
Abstract
Background
l-Methionine is the only bulk amino acid that has not been industrially produced by the fermentation method. Due to highly complex and strictly regulated biosynthesis, the development of microbial strains for high-level l-methionine production has remained challenging in recent years.
Results
By strengthening the l-methionine terminal synthetic module via site-directed mutation of l-homoserine O-succinyltransferase (MetA) and overexpression of metAfbr, metC, and yjeH, l-methionine production was increased to 1.93 g/L in shake flask fermentation. Deletion of the pykA and pykF genes further improved l-methionine production to 2.51 g/L in shake flask fermentation. Computer simulation and auxotrophic experiments verified that during the synthesis of l-methionine, equimolar amounts of l-isoleucine were accumulated via the elimination reaction of cystathionine γ-synthetase MetB due to the insufficient supply of l-cysteine. To increase the supply of l-cysteine, the l-cysteine synthetic module was strengthened by overexpression of cysEfbr, serAfbr, and cysDN, which further increased the production of l-methionine by 52.9% and significantly reduced the accumulation of the byproduct l-isoleucine by 29.1%. After optimizing the addition of ammonium thiosulfate, the final metabolically engineered strain MET17 produced 21.28 g/L l-methionine in 64 h with glucose as the carbon source in a 5 L fermenter, representing the highest l-methionine titer reported to date.
Conclusions
In this study, a high-efficiency strain for l-methionine production was derived from wild-type Escherichia coli W3110 by rational metabolic engineering strategies, providing an efficient platform for the industrial production of l-methionine.
Similar content being viewed by others

Background
l-Methionine is an essential amino acid and is widely used in food, medicine, feed, and other industries [1, 2]. Currently, l-methionine is mainly synthesized by chemical methods, which use volatile and toxic hydrocyanic acid, acrolein, and methanethiol as substrates and cause severe environmental pollution [3]. Therefore, there has been much interest in the production of l-methionine via environmentally friendly and reproducible fermentation by employing metabolically engineered microorganisms.
Several metabolic engineering efforts to efficiently produce l-methionine from glucose in Escherichia coli or Corynebacterium glutamicum have been reported (Table 1). The major strategies for constructing l-methionine producers include alleviating feedback inhibition, overexpressing key enzymes, blocking competing pathways, increasing the supply of cofactors, and enhancing l-methionine transport. For instance, inactivating the l-methionine transcriptional repressor MetJ and releasing the feedback inhibition of l-homoserine O-succinyltransferase MetA result in the initial accumulation of l-methionine [4, 5]. Overexpressing the l-methionine efflux protein YjeH or BrnFE and deleting the l-threonine and l-lysine pathways significantly increased the production of l-methionine [6, 7]. Enhancing the NADPH supply by modifying the pentose phosphate pathway increased l-methionine production [8, 9].
Despite the above efforts to improve l-methionine production by fermentation, the highest titer of l-methionine produced by C. glutamicum was 6.85 g/L [9] and E. coli was 17.74 g/L [10]. There remain some limitations to these metabolic engineering strategies, for example, the exogenous addition of l-lysine and l-threonine was needed during fermentation due to auxotroph, which increased the cost of l-methionine production and process complexity [6, 7]. Moreover, a recombinant plasmid was used to express key genes in those strains. However, antibiotics and inducers are not conducive to scale-up fermentation [7, 10]. Therefore, developing a non-auxotrophic and plasmid-free strain will be beneficial for the biological industrial production of l-methionine.
In this study, a highly efficient l-methionine producer was derived from E. coli W3110 using rational metabolic engineering strategies. The CRISPR/Cas9 system was used to modify the related modules for l-methionine synthesis, and the specific reasons for the accumulation of the byproduct l-isoleucine were analyzed and discussed herein. The enhancement of the l-cysteine synthetic module further increased l-methionine production and significantly reduced l-isoleucine accumulation. The final engineered strain MET17 produced 21.28 g/L l-methionine in a 5 L bioreactor.
Results and discussion
Strengthening the l-methionine terminal synthetic module
In E. coli, the biosynthesis pathway from l-homoserine to l-methionine forms the l-methionine terminal synthetic module, where the components required (including the carbon skeleton, sulfur, and methyl group) are assembled into l-methionine and transported out of the cell (Fig. 1). Removing the feedback inhibition of key enzymes in metabolic pathways is usually the first step in the construction of genetically engineered strains [11]. As the first key enzyme in the l-methionine terminal synthetic module, l-homoserine O-succinyltransferase (MetA, encoded by metA) is feedback inhibited by l-methionine and S-adenosylmethionine (SAM) [12] and is extremely sensitive to many stress conditions (e.g., thermal, oxidative, or acidic stress) [13,14,15]. To optimize MetA performance, we combined the previously reported thermostable mutations I124L and I229Y [16] with the feedback-resistant (fbr) mutations I296S-P298L-R27C [5] by site-directed mutagenesis (Fig. 2A). Seven MetA mutants were constructed by plasmid pBR322 and overexpressed in MET1 (in which a transcriptional repressor of the l-methionine biosynthesis genes, encoded by metJ, was deleted from the wild-type E. coli W3110). The expression of different MetA mutants was detected by SDS-PAGE (Additional file 1: Figure S1). Shake flask fermentation showed that except for MetAI124L, I229L, R27C-I296S-P298L, all the MetA mutants exhibited significantly increased l-methionine production, and the MetA R27C-I296S-P298L (metAfbr) mutant exhibited the highest relative titer of l-methionine, which was 2.3-fold higher than that of the wild type (Fig. 2B). These results demonstrated that combining all of these mutations was not suitable for l-methionine production and significantly inhibited l-methionine synthesis. Then, we introduced the R27C, I296S, and P298L mutations into the chromosome of MET1 to obtain the MET2 strain (MET1metAfbr). As shown in Fig. 2C, MET2 produced 0.58 g/L l-methionine at 36 h, and its growth was slightly reduced compared with that of MET1, while MET1 did not exhibit l-methionine accumulation. When we drafted this manuscript, Hye-Young Sagong et al. [17] reported the crystal structure of MetA from E. coli and identified a putative l-methionine inhibitor binding site T242 located in the vicinity of the substrate binding site. The T242A variant retained more than 70% of its activity with the addition of 5 mM l-methionine and the Ki was 17.4 mM, which significantly reduced the feedback inhibition compared with the wild type (Ki was 2.44 mM). This provides valuable knowledge for the improvement of l-methionine production. Especially, the combination of T242A with I296S-P298L-R27C could be useful for further study of the effect on l-methionine synthesis.

