022;291:132691. https://doi.org/10.1016/j.chemosphere.2021.132691.
Lin Z, Zhen Z, Luo SW, Ren L, Chen YJ, Wu WJ, Zhang WJ, Liang YQ, Song ZG, Li YT, Zhang DY. Effects of two ecological earthworm species on tetracycline degradation performance, pathway and bacterial community structure in laterite soil. J Hazard Mater. 2021;412:125212. https://doi.org/10.1016/j.jhazmat.2021.125212.
Wang Q, Li XN, Yang QX, Chen YL, Du BB. Evolution of microbial community and drug resistance during enrichment of tetracycline-degrading bacteria. Ecotoxicol Environ Saf. 2019;171:746–52. https://doi.org/10.1016/j.ecoenv.2019.01.047.
Fang HT, Oberoi AS, He ZQ, Khanal SK, Lu H. Ciprofloxacin-degrading Paraclostridium sp isolated from sulfate-reducing bacteria-enriched sludge: optimization and mechanism. Water Res. 2021;191:116808. https://doi.org/10.1016/j.watres.2021.116808.
Liang CY, Ye QH, Huang Y, Wang Y, Zhang ZT, Wang H. Shifts of the new functional marker gene (pahE) of polycyclic aromatic hydrocarbons (PAHs) degrading bacterial population and its relationship with PAHs biodegradation. J Hazard Mater. 2022;437:129305. https://doi.org/10.1016/j.jhazmat.2022.129305.
Gao YC, Du JH, Bahar MM, Wang H, Subashchandrabose S, Duan LC, Yang XD, Megharaj M, Zhao QQ, Zhang W, Liu YJ, Wang JN, Naidu R. Metagenomics analysis identifies nitrogen metabolic pathway in bioremediation of diesel contaminated soil. Chemosphere. 2021;271:129566. https://doi.org/10.1016/j.chemosphere.2021.129566.
Wang MS, Yu Y, Ren YC, Wang JY, Chen H. Effect of antibiotic and/or heavy metal on nitrogen cycle of sediment-water interface in aquaculture system: Implications from sea cucumber culture. Environ Pollut. 2023;325:121453. https://doi.org/10.1016/j.envpol.2023.121453.
Zhai WJ, Jiang WQ, Guo QQ, Wang ZX, Liu DH, Zhou ZQ, Wang P. Existence of antibiotic pollutant in agricultural soil: exploring the correlation between microbiome and pea yield. Sci Total Environ. 2023;871:162152. https://doi.org/10.1016/j.scitotenv.2023.162152.
Zhang XL, Li XJ, Zhao XD, Chen XD, Zhou B, Weng LP, Li YT. Bioelectric field accelerates the conversion of carbon and nitrogen in soil bioelectrochemical systems. J Hazard Mater. 2020;388:121790. https://doi.org/10.1016/j.jhazmat.2019.121790.
Zhang XL, Li RX, Wang JN, Liao CM, Zhou L, An JK, Li T, Wang X, Zhou QX. Construction of conductive network using magnetite to enhance microflora interaction and petroleum hydrocarbons removal in plant-rhizosphere microbial electrochemical system. Chem Eng J. 2022;433:133600. https://doi.org/10.1016/j.cej.2021.133600.
Li XJ, Wang X, Zhao Q, Wan LL, Li YT, Zhou QX. Carbon fiber enhanced bioelectricity generation in soil microbial fuel cells. Biosens Bioelectron. 2016;85:135–41. https://doi.org/10.1016/j.bios.2016.05.001.
Zhou EZ, Lekbach Y, Gu TY, Xu DK. Bioenergetics and extracellular electron transfer in microbial fuel cells and microbial corrosion. Curr Opin Electrochem. 2022;31:100830. https://doi.org/10.1016/j.coelec.2021.100830.
Zhang PB, Zhou X, Wang XY, Li ZP. Enhanced bidirectional extracellular electron transfer based on biointerface interaction of conjugated polymers-bacteria biohybrid system. Coll Surf B: Biointerfaces. 2023;228:113383. https://doi.org/10.1016/j.colsurfb.2023.113383.
