
Abstract
Keratin is a recalcitrant protein and can be decomposed in nature. However, the mechanism of keratin degradation is still not well understood. In this study, Bacillus sp. 8A6 can completely degrade the feather in 20 h, which is an efficient keratin degrader reported so far. Comprehensive transcriptome analysis continuously tracks the metabolism of Bacillus sp. 8A6 throughout its growth in feather medium. It reveals for the first time how the strain can acquire nutrients and energy in an oligotrophic feather medium for proliferation in the early stage. Then, the degradation of the outer lipid layer of feather can expose the internal keratin structure for disulfide bonds reduction by sulfite from the newly identified sulfite metabolic pathway, disulfide reductases and iron uptake. The resulting weakened keratin has been further proposedly de-assembled by the S9 protease and hydrolyzed by synergistic effects of the endo, exo and oligo-proteases from S1, S8, M3, M14, M20, M24, M42, M84 and T3 families. Finally, bioaccessible peptides and amino acids are generated and transported for strain growth. The keratinase has been applied for soybean hydrolysis, which generates 2234 peptides and 559.93 mg/L17 amino acids. Therefore, the keratinases, inducing from the poultry waste, have great potential to be further applied for producing bioaccessible peptides and amino acids for feed industry.
Similar content being viewed by others


Introduction
More than 10 million tons of feather wastes are annually accumulated from the poultry farm [1]. These 90% underexploited and recalcitrant feather proteins have great potential to be converted to bioaccessible peptides and amino acids for supplementing the animal feed [2, 3]. Meanwhile, feathers are also the worth substrate to induce and produce the high-performance microbial keratinases, which can be further used in agriculture and diverse industries relating to leather, textile, pulp and paper, textile, detergent, fine chemical production, feed and food, cosmetic and medicine [4, 5]. Both the keratinolytic bioaccessible products and the multi-functional keratinases are in highly demand in the future sustainable and new bioeconomy era.
Feathers (composed of 32.2% of α-helix, 53.6% of β-sheets and 14.2% turns) [6] are classified as hard keratins with diversified morphological structures and numerous disulfide bonds [7]. Currently, keratinases such as alcalase, esperase, savinase, versazyme and prionzyme from S8 family are commercially available. However, a single type of protease cannot degrade keratin sufficiently. Meanwhile, keratinases from different microbial origins have related specific cleaving preferences. Qiu et al. [8] have concluded the bacterial and fungal keratinases including the endoproteases from S1, S8, S16, M4, M16, M36 families, exoproteases from S10, M14, M28, M38, M55 families and oligopeptidases from M3 and M32 families. AA11 lytic polysaccharide monooxygenases (LPMOs), found in fungal degradation systems, were identified to break down the glycosylation bonds between N-acetylglucosamine and serine or threonine in the non-coiled head structure of the keratin filament dimer [9, 10].
Unfortunately, feathers cannot be completely degraded by the sole synergistic effect of different types of keratinases. It has been reported that the blending of the purified recombinant keratinases cannot well degrade the feather [11, 12]. The microbial cells, on the other hand, can completely degrade the feather when using the feather as the carbon and nitrogen source [13,14,15,16,17]. Therefore, the microbial metabolites and the cell membrane system might have significant functions in the keratin degradation. Grumbt et al. [18] were the first to propose the mechanism of keratin degradation by dermatophytes which relied on the sulfite efflux pump to secrete the reducing agent sulfite from environmental cysteine via the action of the enzyme cysteine dioxygenase Cdo1. Then the sulphite released can weaken the keratin structure for further degradation by keratinases. In Jian Chen’s group, the fungal and bacterial keratin degradation process has been concluded into the factors of cell membrane, mechanical pressure, thiolysis and enzyme hydrolysis [19]. The thiolysis proposal has been further confirmed in the keratin degradation process by the engineered B. licheniformis BBE11-1 and its mutants, which showed that cysteine catabolism can mediate the catalytic procedure of keratinases via the secreting the sulfite [20]. The supplement of sodium sulfite to the cell-free keratinases can significantly improve the recycled keratin waste to amino acids [21]. Li et al. [22] were the first to analyze the transcriptome when the Streptomyces sp. SCUT-3 grew in the feather and indicated that the disulfide bond reduction is the first key step for feather degradation. Then the proteases hydrolyzed the feather to peptides and amino acids for the bacteria nutrients. Iron addition and aeration have been discussed to improve the feather degradation efficiency. These studies have mainly elucidated the thiolysis mechanism of keratin degradation. However, it is still unclear how the keratin degraders acquire nutrients and energy in an oligotrophic feather medium for rapid adaptation and proliferation in the early stage. Meanwhile, the full elucidation of the mechanism of sufficient keratin degradation is still needed for efficiently refining the keratin waste.
The efficient keratin degrader, Bacillus sp. 8A6, was recently identified to completely degrade feathers in our previous work [2]. This study was the first to comprehensively analyze the metabolic regulation process when this efficient keratin degrader Bacillus sp. 8A6 grew in the feather medium. In addition, soybeans are the major sources for food and feed proteins [23, 24]. However, the bioaccessibility of soybean proteins is limited by the trypsin inhibitors, antinutritional factors, glycinin and β-conglycinin and the cell wall structural integrity [25, 26]. Alkaline protease hydrolysis has been confirmed with the function to change the structure and conformation of soybean protein [27]. Therefore, the resulting stable and efficient keratinases from Bacillus sp. 8A6 were further applied for soybean protein hydrolysis. Relatively high amounts of bioaccessible peptides and amino acids after the degradation revealed the potential for applying the keratinase in feed industry.
Materials and method
Strain and culture condition
Bacillus sp. 8A6 was obtained from the Bacillus Genetic Stock Centre at Ohio State University (Biological Sciences 556 484 W. 12th Ave Columbus, OH 43210-1214). The LB medium (10 g/L tryptone, 5 g/L yeast extract and 10 g/L NaCl) was used for cell growth. The feather medium contained 0.75 g/L NaCl, 1.75 g/L K2HPO4, 10 mM MOPS, 0.25 g/L MgSO4·7H2O, 0.055 g/L CaCl2, 0.010 g/L FeSO4·7H2O, 0.005 g/L ZnSO4·7H2O and different concentration of feathers (0.5, 1, 2, 3, 4, 5, and 10% (w/v)).
RNA extraction and synthesis of cDNA
After the single-factor and response surface method (RSM) optimization, the optimal conditions were inoculation amount of 1.83 mL, feather concentration of 3.905% (w/v) and pH 9.21 (see Additional file 1: Supplemental Materials and Method; Supplemental Results and Discussion), Bacillus sp. 8A6 was inoculated into the sterile feather medium and growth under the optimal conditions for 0, 8, 20 h. Cells were collected for RNA extraction. The cells from three time points were mixed and RNA was extracted from the mixture. After grinding cells into powder with liquid nitrogen, RNA was extracted by the instruction of the RNAprep pure Bacteria Kit (DP430, Tian gen). Then, the RNA was reverse transcribed to cDNA using reverse transcriptase (TAKARA, RR036A) according to the manufacturer's instructions. The reaction system was as follows: 4 μl 5X PrimeScript RT Master Mix (Perfect Real Time), 10 μl total RNA, and 6 μl RNase Free ddH2O. The reaction conditions were as follows: 37 °C for 15 min, 85 °C for 5 s and then 4 °C.
Selection of internal reference genes and design of real-time fluorescence quantitative PCR (RT-qPCR) primers
According to Chen’s research [28], 16sRNA was selected as the internal reference gene. RT-qPCR was used to determine the transcript levels of 7 differentially expressed genes (DEGs) from the S8, S9 and S1A families. Primer Premier 5.0 was used to design RT-qPCR primers, which were synthesized by Sangon Biotech (Shanghai) Co. (Additional file 1: Table S1).