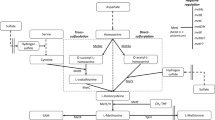
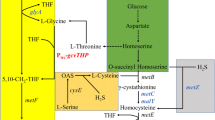
Metabolic strategies for enhancing l-methionine production in E.coli. Glc, glucose; 3-PG, glyceraldehyde-3-phosphate; PEP, phosphoenolpyruvate; PYR, pyruvate; Ac-CoA, acetyl coenzyme A; OAA, oxaloacetate; ASP, l-aspartate; ASP-P, aspartyl-β-phosphate; ASP-SA, aspartyl-β-semialdehyde; HOM, l-homoserine; O-SUCC-HOM, O-succinyl-l-homoserine; CYSTA, l-cystathionine; HOMCYS, l-homocysteine; MET, l-methionine; 3-PHP, 3-Phosphonooxypyruvate; SER-P, 3-phosphoserine; SER, l-serine; O-AC-SER, O-acetyl-l-serine; CYS, l-cysteine; APS, adenosine 5-phosphosulfate; PAPS, 3-phosphoadenosine 5-phosphosulfate; GLY, l-glycine; M-THF, 5-methyltetrahydrofolate; THF, tetrahydrofolate; THR, l-threonine; 2-KB, 2-ketobutyrate; ILE, l-isoleucine; pykFA, pyruvate kinase; metL, fused aspartate kinase/homoserine dehydrogenase 2; metA, l-homoserine O-succinyltransferase; metB, cystathionine γ-synthase; metC, cystathionine β-lyase; metE/H, homocysteine methyltransferase; yjeH, l-methionine exporter; MetD, l-methionine absorption transporter operon; RhtA, l-homoserine exporter; serA, phosphoglycerate dehydrogenase; serC, phosphoserine aminotransferase; serB, phosphoserine phosphatase; cysE, serine O-acetyltransferase; cysK, cysteine synthase; cysN, sulfate adenylyltransferase subunit 1; cysD, sulfate adenylyltransferase subunit 2; cysC, adenylylsulfate kinase; cysH, phosphoadenosine phosphosulfate reductase; cysJ, sulfite reductase (NADPH) flavoprotein alpha-component; cysI, sulfite reductase (NADPH) flavoprotein beta-component

Metabolic engineering modification of the l-methionine terminal synthetic module. A Schematic diagram of MetA mutation sites. B The relative l-methionine titer of different mutant MetA-overexpressing strains in shake flask fermentation. C Production of l-methionine and growth of the MET1 and MET2 strains. D Flow chart of chromosomal integration for the metAfbr, metC, and yjeH genes. E l-Methionine and l-isoleucine production and OD600 of MET3, MET4, MET6, MET8, MET9, and MET10. Data are presented as the mean values with the standard deviation from three biological replicates. One-way analysis of variance (ANOVA) was used to determine significant differences (* p < 0.05, ** p < 0.01)
To further improve l-methionine production, a copy of metAfbr driven by the strong Ptrc promoter was integrated into the rhtA site (encoding an effective exporter of l-homoserine, a precursor for l-methionine synthesis) of MET2 to obtain the MET4 strain (Fig. 2D). The rhtA gene was deleted from strain MET2 to generate MET3 as a control. Shake flask fermentation results showed that MET3 and MET4 produced 0.63 g/L and 0.86 g/L l-methionine, which were 8.6% and 48.3% higher than the amount produced by MET2, respectively (Fig. 2E). While MET3 and MET4 also accumulated 0.39 g/L and 0.61 g/L l-isoleucine, respectively. To investigate other key genes that affect l-methionine synthesis in the l-methionine terminal synthetic module, the metBL, metC, and metF genes of MET4 were individually upregulated by replacing the native promoter with the strong promoter Ptrc to obtain the MET5, MET6, and MET7 strains. As shown in Additional file 1: Figure S2, the relative l-methionine titer of the MET6 strain (with upregulation of metC) was 29.1% higher than that of the MET4 strain. Upregulation of metBL (MET5) and metF (MET7) expression significantly reduced the relative l-methionine titer by 84.9% and 22.1%, respectively, which might be due to the upregulation of these genes affecting the balance of intracellular metabolism, which is consistent with the results of previous studies [18]. Then, we integrated a copy of metC driven by the strong promoter Ptrc into the rhtA site of MET6 to obtain strain MET8. Shake flask fermentation results showed that MET8 produced 1.36 g/L l-methionine, which was 21.1% higher than the amount produced by MET6 (Fig. 2E). Notably, MET8 also accumulated 1.14 g/L l-isoleucine. The l-isoleucine pathway is a competing pathway for l-methionine synthesis in E. coli, and the overexpression of metAfbr and metC in the MET8 strain would enlarge the carbon flux from l-homoserine to l-methionine and reduce the metabolic flux to l-isoleucine pathway (Fig. 2D). However, the accumulation of l-isoleucine was also increased, so we speculated that there might be other potential pathways for l-isoleucine synthesis.
Finally, l-methionine efflux was enhanced by blocking the absorption transporter MetD (encoded by the metN, metI, and metQ genes) [19] and overexpressing the efflux protein YjeH (encoded by yjeH) [20] in MET8 strain. The MET9 strain (MET8ΔmetD) and MET10 strain (MET8ΔmetD::PL-yjeH) were constructed, respectively (Fig. 2D). Shake flask fermentation results showed that MET9 and MET10 produced 1.68 g/L and 1.93 g/L l-methionine, which were 23.5% and 41.9% higher, respectively, than the amount produced by the MET8 strain. As YjeH can also transport branched-chain amino acids [20], the accumulation of the byproduct l-isoleucine also increased to 1.24 g/L and 1.63 g/L in MET9 and MET10, respectively (Fig. 2E).