Yi Y, Zhao T, Zang YX, Xie BZ, Liu H. Different mechanisms for riboflavin to improve the outward and inward extracellular electron transfer of Shewanella loihica. Electrochem Commun. 2021;124:106966. https://doi.org/10.1016/j.elecom.2021.106966.
Chen XD, Han T, Miao XY, Zhang XL, Zhao LX, Sun Y, Ye HK, Li XJ, Li YT. Ferrihydrite enhanced the electrogenic hydrocarbon degradation in soil microbial electrochemical remediation. Chem Eng J. 2022;446:136901. https://doi.org/10.1016/j.cej.2022.136901.
Zhang JR, Huang S, Yin YG, Yang LQ, Li XN, Jiao WT, Sakamaki T. Fe2O3 micron particles are critical for electron transfer and the distribution of electrochemically active bacteria in soil MFCs. Sci Total Environ. 2023;893:164909. https://doi.org/10.1016/j.scitotenv.2023.164909.
Wang LX, Liang DM, Shi YQ. Profiling of co-metabolic degradation of tetracycline by the bio-cathode in microbial fuel cells. RSC Adv. 2022;12:509–16. https://doi.org/10.1039/D1RA07600K.
Chen JF, Yang YY, Ke YC, Chen XL, Jiang XS, Chen C, Xie SG. Sulfonamide-metabolizing microorganisms and mechanisms in antibiotic-contaminated wetland sediments revealed by stable isotope probing and metagenomics. Environ Int. 2022;165:107332. https://doi.org/10.1016/j.envint.2022.107332.
Holochová P, Mašlaňová I, Sedláček I, Švec P, Králová S, Kovařovic V, Busse H-J, Staňková E, Barták M, Pantůček R. Description of Massilia rubra sp. nov., Massilia aquatica sp. nov., Massilia mucilaginosa sp. nov., Massilia frigida sp. nov., and one Massilia genomospecies isolated from Antarctic streams, lakes and regoliths. Syst Appl Microbiol. 2020;43:126112. https://doi.org/10.1016/j.syapm.2020.126112.
Dou RN, Sun JT, Lu J, Deng FC, Yang C, Lu GN, Dang Z. Bacterial communities and functional genes stimulated during phenanthrene degradation in soil by bio-microcapsules. Ecotoxicol Environ Saf. 2021;212:111970. https://doi.org/10.1016/j.ecoenv.2021.111970.
Zeng QZ, Xiang JX, Yang CY, Wu JX, Li YX, Sun YN, Liu QW, Shi SN, Gong Z. Microplastics affect nitrogen cycling and antibiotic resistance genes transfer of sediment. Chem Eng J. 2023;454:140193. https://doi.org/10.1016/j.cej.2022.140193.
Bacosa HP, Erdner DL, Rosenheim BE, Shetty P, Seitz KW, Baker BJ, Liu Z. Hydrocarbon degradation and response of seafloor sediment bacterial community in the northern Gulf of Mexico to light Louisiana sweet crude oil. ISME J. 2018;12:2532–43. https://doi.org/10.1038/s41396-018-0190-1.
Lovley DR. Bug juice: harvesting electricity with microorganisms. Nat Rev Microbiol. 2006;4:497–508. https://doi.org/10.1038/nrmicro1442.
Logan BE, Rossi R, Aa R, Saikaly PE. Electroactive microorganisms in bioelectrochemical systems. Nat Rev Microbiol. 2019;17:307–19. https://doi.org/10.1038/s41579-019-0173-x.
Sreelekshmy BR, Basheer R, Shibli SMA. Exploration of bifurcated electron transfer mechanism in Bacillus cereus for enhanced power generation in double-chambered microbial fuel cells. J Environ Chem Eng. 2022;10:107601. https://doi.org/10.1016/j.jece.2022.107601.