RT-qPCR reaction
The prepared cDNA was used as a template for the RT-qPCR reaction using TB Green® Premix Ex Taq™ II (Tli RNaseH Plus) (TAKARA, RR820A). The reaction system was as follows: 10 μl TB Green® Premix Ex Taq™ II, 1 μl each of forward and reverse primers, 1 μl cDNA, and 7 μl ddH2O. RT-qPCR cycling conditions were 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s, 55 ℃ for 30 s and 72 °C for 20 s, at last, the melting curve was generated at 95 °C for 15 s, 65 °C for 60 s and 95 °C for 15 s. Each reaction was performed 3 times. Using 16sRNA as an internal reference gene, the relative expression level of each gene was calculated using 2−ΔΔCT method.
Transcriptome analysis
The RNA was sent to Genewiz company (Suzhou, China) for Illumina sequencing. The raw data were filtered by Cutadapt (v 1.9.1) and mapped to the Bacillus sp. 8A6 genome sequences (Genbank: accession number QFZE00000000) via software Bowtie2 (v2.2.6) [29]. Then HTSeq (v0.6.1p1) [30] was used to estimate gene expression levels from the pair-end clean data. Differential expression analysis used the DESeq2 Bioconductor package, a model based on the negative binomial distribution [31, 32], and sequencing depths at the 0 h, 8 h, and 20 h were 1227X, 1391X and 1283X, respectively. After being adjusted by Benjamini and Hochberg’s approach for controlling the false discovery rate, Padj of genes were set < 0.05 to detect differential expressed ones. Sequencing data quality assessment was analyzed using the software FastQC (v0.10.1). In general, the sequencing error rate should be less than 0.5% at each base position. If the sequencing error rate was denoted by e and the base quality value of Illumina sequencing was denoted by Qphred, the Qphred = − 10log10(e), low-quality data were then filtered and contamination and splice sequences were removed by the software Cutadapt (version 1.9.1). GO enrichment analysis method was Goseq [33], which was based on Wallenius non-central hyper-geometric distribution. Compared with the ordinary hyper-geometric distribution, this distribution was characterized by the difference between the probability of extracting an individual from a certain category and the probability of extracting an individual from outside a certain category. The difference in this probability was obtained by estimating the preference of gene length. Thus, the probability of GO term enrichment by differential gene can be calculated more accurately. The GOSeq (v1.34.1) was used to identify Gene Ontology (GO) terms that annotate a list of enriched genes with a significant p-value less than 0.05. And topGO was used to plot DAG. The Kyoto Encyclopedia of Genes and Genomes (KEGG) web server was used to extract enzyme codes and the KEGG pathways. The software used in this analysis was EdgeR. The results were measured according to the difference significance criteria (differential gene expression changes of more than 2 times and q-value less than 0.05) for screening. Cluster of Orthologous Groups of proteins (COG) database was used to predict the function of proteins or protein assemblies [34]. COG classification of the major distribution of differential genes was obtained through annotation and classification analysis of COG database [35].
Analysis of protease activity
The protease activity was analyzed by using Azo-casein as substrate as described with few modifications [2]. 20 μl 1.5% (w/v) azo-casein in 50 mM Tris–HCl buffer (pH 9) as substrate and 20 μl crude enzyme solution (diluted fivefold with 50 mM Tris–HCl buffer pH 9.0) were mixed and incubated at 60 °C for 15 min at 500 rpm. One arbitrary unit (U) of protease activity is defined as the amount of enzyme that causes an increase in absorbance of 0.01 between the sample and the control at 405 nm under the assay conditions [2].
Analysis of keratinase activity
The keratinase activity was detected as described [36]. 0.4 g keratin azure were cut into pieces and suspended in 100 mL 50 mM sodium carbonate buffer (pH 9.0). 500 μl keratin azure suspension and 500 μl enzyme solution were then mixed to react at 50 °C for 24 h at 1000 rpm. One unit of keratinase activity was defined as the amount of enzyme that caused an increase of 0.01 absorbance at 595 nm.
Optimum pH and temperature of protease
The optimum pH of protease was determined according to the method of protease activity analysis described above with some modifications that crude enzyme solution was diluted by buffer at a pH range from 6 to 10 (50 mM sodium acetate buffer pH 6, 50 mM Tris–HCl buffer pH 7–9, and 50 mM sodium carbonate buffer pH 10). And the substrate Azo-casein (50 mM) was also prepared with buffers at pH 6–10.
Similarly, in the determination of the optimum temperature of enzymes, diluted crude enzyme solution and azo-casein were prepared by above optimum pH buffer. The reaction system was incubated at the range of 30–65 °C.
Tolerance of pH and temperature on protease activity
The crude enzyme solution was diluted 2.5 times with buffers of pH 6–11 (50 mM sodium acetate buffer pH 6, 50 mM Tris–HCl buffer pH 7–9, 50 mM sodium carbonate buffer pH 10, and 50 mM sodium carbonate buffer pH 11) and incubated in the 4 °C for 1 h or 4 h. And then diluted it 2 times again for the analysis of the residual activity of protease by Azo-casein as substrate. Similarly, the crude enzyme solution diluted 2.5 times with 50 mM pH 9 Tris–HCl buffer was incubated at the temperature range of 30–65 °C for 1 h or 4 h and the residual activity of protease was measured as above.
Morphological analysis of the degraded feathers by scanning electron microscopy (SEM)
The surface morphology at different times in the degradation process of feathers was investigated by SEM, including 0 h, 8 h and 20 h. The feather culture was harvested at 0 h, 8 h and 20 h, and the feather was washed three times with phosphate buffer saline (50 mM, pH 7.2). The remaining steps of sample preparation follow the literature [37].
Degradation of soy protein isolate (SPI), peptides and free amino acids analysis
The biodegradability of SPI was investigated. The SPI degradation system contained 1 g SPI and 2 mL Bacillus sp. 8A6 keratinase or NY100 with the same enzyme activity, then the system was suspended with 10 mL sodium carbonate buffer (pH 9) and incubated at 60 °C for 1 h with shaking at 250 rpm. Besides, the system without Bacillus sp. 8A6 keratinase or NY100 was used as a negative control. After centrifugation at 12,000 rpm for 10 min, the supernatant was analyzed for amino acids and short chain peptides by Nano LC–MS/MS in Standard Testing Group Co. Ltd. (Qingdao, China).
Statistical analysis
The statistical analyses were executed by GraphPad Prism 8.3.0. In this experiment, the mean ± standard deviation (SD) of more than three sets of data were compared, and checked the significance between each of the two groups by one-way ANOVA. *p < 0.05 was regarded as statistically significant.
Results and discussion
Comprehensive transcriptome analysis for Bacillus sp. 8A6 grown in feather medium
RNA preparation and transcriptome sequencing
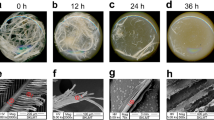
After single-factor and RSM optimization of the growth conditions of Bacillus sp. 8A6 in the medium with feather as the sole carbon and nitrogen source (Additional file 1: Supplemental Results and Discussion, Table S2, Fig S1). The feathers were all decomposed within 20 h (cultures in flask in Fig. 1, Additional file 1: Supplemental Results and Discussion), which is the efficient keratin degrader reported so far when compared with the talented keratin degraders Amycolatopsis keratiniphila subsp. keratiniphila D2T, which can degrade feathers within 3 days [38, 39]; and Ectobacillus sp. JY-23 [40], Bacillus species such as Bacillus sp. CN2 (48 h) [41], B. licheniformis, B. subtilis, B. cereus, B. thuringiensis, B. aerius, Bacillus sp. CL18, etc. (24–84 h degradation with extra nitrogen source in the medium) [42,43,44,45,46,47], Pseudochrobactrum sp. IY-BUK1 (3 days), and even microbial consortia KMCG6 (3 days) [48], etc. Meanwhile, the keratinase activity of Bacillus sp. 8A6 achieved 136.5 U/mL and it was considerably higher than that of other strains [36, 49,50,51]. SEM showed that most of the feathers’ barbules and barbs have been degraded after 8 h of inoculation (Fig. 1A–F). It was further shown that the strains can work on the fracture section of the rachis for the feather degradation (Fig. 1E–F). After 20 h inoculation, the strains were enriched on the small feather chips to achieve the comprehensive degradation of the feather (Fig. 1G–I). According to the morphological changes of feathers when Bacillus sp. 8A6 grown in the feather medium and the growing properties in LB medium (Additional file 1: Fig S2). Therefore, the samples when Bacillus sp. 8A6 grew in feather medium for 0, 8, and 20 h were prepared for RNA extraction and transcriptome sequencing for revealing the keratin degradation mechanism.