Coupling of the central carbon metabolism module with the l-methionine terminal synthetic module
The l-methionine synthesis pathway in E. coli is complex and is regulated by multiple factors [18]. Some metabolic pathways share the same substrate and product. For instance, succinyl-CoA and l-homoserine are converted to O-succinyl-l-homoserine, and succinic acid is released during the process of O-succinyl-l-homoserine being converted to cystathionine, catalyzed by cystathionine γ-synthase (encoded by metB), which is consistent with the conversion of succinyl-CoA to succinic acid (catalyzed by succinyl-CoA synthetase, encoded by sucCD) in the TCA cycle of the central carbon metabolism module (Fig. 3A). On the other hand, the decomposition of cystathionine to homocysteine by cystathionine β-lyase (encoded by metC) releases pyruvate, the same reaction product formed by pyruvate kinase (encoded by pykF and pykA) in glycolysis in the central carbon metabolism module (Fig. 3B). Kind et al. [21] and Ruan et al. [22] used a metabolic coupling strategy to delete the sucCD genes, which coupled the TCA cycle with the l-lysine and SAM synthesis pathways in C. glutamicum and Bacillus amyloliquefaciens, respectively, and significantly increased the production of these compounds. To verify the effect of these couplings on l-methionine production, the sucCD, pykA, and pykF genes of MET10 were deleted to obtain the MET11 (sucCD), MET12 (pykA), and MET13 (pykApykF) strains, respectively. As shown in Fig. 3C, the growth of the MET11 strain was significantly inhibited, and l-methionine accumulation was not detected. This might be attributed to the l-methionine terminal synthetic module not compensating for the TCA cycle to maintain normal cell growth in the sucCD-deleted strain, which was also demonstrated in B. amyloliquefaciens [22]. In contrast, MET12 and MET13 produced 2.38 g/L and 2.51 g/L l-methionine, respectively, which were 23.3% and 30.1% higher than the amount produced by MET10. In addition, 1.97 g/L and 2.27 g/L l-isoleucine as a byproduct also accumulated in the MET12 and MET13 strains. Deletion of the pykA and pykF genes increased l-methionine precursor synthesis and coupled l-methionine synthesis with cell growth. Due to the enhancement of the l-methionine terminal synthetic module and pyruvate release being commonplace in synthetic pathways [23, 24], cell growth was not affected.

Effect of metabolic coupling strategy on l-methionine production. A Coupling of the central carbon metabolism module to the l-methionine terminal synthetic module by deleting the succinyl-CoA synthetase sucCD. B Coupling of the central carbon metabolism module with the l-methionine terminal synthetic module by deleting the pykF and pykA genes. C l-Methionine and l-isoleucine production and OD600 of MET10, MET11, MET12, and MET13. Data are presented as the mean values with the standard deviation from three biological replicates. One-way analysis of variance (ANOVA) was used to determine significant differences (* p < 0.05, ** p < 0.01)
In silico simulation of flux distribution for l-isoleucine accumulation
Although the production of l-methionine was significantly promoted by modifying the l-methionine terminal synthetic module, considerable l-isoleucine (2.27 g/L, Fig. 3C) was accumulated by MET13, indicating that we should shift the metabolic flux from l-isoleucine to the synthesis of l-methionine. In wild-type E. coli, the metabolic flux in the l-homoserine node was partitioned partially to O-succinyl-l-homoserine for l-methionine synthesis and partially to l-threonine for l-isoleucine formation (Fig. 1). Since a common strategy to block the l-isoleucine pathway is to delete ilvA, we deleted the ilvA gene in the MET13 strain to obtain the MET13ΔilvA strain, and deletion of ilvA in the wild-type E. coli W3110 (WTΔilvA) was performed as a control. Surprisingly, WTΔilvA was found to be l-isoleucine auxotrophic in M9 minimum medium without l-isoleucine, but MET13ΔilvA did not exhibit this trait (Fig. 4C, D). These results indicated that the synthesis of l-isoleucine occurs via other pathways.

In silico simulation of the metabolic flux distribution in the wild-type strain and l-methionine overproducer by flux balance analysis. A The metabolic flux distribution in the WT strain (blue) and l-methionine overproducer (red). B The impacts of increased CysE reaction flux on intracellular metabolic flux by FBA using l-methionine synthase as the objective function with 20% theoretical maximum biomass constraint. The CysE reaction flux was set to 1.0-, 3.0-, 5.0-, 10.0-, and 20.0-fold (from left to right) compared to that in the WT strain. C Strains WTΔilvA and MET13ΔilvA on solid M9 medium with or without 0.5 g/L l-isoleucine supply. D Growth of WT, WTΔilvA, MET13, and MET13ΔilvA in M9 medium with or without 0.5 g/L l-isoleucine supply. E Growth of MET13 and MET13ΔmetB in fermentation medium with or without 0.5 g/L l-methionine supply. F l-methionine and l-isoleucine production and OD600 of MET13 and MET13 with l-cysteine supply. Data are presented as the mean values with the standard deviation from three biological replicates. One-way analysis of variance (ANOVA) was used to determine significant differences (* p < 0.05, ** p < 0.01)
To investigate the underlying reason, the relationships between l-methionine flux and biomass as well as l-isoleucine accumulation were investigated by flux balance analysis (FBA) [25] based on the iY75_1357 model of E. coli W3110 [26]. The intracellular metabolic fluxes in the WT strain were calculated using biomass as the objective function. For the l-methionine producer, the lowest biomass was restricted to 20% of the theoretical maximum, and the metabolic fluxes were calculated using l-methionine synthase as the objective function. The comparison of metabolic flux distribution between the WT strain and l-methionine producer showed the most important difference in the flux redistribution as being at the l-cysteine synthetic module and l-methionine terminal synthetic module. The flux from O-acetyl-l-serine entering into l-cysteine increased 6.2-fold in the l-methionine producer compared to the WT strain. In contrast, the flux from l-homoserine to l-threonine in the l-methionine producer dropped nearly twofold, and there was a 9.7-fold increase in the flux redirected toward the l-methionine terminal synthetic module (Fig. 4A). As previously reported, when the l-cysteine supply is insufficient, cystathionine γ-synthase (encoded by metB) catalyzes the conversion of O-succinyl-l-homoserine (OSHS) to 2-ketobutyrate (2-KB) via an elimination reaction and then forms l-isoleucine [27, 28]. The flux from OSHS to 2-KB also increased 9.7-fold in the l-methionine producer (Fig. 4A). Considering that the cysE-catalyzed reaction is a major control switch in the l-cysteine synthesis pathway of E. coli [29, 30], the effects of the cysE-catalyzed reaction flux change on the carbon flux distribution and l-methionine synthesis were investigated using FBA under 20% of the theoretical maximum biomass constraint. When the cysE-catalyzed reaction flux improved tenfold, the l-methionine productivity reached a relative maximum (Fig. 4B). Meanwhile, the flux toward 2-KB decreased, which indicated the diversion of more OSHS toward l-methionine synthesis.