Rushimisha IE, Xj Li, Han T, Chen XD, Abdoul Magid ASI, Sun Y, Li YT. Application of biochar on soil bioelectrochemical remediation: behind roles, progress, and potential. Crit Rev Biotechnol. 2022. https://doi.org/10.1080/07388551.2022.2119547.
Lovley DR, Holmes DE. Electromicrobiology: the ecophysiology of phylogenetically diverse electroactive microorganisms. Nat Rev Microbiol. 2022;20:5–19. https://doi.org/10.1038/s41579-021-00597-6.
Zheng Y, Quan XC, Zhuo MH, Zhang XF, Quan YP. In-situ formation and self-immobilization of biogenic Fe oxides in anaerobic granular sludge for enhanced performance of acidogenesis and methanogenesis. Sci Total Environ. 2021;787:147400. https://doi.org/10.1016/j.scitotenv.2021.147400.
Li XJ, Wang X, Weng LP, Zhou QX, Li YT. Microbial fuel cells for organic-contaminated soil remedial applications: a review. Energy Technol. 2017;5:1156–64. https://doi.org/10.1002/ente.201600674.
Leng YF, Bao JG, Chang GF, Zheng H, Li XX, Du JK, Snow D, Li X. Biotransformation of tetracycline by a novel bacterial strain Stenotrophomonas maltophilia DT1. J Hazard Mater. 2016;318:125–33. https://doi.org/10.1016/j.jhazmat.2016.06.053.
Shao SC, Hu YY, Cheng JH, Chen YC. Biodegradation mechanism of tetracycline (TEC) by strain Klebsiella sp. SQY5 as revealed through products analysis and genomics. Ecotoxicol Environ Saf. 2019;185:109676. https://doi.org/10.1016/j.ecoenv.2019.109676.
Tan H, Kong DL, Li QQ, Zhou YQ, Jiang X, Wang ZY, Parales RE, Ruan ZY. Metabolomics reveals the mechanism of tetracycline biodegradation by a Sphingobacterium mizutaii S121. Environ Pollut. 2022;305:119299. https://doi.org/10.1016/j.envpol.2022.119299.
Tan ZW, Chen JC, Liu YL, Chen L, Xu YQ, Zou YX, Li YT, Gong BN. The survival and removal mechanism of Sphingobacterium changzhouense TC931 under tetracycline stress and its’ ecological safety after application. Biores Technol. 2021;333: 125067. https://doi.org/10.1016/j.biortech.2021.125067.
Liu ZZ, Yang YB, Liu G, Fang J. Study on a novel immobilized microbe pellets constructed with Alcaligenes sp R3 and its ability to remove tetracycline. J Environ Chem Eng. 2023;11:109378. https://doi.org/10.1016/j.jece.2023.109378.
Yin ZF, Xia D, Shen M, Zhu DW, Cai HJ, Wu M, Zhu QR, Kang YJ. Tetracycline degradation by Klebsiella sp. strain TR5: Proposed degradation pathway and possible genes involved. Chemosphere. 2020;253:126729. https://doi.org/10.1016/j.chemosphere.2020.126729.
Ma YC, Yu HM, Pan WY, Liu CC, Zhang SL, Shen ZY. Identification of nitrile hydratase-producing Rhodococcus ruber TH and characterization of an amiE-negative mutant. Bioresour Technol. 2010;101:285–91. https://doi.org/10.1016/j.biortech.2009.07.057.
Banerjee A, Sharma R, Banerjee UC. The nitrile-degrading enzymes: current status and future prospects. Appl Microbiol Biotechnol. 2002;46:33–44. https://doi.org/10.1007/s00253-002-1062-0.
Zhou QX, Song CL, Wang PF, Zhao ZY, Li Y, Zhan SH. Generating dual-active species by triple-atom sites through peroxymonosulfate activation for treating micropollutants in complex water. Proc Natl Acad Sci. 2023;120:e2300085120. https://doi.org/10.1073/pnas.2300085120.
Kamerbeek NM, Moonen MJH, van der Ven JGM, van Berkel WJH, Fraaije MW, Janssen DB. 4-Hydroxyacetophenone monooxygenase from Pseudomonas fluorescens ACB. Eur J Biochem. 2001;268:2547–57. https://doi.org/10.1046/j.1432-1327.2001.02137.x.