The morphological changes of feather degradation by Bacillus sp. 8A6 under the optimized conditions were observed by scanning electron microscope. The first row is initial incubation (0 h) (A–C), the second row is 8 h incubation (D–F) and the third row is 20 h incubation (G–I)
After transcriptome sequencing, the raw reads (Additional file 1: Table S3) were cleaned. 4.446, 5.063 and 4.169 Gbp of data in total were obtained and 98.57%, 97.49% and 98.71% reads were mapped to the genome sequences for the Bacillus sp. 8A6 before (0 h) and after being inoculated in the feather medium for 8 h and 20 h, respectively (Additional file 1: Table S4). Finally, 3863 genes were identified. The Pearson correlation coefficient and principal component analysis indicated that after inoculation in the feather medium for 8 h and 20 h, the transcripts changed greatly (Additional file 1: Fig S4A and S4B). According to FPKM statistics, the density of the 8 h inoculation of Bacillus sp. 8A6 was higher than that of the 20 h inoculation followed by the original culture. 1006 genes up-regulated and 790 genes down-regulated after 8 h incubation (Additional file 1: Fig S5A). Then 412 genes were up-regulated and 503 genes down-regulated after 20 h incubation (Additional file 1: Fig S5B). The hierarchical clustering showed an obvious change trend that most of the genes of the first two clusters were up-regulated obviously when Bacillus sp. 8A6 was grown in feather medium for 8 h. These genes should be important for strain inoculation on the feather, keratin decomposition, nutrients transportation and absorption (Additional file 1: Fig S6).
Overall analysis of transcriptome
After inoculation, the Bacillus sp. 8A6 needs to survive in the oligotrophic and insoluble feather medium without any readily accessible carbon and nitrogen source, which is different from the nutrition-rich LB medium. GO analysis (Additional file 1: Fig S7) showed that functional genes related to sporulation and cell wall regulation were expressed and enriched in the following genes (Fig. 2A). During growth and proliferation, Bacillus sp. 8A6 cells had high energy requirements, as shown by the log10(p-value) of fatty acid β-oxidation, proton-transporting ATP synthase, inorganic phosphate transmembrane transporter and iron uptake. According to the FPKM statistics, the sulfur cluster binding functional genes were also enriched during this period, which might relate to the sulfite synthesis and exportation for breaking down the feather disulfide bonds. The resulting loose feather could be easily accessed and further hydrolyzed by the induced proteases/peptidases. When the cells grew in the feather medium for 20 h, the GO terms changed greatly (Fig. 2B). The cells continuously required tremendous energy and oxygen for cell growth, metabolism and proliferation through the oxidoreductase, proton-transporting ATP synthase and complex, cytochrome, iron binding and aerobic respiration. Meanwhile, the serine endopeptidases and aminopeptidases were enriched for complete feather degradation. The genes for mass-transportation were enriched obviously during this stage, such as the protein, amino acid, proton, transmembrane transport and nitrate assimilation, which are mainly responsible for up-taking the feather degraded soluble peptides and amino acids.

Gene Ontology (GO) analysis of the molecular function, cellular component and biological process for the groups 0 h VS 8 h (A) and 8 h VS 20 h (B) according to the -log10(p-value) when Bacillus sp. 8A6 grew in feather medium. The higher −log10(p-value) indicates the more significant enrichment of the GO term. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of the pathways for the groups 0 h VS 8 h (C) and 8 h VS 20 h (D) according to the −log10(p-value) when Bacillus sp. 8A6 grew in feather medium. Meanwhile, the group 0 h VS 8 h meant the comparisons of gene expression between 0 and 8 h. The group 8 h VS 20 h meant the comparisons of gene expression between 8 and 20 h. The most significantly different pathways are shown according to the Q-value. Other significantly different pathways are shown in Additional file 1: Fig. S7
The KEGG analysis showed that after Bacillus sp. 8A6 inoculation for 8 h, the cells were mainly colonized on the feather for growth. The result showed that the sulfur relay system was active when the cells grew in the feather medium (Fig. 2C). The up-regulated ThiS ubiquitin-like proteins (Ubls) were a key sulfur carrier protein and signaling messenger that control cell proliferation [52]. When the cells grew on the feather medium from 8 to 20 h, the feather has been degraded and the amino acids metabolism has been up-regulated obviously, including the phenylalanine, tyrosine, tryptophan, histidine, D-alanine, together with the nitrogen metabolism (Fig. 2D). The substantial difference was that the gene for protein export and ABC transporters were highly induced during this stage. It indicated that the cells were extremely active to consume the degraded soluble peptides and amino acids for growth. The COG analysis further identified 113 up-regulated genes for amino acids transport and metabolism after 8 h inoculation, which was followed by 16 related up-regulated genes after 20 h inoculation (Additional file 1: Figs. S8 A and B).
Mechanism for Bacillus sp. 8A6 growth and feather degradation
Energy and nutrients for Bacillus sp. 8A6 surviving in feather medium at the early stage
The Bacillus sp. 8A6 can grow in the optimized feather medium only including feather and trace minerals. The question of how the strain starts to use such resistant polymers as carbon and nitrogen source for growth and proliferation is still unelucidated. In this study, all the genes for fatty acid (hexadecanoate) degradation were identified and all of them were up-regulated (Fig. 3). It has been reported that the surface of feathers is rich in lipids (5%) [53]. The outer lipid layer might consist of hexadecanoate along with other fatty acids to bound through thioester linkages to the keratin proteins, which was similar to the wool keratin as described by Ghosh et al. [54]. Li et al. [55] reported that fatty acid metabolism had significant up-regulated expression. This study confirmed that when Bacillus sp. 8A6 inoculated in the feather medium for 8 h, the strain up-regulated the fatty acid degradation pathways (Fig. 3A). The resulting acetyl-CoA was then involved in the TCA cycle, which was also highly active for all the enzymes (Fig. 3A). The intermediates of the active TCA cycle can further stimulate other metabolic pathways, providing necessary energy and nutrients for the growth and proliferation of the Bacillus sp. 8A6. During the same period, the genes of the oxidative phosphorylation in complex II (succinate dehydrogenase, SdhA & SdhB), complex IV (cytochrome c oxidase, CoxA, CoxB, CoxC & CoxD), complex V (F-type ATPase, F1 unit (α, β, γ, δ, ε) and F0 unit (a & b)) were also up-regulated when the strain grew in the feather medium for 8 h. The complex II & IV can push the protons across the cell membrane into the cytoplasm, resulting in the formation of electromotive force which can further provide the free energy for ATP synthesis by the up-regulated complex V. Therefore, when the Bacillus sp. 8A6 started to grow in the feather medium, the oxidative phosphorylation was stimulated to provide sufficient ATP for cell growth and metabolism (Fig. 3C). After the breakdown of outer lipid layer and the exposure of internal keratin, the fatty acid regulation, TCA cycle and oxidative phosphorylation were down-regulated when the strains grew in the feather medium for 20 h (Fig. 3B and D). Therefore, the fatty acid degradation, TCA cycle and oxidative phosphorylation were the key first steps for providing energy and nutrients for strain survival and growth in the feather medium. Meanwhile, the degradation of the outer lipid layer of feather can assist in exposing the internal keratin structure to further degradation.