To verify the simulation, we performed fermentation experiments with MET13 lacking the metB (MET13ΔmetB) gene and with the exogenous addition of l-cysteine. As shown in Fig. 4E, when the metB gene of MET13 was deleted, the cells showed l-methionine auxotroph and l-methionine supplementation restored the growth of MET13ΔmetB. l-Isoleucine was also not detected in MET13ΔmetB after fermentation (data not shown). After l-cysteine was added during the fermentation process of MET13, the accumulation of l-isoleucine decreased by 18.5%, and the production of l-methionine increased by 16.5% (Fig. 4F). These results indicated that the l-cysteine supply was insufficient during l-methionine fermentation and that the accumulation of l-isoleucine occurred mainly due to the elimination reaction of MetB in the l-methionine terminal synthetic module rather than the l-threonine synthetic pathway.
Strengthening the l-cysteine synthetic module
In E. coli, the l-cysteine synthetic module involves both carbon metabolism and sulfur metabolism [31]. In terms of carbon metabolism, glyceraldehyde 3-phosphate (3-PG) is catalytically acted upon by phosphoglycerate dehydrogenase (PGDH, encoded by serA), phosphoserine aminotransferase (encoded by serC), phosphoserine phosphatase (encoded by serB), and l-serine acetyltransferase (SAT, encoded by cysE) to form O-acetyl-l-serine (OAS) (Fig. 1). SAT and PGDH are the two rate-limiting enzymes of l-cysteine synthesis, which are inhibited by l-cysteine and l-serine, respectively [30, 32]. To increase the l-cysteine supply, we first integrated a copy of feedback-resistant cysEfbr (cysEM201R) [33, 34] which was controlled by the P23119 promoter into the chromosome of MET13, with wild-type cysE as a control, to obtain the MET14 (Ptrc-cysE) and MET15 (Ptrc-cysEfbr) strains, respectively (Fig. 5A). As shown in Fig. 5B, MET14 and MET15 produced 2.82 g/L and 3.06 g/L l-methionine, increasing by 18.5% and 28.6% than MET13, respectively. While the accumulation of l-isoleucine was 2.02 g/L and 1.95 g/L, decreasing by 21.1% and 23.8% than MET13, respectively. Then, we integrated a copy of feedback-resistant serAfbr (serAH344A−N346A) [35] into the chromosome of MET15. Although we initially attempted to integrate serAfbr with the strong promoter P23119 control, the experiment consistently failed. The failure might have been due to the excessive activity of SAT and PGDH resulting in the overconsumption of carbon sources and destruction of the balance of the glycolysis and l-cysteine synthetic module. Therefore, we used a relatively weak promoter, P23105 (Fig. 5A). The resultant MET16 strain produced 3.49 g/L l-methionine and 1.78 g/L l-isoleucine, representing a 14.1% increase in l-methionine production compared to that in MET15 and a 12.4% decrease in l-isoleucine production, with a slight decrease in biomass (Fig. 5B). Since the expression of sulfur assimilation-related genes in E. coli is activated by OAS [29], we confirmed the expression of these genes in MET16 by RT‒PCR, and the results showed that the expression of sulfur reduction genes was significantly upregulated (Fig. 5C).

Metabolic engineering modification of the l-cysteine synthetic module. A A schematic representation of the enhancement of the l-cysteine synthetic module. B l-methionine and l-isoleucine production and OD600 of MET13, MET14, MET15, MET16, and MET17. C Relative transcript levels of related genes of MET15 in shake flask cultivation. D Effect of overexpression of key genes in the sulfur assimilation pathway on l-methionine production. Data are presented as the mean values with the standard deviation from three biological replicates. One-way analysis of variance (ANOVA) was used to determine significant differences (* p < 0.05, ** p < 0.01)
In terms of sulfur metabolism, sulfate is often used as a sulfur source for E. coli [36]. It can only be assimilated by the sulfate assimilation pathway which includes sulfate adenylyltransferase (encoded by cysD and cysN), adenylyl-sulfate kinase (encoded by cysC), phosphoadenosine phosphosulfate reductase (encoded by cysH) and sulfite reductase (encoded by cysJ and cysI) [37] (Fig. 5). To investigate the effect of key enzymes in the sulfate assimilation pathway on l-methionine production, we overexpressed the key genes cysD, cysN, cysH, cysC, cysJ, and cysI from the pACYC184 plasmid (Fig. 5A). Simultaneously, we also designed constructs for the coexpression of cysDN and cysJI, the two enzyme subunits. Then, the eight constructed plasmids were transferred into MET16, with the pACYC184 plasmid used as the control. The effects of these genes on l-methionine production were compared by shake flask fermentation. As shown in Fig. 5D, CysN, CysDN, CysH, CysI, and CysJI significantly increased the accumulation level of l-methionine. The overexpression of cysDN produced the highest relative titer of l-methionine, a 1.86-fold improvement in comparison with the control strain. These results indicate that cysDN plays an important role in the sulfur assimilation pathway. Then, we integrated a copy of cysDN under the control of the Ptrc promoter into the chromosome of MET16 to obtain strain MET17. The results of shake flask fermentation showed that MET17 produced 3.84 g/L l-methionine and 1.61 g/L l-isoleucine, representing a 10.0% increase in l-methionine production compared to that in MET16 and a 9.0% decrease in l-isoleucine production, with a significant decrease in biomass (Fig. 6C). The decrease in biomass might have been due to the increased consumption of ATP and NADPH by the enhanced sulfur assimilation pathway [38]. Enhancement of the l-cysteine synthetic module increased l-methionine production by 52.9% and decreased l-isoleucine accumulation by 29.1% compared with that in MET13, indicating that sulfur metabolism plays a crucial role in l-methionine synthesis.