Kuypers MMM, Marchant HK, Kartal B. The microbial nitrogen-cycling network. Nat Rev Microbiol. 2018;16:263–76. https://doi.org/10.1038/nrmicro.2018.9.
Pulicharla R, Zolfaghari M, Brar SK, Drogui P, Auger S, Verma M, Surampalli RY. Acute impact of chlortetracycline on nitrifying and denitrifying processes. Water Environ Res. 2018;90:604–14. https://doi.org/10.2175/106143017X15131012153095.
Yin GY, Hou LJ, Liu M, Zheng YL, Li XF, Lin XB, Gao J, Jiang XF, Wang R, Yu CD. Effects of multiple antibiotics exposure on denitrification process in the Yangtze Estuary sediments. Chemosphere. 2017;171:118–25. https://doi.org/10.1016/j.chemosphere.2016.12.068.
Shi ZJ, Hu HY, Shen YY, Xu JJ, Shi ML, Jin RC. Long-term effects of oxytetracycline (OTC) on the granule-based anammox: process performance and occurrence of antibiotic resistance genes. Biochem Eng J. 2017;127:110–8. https://doi.org/10.1016/j.bej.2017.08.009.
Li WB, Shi CZ, Yu YW, Ruan YJ, Kong DD, Lv XF, Xu P, Awasthi MK, Dong M. Interrelationships between tetracyclines and nitrogen cycling processes mediated by microorganisms: a review. Bioresour Technol. 2021;319:124036. https://doi.org/10.1016/j.biortech.2020.124036.
Wang H, Zheng Y, Liu JW, Zhu BL, Qin W, Zhao F. An electrochemical system for the rapid and accurate quantitation of microbial exoelectrogenic ability. Biosens Bioelectron. 2022;215:114584. https://doi.org/10.1016/j.bios.2022.114584.
Zhao XD, Qin XR, Li TL, Cao HB, Xie YH. Effects of planting patterns plastic film mulching on soil temperature, moisture, functional bacteria and yield of winter wheat in the Loess Plateau of China. J Integr Agric. 2023;22:1560–73. https://doi.org/10.1016/j.jia.2023.02.026.
Zhou SH, Song Z, Li ZB, Qiao RY, Li MJ, Chen YF, Guo H. Mechanisms of nitrogen transformation driven by functional microbes during thermophilic fermentation in an ex situ fermentation system. Bioresour Technol. 2022. https://doi.org/10.1016/j.biortech.2022.126917.
Sun XD, Ye YQ, Ma QX, Guan QW, Jones DL. Variation in enzyme activities involved in carbon and nitrogen cycling in rhizosphere and bulk soil after organic mulching. Rhizosphere. 2021;19:100376. https://doi.org/10.1016/j.rhisph.2021.100376.
Zhou Q, Feng FL, Li FL, Liu JL, Wang MZ, Huang SJ, Sun YX. Acylated homoserine lactones regulate the response of methane metabolism and nitrogen metabolism to florfenicol in anaerobic fermentation. Sci Total Environ. 2022;832:155035. https://doi.org/10.1016/j.scitotenv.2022.155035.
Zhu X, Shen CF, Huang JX, Wang LM, Pang QQ, Peng FQ, Hou J, Ni LX, He F, Xu B. The effect of sulfamethoxazole on nitrogen removal and electricity generation in a tidal flow constructed wetland coupled with a microbial fuel cell system: microbial response. Chem Eng J. 2022;431:134070. https://doi.org/10.1016/j.cej.2021.134070.
Huang R, Meng TY, Liu GG, Gao SS, Tian JY. Simultaneous nitrification and denitrification in membrane bioreactor: effect of dissolved oxygen. J Environ Manag. 2022;323:116183. https://doi.org/10.1016/j.jenvman.2022.116183.