Fatty acid degradation, TCA cycle (A, B) and oxidative phosphorylation (C, D) analyses for the groups 0 h VS 8 h and 8 h VS 20 h when Bacillus sp. 8A6 grew in feather medium, respectively. Meanwhile, the group 0 h VS 8 h meant the comparisons of gene expression between 0 and 8 h. The group 8 h VS 20 h meant the comparisons of gene expression between 8 and 20 h. The up-regulated, down-regulated and unchanged genes are indicated as red, blue and green colors, respectively
New sulfite metabolic pathway for breaking down the disulfide bonds
The main challenge for sufficient keratin decomposition is the abundant cross-linked disulfide bonds in/among the keratin filaments [56]. It is necessary to reduce the disulfide bridges to weaken the rigid structure. This study was the first to identify a different sulfite metabolism pathway after the Bacillus sp. 8A6 being inoculated in the feather medium for 8 h (Fig. 4A). l-Cysteine was converted to l-cystathionine by cystathionine-γ-synthase. Then the key enzymes including the β-cystathionase (EC4.4.1.13), homocysteine S-methyltransferase (EC2.1.1.10), methionine synthase (EC2.1.1.13) and 5-methyltetrahydropteroyltriglutamate-homocysteine S-methyltransferase (EC2.1.1.14) were highly up-regulated for l-methionine synthesis. The l-methionine was further converted to methylmercaptan and sulfite by the up-regulated alkanesulfonate monooxygenase (EC1.14.14.5). Meanwhile, the sulfite reductase (NADPH) (EC1.8.1.2) which was down-regulated for sulfite accumulation. The sulfite was then exported to exocellular to lose the feather structure. When the cells were inoculated into the feather medium for 20 h, the sulfite metabolism was down-regulated and the sulfite was converted to sulfide (Fig. 4B). Therefore, the sulfite accumulation and exportation were crucial during the beginning stage of keratin degradation. The synergistic factor sulfite for keratin degradation has been confirmed by mixing keratinase and sodium sulfite in vitro for complete feather degradation [20, 22]. Grumbt et al. [18] have proposed the model for keratin degradation by dermatophytes which were mainly dependent on the cysteine dioxygenase (Cdo1) and sulfite efflux pump (Ssu1). The route further cleared the cysteine catabolism that aspartate aminotransferase (Ast1) was important for deamination after the Cdo1 oxidation [20]. Two cysteine dioxygenase genes (cdo1 and cdo2) were identified and up-regulated. Meanwhile, the sulfite exporter gene tauE instead of sulfite efflux pump gene (ssu) was found when bacteria Streptomyces sp. SCUT-3 grew in feather medium [22]. However, only 4 aspartate aminotransferase genes were identified in the 4 pathways when Bacillus sp. 8A6 grew in feather medium, the 4 pathways are purine metabolism, 2-oxocarboxylic acid metabolism, microbial metabolism in diverse environments and glycine, serine and threonine metabolism. The new up-regulated sulfite metabolic pathways for Bacillus sp. 8A6 in our study could further provide potential strategies for construction efficient engineering Bacillus strains for keratin decomposition.

Sulfite metabolism for the groups 0 h VS 8 h and 8 h VS 20 h when Bacillus sp. 8A6 grew in feather medium. Meanwhile, the group 0 h VS 8 h meant the comparisons of gene expression between 0 and 8 h. The group 8 h VS 20 h meant the comparisons of gene expression between 8 and 20 h. The up-regulated and down-regulated genes are indicated as red and blue color, respectively
It has been reported that disulfide reductases can degrade disulfide bonds in keratin [8, 57]. In this study, 4 disulfide reductases (3 from TlpA family and 1 from CoA family) were up-regulated when the Bacillus sp. 8A6 grew in the feather medium for 8 h (Table 1 and Additional file 1: Table S5). After 20 h growth, 1 TusA family sulfurtransferase was up-regulated (Additional file 1: Table S6). The synergistic effect of disulfide reductases and keratinases has been confirmed by blending these two types of enzymes [58, 59]. However, the keratinolytic disulfide reductases from the efficient degraders have not yet been well exploited and characterized currently.
Diverse proteases for keratin decomposition
After Bacillus sp. 8A6 growing in feather medium for 8 h, 52 up-regulated proteases were identified from metalloproteases (M3, 2M14, M15, 4M20, 3M23, M24, M42, M56, 2M78, M84), cysteine proteases (C39, 2C40, C60, C82), serine proteases (S1, 8S8, 3S9, 2S11, S13, S14, S16, S41, 2S66), unknown proteases (U57), glutamic proteases (2G5), aspartic proteases (2A25, A36), threonine proteases (T3), and 3 unclassified proteases (Additional file 1: Table S5). When the strain grew in the feather medium for 20 h, 15 proteases from metalloproteases (M24, M50, M78), cysteine proteases (C40, 2C56, C60), serine proteases (3S8, S24, S26), and 3 unclassified proteases (Additional file 1: Table S6). Seven genes from different families validated RNA-seq results using RT-qPCR analysis. The results showed that all seven genes were up-regulated at 8 h compared to that of control, and at 20 h, RS16285, RS08615 from S8 family, RS11220 from S9 family, and RS14955 from S1A family were down-regulated at 20 h (Fig. 5). The RT-qPCR statistics of these 7 differential genes were consistent with the RNA-seq sequencing results. Functional analysis further showed that some proteases were mainly related to lysing cell wall (such as proteases from M15, M23, M56, S11, S13, S66, C40, C60, C82 families), sporulation (such as proteases from A25 and A36 families), and cleaving the ImmR protein from M78 family. Therefore, after comprehensive analysis of the function of the up-regulated proteases after the cells growing in the medium for 8 and 20 h, 4 disulfide reductases and 22 proteases from S1, S8, S9, M3, M14, M20, M24, M42, M84, and T3 families have the great potential for synergistic keratinolysis (Table 1). The identified 3 significantly up-regulated S9 peptidase/acetylxylan esterase/chitobiase/beta-hexosaminidase might have function on the glycoprotein groups, especially on the head domain of the polypeptide monomer of the keratin structure. Then the charge of the filament head structure will be changed [9], resulting in the de-assembly of the keratin filaments. The disassembled keratin can be further hydrolyzed by the synergistic proteases as follows. Recently, the commercial and the most studied keratinases were from S8 family [8, 9]. In our study, eight S8 proteases have great potential for keratin decomposition. S1 family proteases are endoproteases with a classic catalytic triad, which also contributed to the keratin degradation. Two keratinolytic S1 serine proteases have been identified by mass spectrometric peptide mapping and purified from A. keratinophila subsp. keratinophila D2T [60]. Our previous secretome analysis of Bacillus sp. 8A6 also indicated that this S1 trypsin-like serine protease was up-regulated when it grew in the pig bristle and feather medium [2]. Other keratinolytic proteases in S1 family originated from Paenarthrobacter nicotinovorans [61], Nocardiopsis sp. TOA-1 [62], Streptomyces fradiae var. k11 [63] and S. albidoflavus [64] were also characterized. This study further identified that the M3 oligoendopeptidase was significantly up-regulated in this bacteria species as the keratinolytic M3 oligopeptidase from the fungus Onygena corvina [3]. The metalloaminopeptidases (M42), exo-metallopeptidase (M14, M20, M24) and endo-metallopeptidase (M84) were the first discovered and have great potential to participate in the keratin decomposition. Meanwhile, the γ-glutamyltransferase from T3 family was both up-regulated significantly in Bacillus sp. 8A6 secretome [2] and transcriptome when the strain grew in the keratin (bristles and feather) medium, indicating that the γ-glutamyltransferase was important to balance the levels of intracellular cysteine [65, 66] for further sulfitolysis of feather [67]. Therefore, this study can provide a shortcut to develop efficient keratinolytic enzymes including serine proteases, metallopeptidase, γ-glutamyltransferase and disulfide reductases. The advanced protein engineering and heterogenous expression might further improve the keratinolytic properties for industrial and commercial application [4].