Fed-batch fermentation process curve in a 5 L bioreactor. A Without sulfate or thiosulfate feeding during fed-batch cultivation of the MET17 strain. B With sodium thiosulfate feeding during fed-batch cultivation of the MET17 strain. C With ammonium thiosulfate feeding during fed-batch cultivation of the MET17 strain. D With ammonium sulfate and sodium thiosulfate feeding during fed-batch cultivation of the MET17 strain. E With ammonium sulfate and ammonium thiosulfate feeding during fed-batch cultivation of the MET13 strain. F With ammonium sulfate and ammonium thiosulfate feeding during fed-batch cultivation of the MET17 strain
Fed-batch fermentation for l-methionine in a 5-L fermenter
Different sulfur sources greatly affect the theoretical yield of l-methionine because sulfur assimilation requires a large amount of ATP and NADPH [39, 40]. As previously reported, thiosulfate is a much more effective sulfur source for l-methionine fermentation [6, 38]. To obtain a better sulfur source, we compared the effects of sodium thiosulfate and ammonium thiosulfate on l-methionine production in the MET17 strain in a 5 L bioreactor (Fig. 6). Without sulfur source feeding, MET17 only produced 3.90 g/L l-methionine and 3.53 g/L l-isoleucine at 36 h, with a maximum biomass of 104.9 (OD600) (Fig. 6A). In contrast, with thiosulfate feeding, the fermentation period was extended from 36 to 64 h and the l-methionine production was significantly increased. With sodium thiosulfate feeding, MET17 produced 12.45 g/L l-methionine and 10.49 g/L l-isoleucine, with a maximum biomass of 111 (Fig. 6B). With ammonium thiosulfate feeding, MET17 produced 14.32 g/L l-methionine and 8.64 g/L l-isoleucine, with a maximum biomass of 99.1 (Fig. 6C). These results indicated that feeding with sulfur sources is essential during l-methionine fermentation and that ammonium thiosulfate is better than sodium thiosulfate for l-methionine production. However, excessive thiosulfate is toxic to cells [41]. As the feeding concentration of ammonium thiosulfate increased, cell growth and l-methionine production were severely inhibited (Additional file 1: Figure S3). Therefore, we subsequently chose the combination of ammonium sulfate and thiosulfate. With ammonium sulfate and sodium thiosulfate feeding, MET17 produced 15.68 g/L l-methionine and 8.46 g/L l-isoleucine, with a maximum biomass of 100.7 (Fig. 6D). With ammonium sulfate and ammonium thiosulfate feeding, MET17 produced 21.28 g/L l-methionine and 7.16 g/L l-isoleucine, with a maximum biomass of 97.5 (Fig. 6F), which represented the highest l-methionine titer ever reported and the yield and productivity were 0.12 mol/mol glucose and 0.33 g/L/h, respectively. Under the same conditions, the maximum biomass of MET17 was 10.6% lower than that of MET13 (109.1), and the maximal l-methionine titer of MET17 was 45.3% higher than that of MET13 (14.65 g/L). The maximal l-isoleucine titer of MET17 was 53.6% lower than that of MET13 (15.42 g/L) (Fig. 6E). These results further indicated that the enhancement of the l-cysteine synthetic module had an important effect on l-methionine biosynthesis. Recently, Niu et al. reported a non-auxotrophic l-methionine-producing strain by dynamically regulating the l-lysine biosynthesis pathway, enhancing the central metabolic pathway and l-cysteine catabolic pathway, and finally producing 17.74 g/L l-methionine [10]. However, their l-methionine-producing strain used recombinant plasmid for overexpressing metAfbr, yjeH, serAfbr, gltA, and malY genes. Recombinant plasmid maintenance is considered to be a metabolic burden [42] and is also not conducive to stability in scale-up fermentation [43]. The characteristics of being plasmid-free, inducer-free, and antibiotic-free made MET17 strain more advantageous in fermentation stability, process simplicity, and economic feasibility. Taken together, developing a non-auxotrophic and plasmid-free strain will be beneficial for the biological industrial production of l-methionine.
Conclusions
In this study, a highly efficient E. coli chassis for producing l-methionine from glucose was constructed by strengthening the l-methionine terminal synthetic module, coupling the l-methionine terminal synthetic module with central carbon metabolism module, and strengthening the l-cysteine synthetic module (Fig. 7). Additionally, the specific cause of l-isoleucine accumulation during l-methionine synthesis was elucidated by computer simulation and experiments. The best-performing strain, MET17, produced 3.84 g/L l-methionine in a shake flask and 21.28 g/L in a 5 L fed-batch fermenter, representing the highest l-methionine titer ever reported using rationally engineered microbial cell factories.