Zhou XL, Bi XJ, Fan X, Yang T, Wang XD, Chen SS, Cheng LH, Zhang Y, Zhao WH, Zhao FC, Nie SC, Deng XY. Performance and bacterial community analysis of a two-stage A/O-MBBR system with multiple chambers for biological nitrogen removal. Chemosphere. 2022;303:135195. https://doi.org/10.1016/j.chemosphere.2022.135195.
Wang JL, Wang X, Yu Z, Huang SQ, Yao DY, Xiao JJ, Chen W, Wang ZP, Zan FX. Using algae bacteria consortia to effectively treat coking wastewater: performance, microbial community, and mechanism. J Clean Prod. 2022;334:130269. https://doi.org/10.1016/j.jclepro.2021.130269.
He X, Zhang SY, Jiang YH, Li M, Yuan JL, Wang GJ. Influence mechanism of filling ratio on solid-phase denitrification with polycaprolactone as biofilm carrier. Bioresour Technol. 2021;337:125401. https://doi.org/10.1016/j.biortech.2021.125401.
Wang QZ, Wu SY, Chu GY, Zhang ZM, She ZL, Zhao YG, Guo L, Jin CJ, Gao MC. Metagenomic analysis and microbial activity shifts reveal the effect of the carbon/nitrogen ratio on the nitrogen removal performance of a moving bed biofilm reactor treating mariculture wastewater. J Water Process Eng. 2023;51:103363. https://doi.org/10.1016/j.jwpe.2022.103363.
Yu LP, Yuan Y, Rensing C, Zhou SG. Combined spectroelectrochemical and proteomic characterizations of bidirectional Alcaligenes faecalis-electrode electron transfer. Biosens Bioelectr. 2018;106:21–8. https://doi.org/10.1016/j.bios.2018.01.032.
Yano T. The energy-transducing NADH: quinone oxidoreductase, complex I. Mol Asp Med. 2002;23:345–68. https://doi.org/10.1016/S0098-2997(02)00011-0.
Kilbride SM, Gluchowska SA, Telford JE, O’Sullivan C, Davey GP. High-level inhibition of mitochondrial complexes III and IV is required to increase glutamate release from the nerve terminal. Mol Neurodegener. 2011;6:53. https://doi.org/10.1186/1750-1326-6-53.
Gille L, Rosenau T, Kozlov AV, Gregor W. Ubiquinone and tocopherol: dissimilar siblings. Biochem Pharmacol. 2008;76:289–302. https://doi.org/10.1016/j.bcp.2008.04.003.
Li HN, Li BX, Zhang ZG, Tian YL, Ye J, Lv XW, Zhu CX. Factors influencing the removal of antibiotic-resistant bacteria and antibiotic resistance genes by the electrokinetic treatment. Ecotoxicol Environ Saf. 2018;160:207–15. https://doi.org/10.1016/j.ecoenv.2018.05.028.
Blair JM, Richmond GE, Piddock LJ. Multidrug efflux pumps in Gram-negative bacteria and their role in antibiotic resistance. Future Microbiol. 2014;9:1165–77. https://doi.org/10.2217/fmb.14.66.
Liu K, Sun MM, Ye M, Chao HZ, Zhao YC, Xia B, Jiao WT, Feng YF, Zheng XX, Liu MQ, Jiao JG, Hu F. Coexistence and association between heavy metals, tetracycline and corresponding resistance genes in vermicomposts originating from different substrates. Environ Pollut. 2019;244:28–37. https://doi.org/10.1016/j.envpol.2018.10.022.
Zhang MQ, Yuan L, Li ZH, Zhang HC, Sheng GP. Tetracycline exposure shifted microbial communities and enriched antibiotic resistance genes in the aerobic granular sludge. Environ Int. 2019;130:104902. https://doi.org/10.1016/j.envint.2019.06.012.
Ehrich S, Behrens D, Lebedeva E, Ludwig W, Bock E. A new obligately chemolithoautotrophic, nitrite-oxidizing bacterium, Nitrospira moscoviensis sp. nov. and its phylogenetic relationship. Arch Microbiol. 1995;164:16–23. https://doi.org/10.1007/BF02568729.