Comparison of seven DEGs and RT-qPCR. The data from this study are expressed as mean ± SD of three parallel determinations
Further absorption of peptides and amino acids after keratin degradation
The soluble peptides and amino acids were generated after the synergistic effects of keratinolytic proteases. These bioaccessible peptides and amino acids can be further absorbed into the cell for growth and metabolism by specific transporters. During the early stage of Bacillus sp. 8A6 grew in the feather medium, all the genes (Peb1A, Peb1B and Peb1C) for the aspartate/glutamate/glutamine transporter, gene (TcyM) for cystine transporter and genes (DppE, DppC and DppD) for dipeptide transporter were up-regulated. Meanwhile, the biotin transporter (BioY, EcfT and EcfA1) and the iron transporter (MtsA, MtsC and MtsB) were also up-regulated. Li et al. [22] have confirmed that the iron uptake can improve keratin degradation. Amino acid, nitrogen and urea metabolism have been further comprehensively analyzed in this study. The pathways for aromatic amino acids and histidine metabolism were analyzed when Bacillus sp. 8A6 inoculated in the feather medium for 8 h. The enzymes such as 3-dehydroquinate dehydratase (EC4.2.1.10) and 3-phosphoshikimate 1-carboxyvinyltransferase (EC2.5.1.19) were up-regulated for shikimate and chorismate synthesis after the active pentose phosphate pathway. Then the key enzymes for all the aromatic amino acids (phenylalanine, tyrosine and tryptophan) were equally up-regulated (Fig. 6A and D). Meanwhile, the alkaline histidine biosynthesis pathway was also up-regulated obviously (Fig. 6B and E). The early stage of the inoculation on feather medium can also activate the intermediates synthesis of pyruvate group amino acids including valine, leucine and isoleucine. The cysteine and methionine metabolisms have been active at the early stage to provide enough sulfite for weakening the keratin structure as discussed above.

Amino acid, nitrogen and urea metabolism for the groups 0 h VS 8 h and 8 h VS 20 h when Bacillus sp. 8A6 grew in feather medium. Meanwhile, the group 0 h VS 8 h meant the comparisons of gene expression between 0 and 8 h. The group 8 h VS 20 h meant the comparisons of gene expression between 8 and 20 h. The up-regulated and down-regulated genes are indicated as red and blue color, respectively
Kang et al. [48] have indicated that the urea cycle might boost the keratinolytic efficiency and be involved in the keratin decomposition process through the metagenome analysis. In our study, all the genes for the urea cycle have been identified, however, most of them were down-regulated at the early inoculation stage (Fig. 6C). After the strain grew in the feather medium for 20 h, all the genes for urea cycle were silenced (Fig. 6F). Urea cycle can be active when the excessive amino acids are degraded to produce extra nitrogen. Therefore, the urea cycle was down-regulated as there were no extra amino acids for degradation, especially when the strain was inoculated in the oligotrophic feather medium. In particular, all living cells need to synthesize the aromatic amino acids, histidine and intermediates of pyruvate group amino acids as above for growth and metabolism. Interestingly, nitrate reductase (EC1.7.99-) and nitrite reductase (NADH) (EC1.7.1.15) which can convert nitrate to ammonia were up-regulated during the late period (20 h). The ammonia could be attribute to the pH increase in the feather medium. This is consistent with Sivakumar’s research [68].
Characterization of the keratinolytic proteases
So far, the maximum keratinase production by Bacillus sp. 8A6 has been easily achieved under the optimized fermentation conditions obtained by single-factor optimization and RSM. Further, the catalytic efficiency of keratinolytic protease was evaluated by measuring its activity at different pH (6.0–11.0) and temperatures (30–65 °C) (Fig. 7A and B). As can be seen from Fig. 7A and B, the optimal pH and temperature for the highest protease activity were separately detected at alkaline pH 8.0 (12,712.5 ± 335.9 U/mL) and at 60 °C (13,862.5 ± 17.7 U/mL), which were similar to other reported keratinolytic proteases [5]. Meanwhile, the acidic/alkaline stability and thermostability were also investigated. Results showed that protease was stable over the pH range of 6.0–10.0 after 4 h of incubation (10,800.0–11,450.0 U/mL) with an exception at pH 11.0. Interestingly, protease lost about 75% of its activity after 1 h of incubation at pH 11.0 but retained the activity after 4 h of incubation (Fig. 7C). In the case of thermostability (Fig. 7D), better stability was observed in the temperature range of 30–50 °C after 1 h of incubation (10,137.5–11,537.5 U/mL), which was retained for 4 h of incubation (10,825.0–12,837.5 U/mL). Thus, keratinolytic protease is active over a wider pH and temperature range, which is highly desirable for industrial applications.

A Optimal pH of Bacillus sp. 8A6 keratinolytic protease. B Optimal temperature of Bacillus sp. 8A6 keratinolytic protease. C Alkaline stability of Bacillus sp. 8A6 keratinolytic protease. D Thermostability of Bacillus sp. 8A6 keratinolytic protease. The green and yellow mean the Bacillus sp. 8A6 keratinolytic protease incubating for 1 h and 4 h, respectively. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
Keratinases for producing functional peptides and amino acids from the soybean
Keratinases can be used as additives in animal feed to improve digestibility [69]. The commercial keratinase Cibenza DP100™ and Versazyme from B. licheniformis PWD-1 have been used in animal feed and significantly improved the crude protein digestibility [70, 71]. In this study, the Bacillus sp. 8A6 keratinases were the first attempt to be used for improving soybean protein digestibility. The results showed that efficient keratinase could be applied for soybean protein hydrolysis, resulting in the formation of functional peptides and amino acids. The released functional peptides and amino acids after the degradation by Bacillus sp. 8A6 keratinases and well-known soybean degradation commercial protease NY100 were analyzed. During the hydrolysis of soybean protein, Bacillus sp. 8A6 keratinases produced 2234 short peptides (Supplemental peptide list 1) with a relative concentration of 1.14 × 1011, higher than commercial NY100, which produced 1839 short peptides (Supplemental peptide list 2) at a relative concentration of 8.34 × 1010. On the contrary, negative control (CK) produced 108 short peptides (Supplemental peptide list 3) with a relative concentration of 3.49 × 109. Comparatively, the species and concentration of short peptides produced by Bacillus sp. 8A6 keratinases and commercial NY100 separately increased nearly 20-fold and 1–2 orders of magnitude compared to CK, proofing the superiority of Bacillus sp. 8A6 keratinases. Further analysis of amino acids released during hydrolysis, it was found that 17 functional amino acids, including 6 essential amino acids (lysine, phenylalanine, threonine, isoleucine, leucine, and valine) were produced after 1 h of degradation at 60 °C (Table 2). Among the amino acids, methionine (100.23 mg/L) and phenylalanine (82.81 mg/L) for Bacillus sp. 8A6 keratinases, leucine (82.44 mg/L) and methionine (66.05 mg/L) for NY100 were the two most abundant amino acids, with almost twice the amount of abundant amino acids released by CK. More importantly, the most abundant amino acids produced by Bacillus sp. 8A6 keratinases and NY100 each contained one essential amino acid, namely phenylalanine and leucine. Meanwhile, the amino acid production of Bacillus sp. 8A6 keratinases and NY100 both reached superior levels (559.93 mg/L, 460.83 mg/L) at 1 h of hydrolysis, whereas the amino acid production of CK only reached 105.46 mg/L. In addition, Bacillus sp. 8A6 keratinases degrade soybeans to produce more amino acids than papain [72]. This efficient soybean protein degradation by Bacillus sp. 8A6 keratinase can not only improve the apparent digestibility of the soybean protein, but also have the potential to reduce or eliminate the antigenicity of soybean protein [27]. Therefore, the complete degradation of soybean protein and the released abundant functional peptides and amino acids from the soybean protein indicated that the keratinases might have the potential for feed application.
Conclusions
In this study, we proposed the feather degradation mechanism as follows according to the comprehensive transcriptome analysis (Fig. 8): (1) fatty acid degradation, TCA cycle and oxidative phosphorylation firstly provide energy and nutrients for strain growth in the oligotrophic feather medium during the early stage; (2) the degraded outer lipid layer of feather can be further destroyed the disulfide bonds by sulfite, disulfide reductases and iron uptake; (3) the resulting loosen keratin was further de-assembled by the S9 proteases and hydrolyzed by synergistic effects of the endo, exo and oligo-proteases from the S1, S8, M3, M14, M20, M24, M42, M84, and T3 families; (4) the generated bioaccessible peptides and amino acids were transported by transporters to the cell for strain growth and metabolism. Finally, the efficient keratinases were used for soybean hydrolysis and generated 2234 short peptides and 559.93 mg/L 17 free amino acids, which were much higher than the commercial protease and negative control. Therefore, the keratinase can be produced during the feather degradation, which has great potential to be used for feed application.