Overview of the strain engineering process. The l-methionine titer was significantly improved by metabolic engineering, from 0.58 g/L in the MET2 strain to 21.28 g/L in the final strain MET17. ND, not detected
Materials and methods
Bacterial strains, plasmids, and reagents
All bacterial strains and plasmids used in this study and their relevant characteristics are listed in Table 2 and Additional file 1: Table S1. E. coli EC135 was used as a cloning host for plasmid construction, and E. coli W3110 was used for metabolic engineering. Q5 Hot Start high-fidelity DNA polymerase (New England BioLabs) was used to amplify donor DNA fragments. The DNA Purification Kit, Bacteria DNA Kit, and Mini Plasmid Kit from TIANGEN BIOTECH were used for DNA preparation. A Q5 Site-Directed Mutagenesis Kit from New England BioLabs was used to construct all pTargetF plasmids. In addition, 2 × M5 Super FastTaq PCR Master Mix from Mei5Bio (Beijing) was used for colony PCR. All primers were synthesized by TIANYI HUI YUAN (Beijing), and the primer sequences are listed in Additional file 1: Table S2. Tryptone and yeast extract were supplied by OXOID. Amino acids and antibiotics were acquired from Sigma-Aldrich. All other chemicals were purchased from Beijing Chemical Works unless otherwise stated.
Medium and culture conditions
During strain construction, cultures were grown aerobically at 30 °C or 37 °C in lysogeny broth (LB) medium (5 g/L yeast extract, 10 g/L tryptone, 10 g/L NaCl) [46]. To prepare a solid medium, 20 g/L agar powder was added to the LB medium. If necessary, antibiotics were added at the following concentrations: 50 µg/mL kanamycin or 50 µg/mL spectinomycin. L-(+)-arabinose (10 mM final concentration) was used to induce the CRISPR/Cas9 system and Red homologous recombination system during gene deletion and integration.
To analyze l-methionine production in shake flask fermentation, one loop of cells was inoculated into a 50 mL tube with 5 mL of LB medium and incubated for 8 h at 37 °C with shaking at 220 rpm on a rotary shaker. Then, 1% (v/v) seed culture was inoculated into 500 mL flasks containing 25 mL of shake flask fermentation medium with 20 g/L glucose, 8 g/L (NH4)2SO4, 2 g/L KH2PO4, 2 g/L MgSO4·7H2O, 2.5 g/L yeast extract, 0.02 mg/L VB12, 40 g/L MOPS, and 5 mL/L trace element solution, with the pH brought to 7.0 with ammonium solution. Fermentation samples were taken (500 μL each time) for analysis of the substrate and product concentrations.
For fed-batch fermentation in the 5 L bioreactor, one loop of cells was inoculated into a 50 mL tube with 5 mL of LB medium and incubated overnight at 37 °C with shaking at 220 rpm on a rotary shaker. Then, 1% (v/v) of seed culture was inoculated into 500 mL shake flasks containing 50 mL of LB medium and incubated for 8 h at 37 °C with shaking at 220 rpm on a rotary shaker. Then, 100 mL of seed medium was inoculated into a 5 L bioreactor with a final volume of 2 L. Fed-batch fermentation medium was prepared as follows [47]: glucose 10 g/L, (NH4)2SO4 8 g/L, KH2PO4 3 g/L, MgSO4·7H2O 1 g/L, yeast extract 2 g/L, refined corn pulp (powder) 1 g/L, VB12 0.01 g/L, phosphopyridoxal (PLP) 2 mg/L, and trace element solution 5 mL/L. The pH was maintained at 6.8–7.0 by the addition of 28% (v/v) ammonium. The dissolved oxygen (DO) level was maintained at ~ 30% with agitation at 350 to 800 rpm. When the initial glucose concentration was nearly depleted, the feeding glucose (the stock solution contained 450 g/L glucose, 6 g/L MgSO4⋅7H2O, 3 g/L KH2PO4, 33 g/L (NH4)2SO4, and 33.5 g/L ammonium thiosulfate or sodium thiosulfate) was added at an initial rate of 4 g/L/h, which was then gradually increased by 2 g/L/h every 4 h and, finally, maintained at 8 g/L/h until the fermentation process was completed. The glucose concentration in the broth was approximately 0–1 g/L.
The trace element solution was prepared as follows [47]: FeSO4·7H2O 6 g/L, CaCl2 1.35 g/L, ZnSO4·7H2O 0.8 g/L, MnSO4·4H2O 1.5 g/L, CuSO4·5H2O 0.15 g/L, (NH4)6Mo7O24·4H2O 0.2 g/L, H3BO3 0.1 g/L, CoCl2·6H2O 0.25 g/L, 35% HCl 10 mL.
Construction of strains and plasmids
All pTargetF plasmids with different N20 sgRNA sequences were constructed by the Q5 Site-Directed Mutagenesis Kit. All donor DNA templates were obtained by overlap PCR with Q5 Hot Start high-fidelity DNA polymerase. All E. coli gene knockout, integration, and modulation procedures were performed by the CRISPR/Cas9 genome editing process as reported previously [48]. Transformants were identified by colony PCR, and DNA sequencing was performed at TIANYI HUI YUAN (Beijing).
Analytical methods
The biomass concentration was monitored by measuring the optical density at 600 nm (OD600). Dry cell weight (DCW) was calculated based on the OD600 (1 OD600 = 0.42 g DCW/L) [49].
Fermentation samples were centrifuged at 8000 × g for 5 min, and the supernatants were used for the analysis of the substrate and product concentrations. The concentration of glucose was assayed with an enzyme electrode analyzer (SBA-40D; Institute of Biology, Shandong, China) containing glucose oxidase [47].
The product concentrations in the supernatant of the fermentation culture were determined by the HPLC method as described previously [50] with slight modifications. An AdvanceBio AAA column (4.6 mm × 150 mm, 5 μm; Agilent Technology, USA) was used at 40 °C after automated online derivatization using O-phthalaldehyde (OPA). The elution was performed using a gradient of reagent A (10 mM Na2HPO4 and 10 mM Na2B4O7, pH to 8.2 by HCl) and reagent B (methanol/acetonitrile/water = 45:45:10, by vol.) at 1 mL/min. The elution gradients were as follows: 0 to 0.35 min, 2% B; then to 13.4 min, 57% B; to 13.5 min, 100% B; 15.7 min, 100% B; 15.8 min, 2% B; 18 min, 2% B. The eluate was monitored at 338 nm.