Wang YL, Ma LP, Mao YP, Jiang XT, Xia Y, Yu K, Li B, Zhang T. Comammox in drinking water systems. Water Res. 2017;116:332–41. https://doi.org/10.1016/j.watres.2017.03.042.
van Kessel MA, Speth DR, Albertsen M, Nielsen PH, Op den Camp HJ, Kartal B, Jetten MS, Lücker S. Complete nitrification by a single microorganism. Nature. 2015;528:555–9. https://doi.org/10.1038/nature16459.
Sun W, Jiao LJ, Wu JP, Ye JQ, Wei MK, Hong YG. Existence and distribution of novel phylotypes of Nitrospira in water columnsof the South China Sea. iScience. 2022;25:104895. https://doi.org/10.1016/j.isci.2022.104895.
Zhao XD, Li XJ, Zhang XL, Li Y, Weng LP, Ren TZ, Li YT. Bioelectrochemical removal of tetracycline from four typical soils in China: a performance assessment. Bioelectrochem. 2019;129:26–33. https://doi.org/10.1016/j.bioelechem.2019.04.016.
Li XJ, Li Y, Zhang XL, Zhao XD, Chen XD, Li YT. The metolachlor degradation kinetics and bacterial community evolution in the soil bioelectrochemical remediation. Chemosphere. 2020;248:125915. https://doi.org/10.1016/j.chemosphere.2020.125915.
Zhu N, Yan YC, Bai KY, Zhang JM, Wang C, Wang X, Xu DW, Liu JH, Xin XP, Chen JQ. Conversion of croplands to shrublands does not improve soil organic carbon and nitrogen but reduces soil phosphorus in a temperate grassland of northern China. Geoderma. 2023;432:116407. https://doi.org/10.1016/j.geoderma.2023.116407.
Zheng WL, Zhang LF, Zhang KY, Wang XY, Xue FQ. Determination of tetracyclines and their epimers in agricultural soil fertilized with swine manure by ultra-high-performance liquid chromatography tandem mass spectrometry. J Int Agric. 2012;11:1189–98. https://doi.org/10.1016/S2095-3119(12)60114-2.
Acknowledgements
The authors thank the reviewers for their comments on the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 42007121 and 41977133), Technological Innovation Programs of Higher Education Institutions in Shanxi (No. 2020L0536), and the Central Public-interest Scientific Institution Basal Research Fund (Agro-Environmental Protection Institute, Ministry of Agriculture and Rural Affairs).
Author information
Authors and Affiliations
Contributions
XJL and XDZ designed the research plan; XJL and XDZ initiated this work and performed the experiments; XDZ, XJL, XRQ, XQJ, TW and QQQ performed the data analysis; and XDZ, XJL, YTL and PMY wrote the manuscript with input and discussions from all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors approved the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
HiSeq sequencing data and alpha indices of the groups. Table S2. HiSeq sequencing data and alpha indices of samples. Table S3. Spearman correlation between tetracycline degradation rate and bacterial abundance at the genus level. Table S4. HiSeq sequencing data of the metagenome. Table S5. Physicochemical properties of the experimental soil. Table S6. Experimental design. Figure S1. Sketch map of KEGG metabolic pathways and related reaction equations (pathway: map00627, reaction: R05590). Figure S2. Sketch map of KEGG metabolic pathways and related reaction equations (pathway: map00363, reaction: R06892). Figure S3. The abundance of nitrogen metabolism in different treatments. Figure S4. The abundance of nitrogen cycling functional genes. Figure S5. Changes in the total abundance of ARGs in different treatments. Figure S6. The resistance mechanism of soil microorganisms to tetracycline in the ATC treatment (a) and the MTC treatment (b).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, X., Qin, X., Jing, X. et al. Key genes of electron transfer, the nitrogen cycle and tetracycline removal in bioelectrochemical systems. Biotechnol Biofuels 16, 174 (2023). https://doi.org/10.1186/s13068-023-02430-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-023-02430-z