Summary of keratin degradation mechanism, the resulted keratinolytic bioaccessible products and the multi-functional keratinases can be applied in feed industry
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- RT-qPCR:
-
Real-time fluorescence quantitative PCR
- DEGs:
-
Differentially expressed genes
- RSM:
-
Response surface method
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- COG:
-
Cluster of Orthologous Groups of proteins
- SEM:
-
Scanning electron microscopy
- SPI:
-
Soy protein isolate
- SD:
-
Standard deviation
- Ubls:
-
Ubiquitin-like proteins
- CK:
-
Negative control
References
Wang Z, Chen Y, Yan M, et al. Research progress on the degradation mechanism and modification of keratinase. Appl Microbiol Biotechnol. 2023;107(4):1003–17. https://doi.org/10.1007/s00253-023-12360-3.
Huang Y, Lezyk M, Herbst FA, et al. Novel keratinolytic enzymes, discovered from a talented and efficient bacterial keratin degrader. Sci Rep. 2020;10(1):10033. https://doi.org/10.1038/s41598-020-66792-2.
Huang Y, Busk PK, Herbst FA, et al. Genome and secretome analyses provide insights into keratin decomposition by novel proteases from the non-pathogenic fungus Onygena corvina. Appl Microbiol Biotechnol. 2015;99(22):9635–49. https://doi.org/10.1007/s00253-015-6805-9.
Su C, Gong JS, Qin J, et al. The tale of a versatile enzyme: molecular insights into keratinase for its industrial dissemination. Biotechnol Adv. 2020;45: 107655. https://doi.org/10.1016/j.biotechadv.2020.107655.
Hassan MA, Abol-Fotouh D, Omer AM, et al. Comprehensive insights into microbial keratinases and their implication in various biotechnological and industrial sectors: a review. Int J Biol Macromol. 2020;154:567–83. https://doi.org/10.1016/j.ijbiomac.2020.03.116.
Ma B, Qiao X, Hou X, et al. Pure keratin membrane and fibers from chicken feather. Int J Biol Macromol. 2016;89:614–21. https://doi.org/10.1016/j.ijbiomac.2016.04.039.
Li Q. Perspectives on converting keratin-containing wastes into biofertilizers for sustainable agriculture. Front Microbiol. 2022;13: 918262. https://doi.org/10.3389/fmicb.2022.918262.
Qiu J, Wilkens C, Barrett K, et al. Microbial enzymes catalyzing keratin degradation: classification, structure, function. Biotechnol Adv. 2020;44: 107607. https://doi.org/10.1016/j.biotechadv.2020.107607.
Lange L, Huang Y, Busk PK. Microbial decomposition of keratin in nature-a new hypothesis of industrial relevance. Appl Microbiol Biotechnol. 2016;100(5):2083–96. https://doi.org/10.1007/s00253-015-7262-1.
Lange L, Huang Y. The potential of integrated bio- and chemical-engineering—for a more sustainable world. GreenChE. 2020;1(1):9–15. https://doi.org/10.1016/j.gce.2020.09.002.
Hamiche S, Mechri S, Khelouia L, et al. Purification and biochemical characterization of two keratinases from Bacillus amyloliquefaciens S13 isolated from marine brown alga Zonaria tournefortii with potential keratin-biodegradation and hide-unhairing activities. Int J Biol Macromol. 2019;122:758–69. https://doi.org/10.1016/j.ijbiomac.2018.10.174.
Li Q. Structure, application, and biochemistry of microbial keratinases. Front Microbiol. 2021;12: 674345. https://doi.org/10.3389/fmicb.2021.674345.
Wu W-L, Chen M-Y, Tu IF, et al. The discovery of novel heat-stable keratinases from Meiothermus taiwanensis WR-220 and other extremophiles. Sci Rep. 2017;7(1):4658. https://doi.org/10.1038/s41598-017-04723-4.
Mahatmanto T, Estiningtyas NR, Maharani SK, et al. Feather waste biorefinery using Chryseobacterium sp. A9.9 adapted to feathers as its sole carbon and nitrogen source. Waste Biomass Valor. 2022;13(10):4137–46. https://doi.org/10.1007/s12649-022-01766-1.
Qin X, Xu X, Guo Y, et al. A sustainable and efficient recycling strategy of feather waste into keratin peptides with antimicrobial activity. Waste Manag. 2022;144:421–30. https://doi.org/10.1016/j.wasman.2022.04.017.
Cavello I, Bezus B, Cavalitto S. The keratinolytic bacteria Bacillus cytotoxicus as a source of novel proteases and feather protein hydrolysates with antioxidant activities. J Genet Eng Biotechnol. 2021;19(1):107. https://doi.org/10.1186/s43141-021-00207-1.
Ramakrishna Reddy M, Sathi Reddy K, Ranjita Chouhan Y, et al. Effective feather degradation and keratinase production by Bacillus pumilus GRK for its application as bio-detergent additive. Bioresour Technol. 2017;243:254–63. https://doi.org/10.1016/j.biortech.2017.06.067.
Grumbt M, Monod M, Yamada T, et al. Keratin degradation by dermatophytes relies on cysteine dioxygenase and a sulfite efflux pump. J Invest Dermatol. 2013;133(6):1550–5. https://doi.org/10.1038/jid.2013.41.
Peng Z, Zhang J, Du G, et al. Keratin waste recycling based on microbial degradation: mechanisms and prospects. ACS Sustain Chem Eng. 2019;7(11):9727–36. https://doi.org/10.1021/acssuschemeng.9b01527.
Peng Z, Xu P, Song Y, et al. Cysteine-mediated cyclic metabolism drives the microbial degradation of keratin. ACS Sustain Chem Eng. 2021;9(29):9861–70. https://doi.org/10.1021/acssuschemeng.1c02627.
Peng Z, Mao X, Zhang J, et al. Biotransformation of keratin waste to amino acids and active peptides based on cell-free catalysis. Biotechnol Biofuels. 2020;13:61. https://doi.org/10.1186/s13068-020-01700-4.
Li ZW, Liang S, Ke Y, et al. The feather degradation mechanisms of a new Streptomyces sp. isolate SCUT-3. Commun Biol. 2020;3(1):191. https://doi.org/10.1038/s42003-020-0918-0.
Watanabe D, Lošák T, Vollmann J. From proteomics to ionomics: soybean genetic improvement for better food safety. Genetika. 2018;50(1):335–50. https://doi.org/10.2298/GENSR1801333W.
Dai C, Hou Y, Xu H, et al. Identification of a thermophilic protease-producing strain and its application in solid-state fermentation of soybean meal. J Sci Food Agric. 2022;102(6):2359–70. https://doi.org/10.1002/jsfa.11574.
Zahir M, Fogliano V, Capuano E. Effect of soybean processing on cell wall porosity and protein digestibility. Food Funct. 2020;11(1):285–96. https://doi.org/10.1039/c9fo02167a.
Swallah MS, Yang X, Li J, et al. The pros and cons of soybean bioactive compounds: an overview. Food Rev Int. 2022;39(8):5104–31. https://doi.org/10.1080/87559129.2022.2062763.
Yin H, Zhang X, Huang J. Study on enzymatic hydrolysis of soybean β-conglycinin using alkaline protease from Bacillus subtilis ACCC 01746 and antigenicity of its hydrolysates. Grain Oil Sci Technol. 2021;4(1):18–25. https://doi.org/10.1016/j.gaost.2020.12.001.
陈东. 基于组学的侧孢短芽孢杆菌溶藻机制研究 [D], 广东海洋大学, 2022, https://doi.org/10.27788/d.cnki.ggdhy.2021.000186.