SDS-PAGE analysis
The crude proteins from E. coli cells were extracted by ultrasonication using lysis buffer (50 mM Tris–HCl (pH 7.5), 1 mM EDTA, 5% glycerol, 1 mM DTT). The supernatants were collected by centrifugation, reconstituted in a loading buffer (1:5 ratio), and heated at 95 °C for 5 min, followed by SDS–polyacrylamide gel electrophoresis. The gel was stained with Coomassie Blue R-250 (Ge Healthcare, Chicago, IL, USA) and scanned using an EPSON Expression 11000XL at a resolution of 300 dpi.
Constraint-Based metabolic flux analysis
Constraint-based metabolic flux analysis
The genome-scale metabolic model iY75_1357 of E. coli W3110 was used for the in silico flux metasynthetic reaction. A synthetic reaction route for 2-ketobutyrate by the metB-encoding cystathionine γ-synthase was added to iY75_1357 to synthesize 2-ketobutyrate from O-succinyl-l-homoserine. The following reaction was added to iY75_1357: ‘suchms_c + h2o_c → 2obut_c + succ_c + nh4_c + h_c’. In silico analysis of the modified target for overproduction of l-methionine was performed with MATLAB 2014a (The MathWorks, Natick, MA, USA) and the COBRA Toolbox 2.05 (MathWorks Inc. PortolaValley, CA, USA) with the glpk solver [51]. Constraint-based flux balance analysis was carried out by the linear programming-based optimization of growth or l-methionine biosynthesis flux as an objective cellular function. The glucose uptake rate was set to 100 mmol/gCDW/h. Small-molecular metabolites such as CO2, H2O, SO3, NH3, PO4, and O2 were allowed to be freely transported across the cell membrane.
RNA preparation and quantitative real-time RT-PCR
E. coli strains were cultivated in LB medium as described above. After overnight culture, cells were harvested by centrifugation. Total RNA was isolated using the RNA prep Pure Cell/Bacteria Kit (Tiangen, China). Reverse transcription of approximately 300 ng of RNA was performed using the FastQuant RT Kit (Tiangen, China) with the specific primers listed in Additional file 1: Table S2. GoTaq qPCR Master Mix (Promega, Madison, WI, USA) in a 20 µL volume was used to perform quantitative PCR using the LightCycler® 96 Real-Time PCR System (Roche, Basel, Switzerland). The GAPDH gene was used as the reference gene to normalize the mRNA levels of cysD, cysN, cysH, cysC, cysJ, cysI, and cysP. Negative controls in each PCR were run to exclude DNA and other contaminants. The qPCR products were verified by melting curve analysis. Data were analyzed using LightCycler® 96 software (Roche, Basel, Switzerland) according to the 2−∆∆CT method [52].
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its additional files).
Abbreviations
- CRISPR:
-
Clustered Regularly Interspaced Short Palindromic Repeat
- E. coli :
-
Escherichia coli
- FBA:
-
Flux balance analysis
- HPLC:
-
High-performance liquid chromatography
- WT:
-
Wild type
- ATP:
-
Adenosine triphosphate
- NADPH:
-
Reduced form of nicotinamide adenine dinucleotide phosphate
- OD600 :
-
Optical density at wavelength (λ) 600 nm
References
Sanderson SM, Gao X, Dai Z, Locasale JW. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat Rev Cancer. 2019;19:625–37.
Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1:228–37.
Willke T. Methionine production–a critical review. Appl Microbiol Biotechnol. 2014;98:9893–914.
Nakamori S, Kobayashi S, Nishimura T, Takagi H. Mechanism of L-methionine overproduction by Escherichia coli: the replacement of Ser-54 by Asn in the MetJ protein causes the derepression of L-methionine biosynthetic enzymes. Appl Microbiol Biotechnol. 1999;52:179–85.
Usuda Y, Kurahashi O. Effects of deregulation of methionine biosynthesis on methionine excretion in Escherichia coli. Appl Environ Microbiol. 2005;71:3228–34.
Qin T, Hu X, Hu J, Wang X. Metabolic engineering of Corynebacterium glutamicum strain ATCC13032 to produce L-methionine. Biotechnol Appl Biochem. 2015;62:563–73.
Huang JF, Liu ZQ, Jin LQ, Tang XL, Shen ZY, Yin HH, Zheng YG. Metabolic engineering of Escherichia coli for microbial production of L-methionine. Biotechnol Bioeng. 2017;114:843–51.
Li Y, Cong H, Liu B, Song J, Sun X, Zhang J, Yang Q. Metabolic engineering of Corynebacterium glutamicum for methionine production by removing feedback inhibition and increasing NADPH level. Antonie Van Leeuwenhoek. 2016;109:1185–97.
Liu B, Sun X, Liu Y, Yang M, Wang L, Li Y, Wang J. Increased NADPH supply enhances glycolysis metabolic flux and L-methionine production in Corynebacterium glutamicum. Foods. 2022;11(7):1031.
Niu K, Fu Q, Mei ZL, Ge LR, Guan AQ, Liu ZQ, Zheng YG. High-level production of L-methionine by dynamic deregulation of metabolism with engineered nonauxotroph Escherichia coli. ACS Synth Biol. 2023;12(2):492–501.
Ma Q, Zhang Q, Xu Q, Zhang C, Li Y, Fan X, Xie X, Chen N. Systems metabolic engineering strategies for the production of amino acids. Synth Syst Biotechnol. 2017;2:87–96.
Lee LW, Ravel JM, Shive W. Multimetabolite control of a biosynthetic pathway by sequential metabolites. J Biol Chem. 1966;241:5479–80.