Benjamin AM, Nichols M, Burke TW, et al. Comparing reference-based RNA-Seq mapping methods for non-human primate data. BMC Genomics. 2014;15(1):570. https://doi.org/10.1186/1471-2164-15-570.
Chandramohan R, Wu PY, Phan JH, et al. Benchmarking RNA-Seq quantification tools. Conf Proc IEEE Eng Med Biol Soc. 2013;2013:647–50. https://doi.org/10.1109/EMBC.2013.6609583.
Liu S, Wang Z, Zhu R, et al. Three differential expression analysis methods for RNA sequencing: limma, EdgeR, DESeq2. J Visualized Exp. 2021. https://doi.org/10.3791/62528.
Ching T, Huang SJ, Garmire LX. Power analysis and sample size estimation for RNA-Seq differential expression. RNA. 2014;20(11):1684–96. https://doi.org/10.1261/rna.046011.114.
Young MD, Wakefield MJ, Smyth GK, et al. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. https://doi.org/10.1186/gb-2010-11-2-r14.
Galperin MY, Kristensen DM, Makarova KS, et al. Microbial genome analysis: the COG approach. Brief Bioinform. 2019;20(4):1063–70. https://doi.org/10.1093/bib/bbx117.
Tatusov RL, Galperin MY, Natale DA, et al. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28(1):33–6. https://doi.org/10.1093/nar/28.1.33.
Kamp P, Huang Y. Production and characterization of keratinolytic proteases produced by Onygena corvina. Fungal Genet Biol. 2015. https://doi.org/10.4172/2165-8056.1000119.
Xue Y, Zhao Y, Ji X, et al. Advances in bio-nylon 5X: discovery of new lysine decarboxylases for the high-level production of cadaverine. Green Chem. 2020;22(24):8656–68. https://doi.org/10.1039/d0gc03100c.
Espersen R, Huang Y, Falco FC, et al. Exceptionally rich keratinolytic enzyme profile found in the rare actinomycetes Amycolatopsis keratiniphila D2(T). Appl Microbiol Biotechnol. 2021;105(21–22):8129–38. https://doi.org/10.1007/s00253-021-11579-2.
Falco FC, Espersen R, Svensson B, et al. An integrated strategy for the effective production of bristle protein hydrolysate by the keratinolytic filamentous bacterium Amycolatopsis keratiniphila D2. Waste Manag. 2019;89:94–102. https://doi.org/10.1016/j.wasman.2019.03.067.
Peng S, Li H, Zhang S, et al. Isolation of a novel feather-degrading Ectobacillus sp. JY-23 strain and characterization of a new keratinase in the M4 metalloprotease family. Microbiol Res. 2023;274:127439. https://doi.org/10.1016/j.micres.2023.127439.
Lai Y, Wu X, Zheng X, et al. Insights into the keratin efficient degradation mechanism mediated by Bacillus sp. CN2 based on integrating functional degradomics. Biotechnol Biofuels Bioprod. 2023;16(1):59. https://doi.org/10.1186/s13068-023-02308-0.
Pei X-D, Li F, Gao T-T, et al. Utilization of feather keratin waste to antioxidant and migration-enhancer peptides by Bacillus licheniformis 8–4. J Appl Microbiol. 2023. https://doi.org/10.1093/jambio/lxad005.
Kim JM, Lim WJ, Suh HJ. Feather-degrading Bacillus species from poultry waste. Process Biochem. 2001;37(3):287–91. https://doi.org/10.1016/S0032-9592(01)00206-0.
Macedo AJ, Da Silva WOB, Gava R, et al. Novel keratinase from Bacillus subtilis S14 exhibiting remarkable dehairing capabilities. Appl Environ Microb. 2005;71(1):594–6. https://doi.org/10.1128/AEM.71.1.594-596.2005.
Yusuf I, Garba L, Shehu MA, et al. Selective biodegradation of recalcitrant black chicken feathers by a newly isolated thermotolerant bacterium Pseudochrobactrum sp. IY-BUK1 for enhanced production of keratinase and protein-rich hydrolysates. Int Microbiol. 2020;23(2):189–200. https://doi.org/10.1007/s10123-019-00090-4.
Bhari R, Kaur M, Singh RS. Nutritional enhancement of chicken feather waste by Bacillus aerius NSMk2. Indian J Microbiol. 2020;60(4):518–25. https://doi.org/10.1007/s12088-020-00897-0.
Sobucki L, Ramos RF, Daroit DJ. Protease production by the keratinolytic Bacillus sp. CL18 through feather bioprocessing. Environ Sci Pollut Res Int. 2017;24(29):23125–32. https://doi.org/10.1007/s11356-017-9876-6.
Kang D, Huang Y, Nesme J, et al. Metagenomic analysis of a keratin-degrading bacterial consortium provides insight into the keratinolytic mechanisms. Sci Total Environ. 2021;761: 143281. https://doi.org/10.1016/j.scitotenv.2020.143281.
Cai CG, Chen JS, Qi JJ, et al. Purification and characterization of keratinase from a new Bacillus subtilis strain. J Zhejiang Univ Sci B. 2008;9(9):713–20. https://doi.org/10.1631/jzus.B0820128.
Haq IU, Akram F, Jabbar Z. Keratinolytic enzyme-mediated biodegradation of recalcitrant poultry feathers waste by newly isolated Bacillus sp. NKSP-7 under submerged fermentation. Folia Microbiol (Praha). 2020;65(5):823–34. https://doi.org/10.1007/s12223-020-00793-6.
Kaewsalud T, Yakul K, Jantanasakulwong K, et al. Biochemical characterization and application of thermostable-alkaline keratinase from Bacillus halodurans SW-X to valorize chicken feather wastes. Waste Biomass Valori. 2020;12(7):3951–64. https://doi.org/10.1007/s12649-020-01287-9.
Li Y, Dammer EB, Gao Y, et al. Proteomics links ubiquitin chain topology change to transcription factor activation. Mol Cell. 2019;76(1):126–37. https://doi.org/10.1016/j.molcel.2019.07.001.
Ganesan S, Sivam S, Elancheziyan M, et al. Novel delipidated chicken feather waste-derived carbon-based molybdenum oxide nanocomposite as efficient electrocatalyst for rapid detection of hydroquinone and catechol in environmental waters. Environ Pollut. 2022;293: 118556. https://doi.org/10.1016/j.envpol.2021.118556.
Ghosh A, Clerens S, Deb-Choudhury S, et al. Thermal effects of ionic liquid dissolution on the structures and properties of regenerated wool keratin. Polymer Degrad Stabil. 2014;108:108–15. https://doi.org/10.1016/j.polymdegradstab.2014.06.007.
李志伟. 高效羽毛降解菌株的筛选鉴定及其降解机制的研究[D].华南理工大学, 2021. https://doi.org/10.27151/d.cnki.ghnlu.2020.001650.
Cao G, Rong MZ, Zhang MQ. Continuous high-content keratin fibers with balanced properties derived from wool waste. ACS Sustain Chem Eng. 2020;8(49):18148–56. https://doi.org/10.1021/acssuschemeng.0c06530.
Jana A, Halder SK, Dasgupta D, et al. Keratinase biosynthesis from waste poultry feathers for proteinaceous stain removal. ACS Sustain Chem Eng. 2020;8(48):17651–63. https://doi.org/10.1021/acssuschemeng.0c04378.
Chen J, Yang S, Liang S, et al. In vitro synergistic effects of three enzymes from Bacillus subtilis CH-1 on keratin decomposition. 3 Biotech. 2020;10(4):159. https://doi.org/10.1007/s13205-020-2143-4.
Zhang B, Peng Z, Zhang J, et al. Heterologous expression and optimization of disulfide reductase, and its application in assisting keratinase to degrade feather waste. Food Ferment Ind. 2023;49(6):42–9. https://doi.org/10.13995/j.cnki.11-1802/ts.032344.
Espersen R, Falco FC, Hagglund P, et al. Two novel S1 peptidases from Amycolatopsis keratinophila subsp. keratinophila D2(T) degrading keratinous slaughterhouse by-products. Appl Microbiol Biotechnol. 2020;104(6):2513–22. https://doi.org/10.1007/s00253-020-10380-x.
Kou F, Zhao J, Liu J, et al. Characterization of a new lysine decarboxylase from Aliivibrio salmonicida for cadaverine production at alkaline pH. J Mol Catal B-Enzym. 2016;133:S88–94. https://doi.org/10.1016/j.molcatb.2016.11.023.
Mitsuiki S, Ichikawa M, Oka T, et al. Molecular characterization of a keratinolytic enzyme from an alkaliphilic Nocardiopsis sp. TOA-1. Enzyme Microb Tech. 2004;34(5):482–9. https://doi.org/10.1016/j.enzmictec.2003.12.011.
Li J, Shi PJ, Han XY, et al. Functional expression of the keratinolytic serine protease gene sfp2 from Streptomyces fradiae var. k11 in Pichia pastoris. Protein Expr Purif. 2007;54(1):79–86. https://doi.org/10.1016/j.pep.2007.02.012.
Park HB, Kamcev J, Robeson LM, et al. Maximizing the right stuff: the trade-off between membrane permeability and selectivity. Science. 2017;356(6343):1137. https://doi.org/10.1126/science.aab0530.
Pompella A, Corti A, Visvikis A. Redox Mechanisms in cisplatin resistance of cancer cells: the twofold role of gamma-glutamyltransferase 1 (GGT1). Front Oncol. 2022;12: 920316. https://doi.org/10.3389/fonc.2022.920316.
Hayashima K, Katoh H. Expression of gamma-glutamyltransferase 1 in glioblastoma cells confers resistance to cystine deprivation-induced ferroptosis. J Biol Chem. 2022;298(3): 101703. https://doi.org/10.1016/j.jbc.2022.101703.
Sharma R, Gupta R. Coupled action of gamma-glutamyl transpeptidase-glutathione and keratinase effectively degrades feather keratin and surrogate prion protein, Sup 35NM. Bioresour Technol. 2012;120:314–7. https://doi.org/10.1016/j.biortech.2012.06.038.
Sivakumar N, Raveendran S. Keratin degradation by bacteria and fungi isolated from a poultry farm and plumage. Br Poult Sci. 2015;56(2):210–7. https://doi.org/10.1080/00071668.2014.996119.
De Menezes CLA, Santos RDC, Santos MV, et al. Industrial sustainability of microbial keratinases: production and potential applications. World J Microbiol Biotechnol. 2021;37(5):86. https://doi.org/10.1007/s11274-021-03052-z.
Chen H, Zhang S, Park I, et al. Impacts of energy feeds and supplemental protease on growth performance, nutrient digestibility, and gut health of pigs from 18 to 45 kg body weight. Anim Nutr. 2017;3(4):359–65. https://doi.org/10.1016/j.aninu.2017.09.005.
Huang C, Ma D, Zang J, et al. Effect of keratinase on ileal amino acid digestibility in five feedstuffs fed to growing pigs. Asian-Australas J Anim Sci. 2018;31(12):1946–55. https://doi.org/10.5713/ajas.17.0815.
Pantaya D, Pamungkas D, Wulandari S, et al. Fermentation of soybean meal-hydrolysates as the medium that treated by papain enzyme with Saccharomyces cerevisiae for biomass production. IOP Conference Series. 2022;980: 012025. https://doi.org/10.1088/1755-1315/980/1/012025.
Acknowledgements
Thanks to Angel Yeast Co., Ltd for providing the soy protein isolate (SPI) and the enzyme NY100, Xianliang Zheng and Chen Yu for partial discussion of this work.
Funding
This work was supported by the National Natural Science Foundation of China under Grant No. 22078024; the Liaoning Revitalization Talents Program No. XLYC2007175; the National Natural Science Foundation of Liaoning Province of China under Grant No. 2021MS316; and the Educational Commission of Liaoning Province of China under Grant No. LQ2020001.
Author information
Authors and Affiliations
Contributions
YH designed the experiments, BZ, YH, YG carried out the experiments, and drafted the manuscript. YX, XJ conducted data analysis, writing and editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Table S1. Primers used in this study. Table S2. Response-surface analysis of the conditions when Bacillus sp. 8A6 grew in feather medium. Table S3. Quality analysis of the raw RNA sequencing data when Bacillus sp. 8A6 grew in feather medium for 0, 8 and 20 h. Table S4. Quality analysis of the clean RNA sequencing data when Bacillus sp. 8A6 grew in feather medium for 0, 8 and 20 h. Table S5. Up-regulated keratinases, disulfide reductases for the group 0 h VS 8 h when Bacillus sp. 8A6 grew in feather medium. Table S6. Up-regulated keratinases, disulfide reductases for the group 8 h VS 20 h when Bacillus sp. 8A6 grew in feather medium. Figure S1. Four factors that affect protease activity when the Bacillus sp. 8A6 grew in feather medium. (A) Effects of different temperatures; (B) Effects of different pH; (C) Effects of different feather concentration; (D) Effects of inoculation amount (*p < 0.05,**p < 0.01, ***p < 0.001, ****p < 0.0001). Response surface 3D plots reveals the interaction of factors affecting protease activity, (E) pH and inoculation amount; (F) inoculation amount and feather concentration; (G) feather concentration and pH. Figure S2. The OD600 of Bacillus sp. 8A6 when grown in feathermedium at different time points. Figure S3. Four factors that affect protein concentration. (A) Effects of different temperatures; (B) Effects of different pH; (C) Effects of different feather concentration; (D) Effects of inoculation amount (*p < 0.05,**p < 0.01, ***p < 0.001, ****p < 0.0001). Figure S4. Pearson correlation coefficient correlation analysis (A) and principal component analysis (B) among the three RNA-seq samples when Bacillus sp. 8A6 grew in feather medium for 0, 8 and 20 h. Figure S5. Volcano plot of the differentially expressed genes for the groups 0 h VS 8 h (A) and 8 h VS 20 h (B) when Bacillus sp. 8A6 grew in feather medium. Up-regulated genes are shown as red dots and down-regulated genes are shown as blue dots. The significant difference means that the expression level of the genes are 2 times different and the qvalue (fdr, pad) ≤0.05. Figure S6. Hierarchical clusters of differentially expressed genes according to the log10(FPKM+1) value when Bacillus sp. 8A6 grew in feather medium for 0, 8 and 20 h. The up-regulated genes were indicated as red color and the down-regulated genes were indicated as blue color based on the color key. Six main clusters were enriched and highlighted by different colors next to the phylogenetic tree. Figure S7. Gene Ontology (GO) analysis of the molecular function, cellular component and biological process for the groups 0 h VS 8 h (A) and 8 h VS 20 h (B) according to the -log10(pvalue) when Bacillus sp. 8A6 grew in feather medium. The higher -log10(p-value) indicates the more significant for enrichment of the GO term. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of the pathways for the groups 0 h VS 8 h (C) and 8 h VS 20 h (D) according to the -log10(p-value) when Bacillus sp. 8A6 grew in feather medium. 30 significantly different pathways are shown according to the Q-value. Figure S8. Cluster of Orthologous Groups of proteins (COG) analysis of the gene function for the groups 0 h VS 8 h (A) and 8 h VS 20 h (B) according to the gene numbers when Bacillus sp. 8A6 grew in feather medium. The up-regulated genes were indicated as red color and the down-regulated genes were indicated as blue color. Supplemental peptide list 1. Short peptides of Bacillus sp. 8A6 keratinases. Supplemental peptide list 2. Short peptides of NY100. Supplemental peptide list 3. Short peptides of CK.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, B., Guo, Y., Xue, Y. et al. Comprehensive insights into the mechanism of keratin degradation and exploitation of keratinase to enhance the bioaccessibility of soybean protein. Biotechnol Biofuels 16, 177 (2023). https://doi.org/10.1186/s13068-023-02426-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-023-02426-9