Biran D, Gur E, Gollan L, Ron EZ. Control of methionine biosynthesis in Escherichia coli by proteolysis. Mol Microbiol. 2000;37:1436–43.
Price-Carter M, Fazzio TG, Vallbona EI, Roth JR. Polyphosphate kinase protects Salmonella enterica from weak organic acid stress. J Bacteriol. 2005;187:3088–99.
Ron EZ, Davis BD. Growth rate of Escherichia coli at elevated temperatures: limitation by methionine. J Bacteriol. 1971;107:391–6.
Mordukhova EA, Kim D, Pan JG. Stabilized homoserine O-succinyltransferases (MetA) or L-methionine partially recovers the growth defect in Escherichia coli lacking ATP-dependent proteases or the DnaK chaperone. BMC Microbiol. 2013;13:179.
Sagong HY, Lee D, Kim IK, Kim KJ. Rational engineering of homoserine O-succinyltransferase from Escherichia coli for reduced feedback inhibition by methionine. J Agric Food Chem. 2022;70:1571–8.
Huang JF, Shen ZY, Mao QL, Zhang XM, Zhang B, Wu JS, Liu ZQ, Zheng YG. Systematic analysis of bottlenecks in a multibranched and multilevel regulated pathway: the molecular fundamentals of L-methionine biosynthesis in Escherichia coli. ACS Synth Biol. 2018;7:2577–89.
Gál J, Szvetnik A, Schnell R, Kálmán M. The metD D-methionine transporter locus of Escherichia coli is an ABC transporter gene cluster. J Bacteriol. 2002;184:4930–2.
Liu Q, Liang Y, Zhang Y, Shang X, Liu S, Wen J, Wen T. YjeH is a novel exporter of L-methionine and branched-chain amino acids in Escherichia coli. Appl Environ Microbiol. 2015;81:7753–66.
Kind S, Becker J, Wittmann C. Increased lysine production by flux coupling of the tricarboxylic acid cycle and the lysine biosynthetic pathway–metabolic engineering of the availability of succinyl-CoA in Corynebacterium glutamicum. Metab Eng. 2013;15:184–95.
Ruan L, Li L, Zou D, Jiang C, Wen Z, Chen S, Deng Y, Wei X. Metabolic engineering of Bacillus amyloliquefaciens for enhanced production of S-adenosylmethionine by coupling of an engineered S-adenosylmethionine pathway and the tricarboxylic acid cycle. Biotechnol Biofuels. 2019;12:211.
Noda S, Shirai T, Oyama S, Kondo A. Metabolic design of a platform Escherichia coli strain producing various chorismate derivatives. Metab Eng. 2016;33:119–29.
Johnson CW, Beckham GT. Aromatic catabolic pathway selection for optimal production of pyruvate and lactate from lignin. Metab Eng. 2015;28:240–7.
Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Nat Biotechnol. 2010;28:245–8.
Monk JM, Charusanti P, Aziz RK, Lerman JA, Premyodhin N, Orth JD, Feist AM, Palsson BO. Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc Natl Acad Sci U S A. 2013;110:20338–43.
Holbrook EL, Greene RC, Krueger JH. Purification and properties of cystathionine gamma-synthase from overproducing strains of Escherichia coli. Biochemistry. 1990;29:435–42.
Cotton CA, Bernhardsgrutter I, He H, Burgener S, Schulz L, Paczia N, Dronsella B, Erban A, Toman S, Dempfle M, et al. Underground isoleucine biosynthesis pathways in E.coli. Elife. 2020;9:e54207.
Takagi H, Ohtsu I. L-Cysteine metabolism and fermentation in microorganisms. Adv Biochem Eng Biotechnol. 2017;159:129–51.
Kredich NM, Tomkins GM. The enzymic synthesis of L-cysteine in Escherichia coli and Salmonella typhimurium. J Biol Chem. 1966;241:4955–65.
Liu H, Hou Y, Wang Y, Li Z. Enhancement of sulfur conversion rate in the production of L-cysteine by engineered Escherichia coli. J Agric Food Chem. 2020;68:250–7.
Denk D, Böck A. L-Cysteine biosynthesis in Escherichia coli: nucleotide sequence and expression of the serine acetyltransferase (cysE) gene from the wild-type and a cysteine-excreting mutant. J Gen Microbiol. 1987;133:515–25.
Wei L, Wang H, Xu N, Zhou W, Ju J, Liu J, Ma Y. Metabolic engineering of Corynebacterium glutamicum for L-cysteine production. Appl Microbiol Biotechnol. 2019;103:1325–38.
Takagi H, Kobayashi C, Kobayashi S, Nakamori S. PCR random mutagenesis into Escherichia coli serine acetyltransferase: isolation of the mutant enzymes that cause overproduction of L-cysteine and L-cystine due to the desensitization to feedback inhibition. FEBS Lett. 1999;452:323–7.
Al-Rabiee R, Zhang Y, Grant GA. The mechanism of velocity modulated allosteric regulation in D-3-phosphoglycerate dehydrogenase. Site-directed mutagenesis of effector binding site residues. J Biol Chem. 1996;271:23235–8.
Rossi E, Motta S, Mauri P, Landini P. Sulfate assimilation pathway intermediate phosphoadenosine 5’-phosphosulfate acts as a signal molecule affecting production of curli fibres in Escherichia coli. Microbiology (Reading). 2014;160:1832–44.
Liu C, Martin E, Leyh TS. GTPase activation of ATP sulfurylase: the mechanism. Biochemistry. 1994;33:2042–7.
Shim J, Shin Y, Lee I, Kim SY. L-Methionine production. Adv Biochem Eng Biotechnol. 2017;159:153–77.
Kromer JO, Wittmann C, Schroder H, Heinzle E. Metabolic pathway analysis for rational design of L-methionine production by Escherichia coli and Corynebacterium glutamicum. Metab Eng. 2006;8:353–69.
Bolten CJ, Schroder H, Dickschat J, Wittmann C. Towards methionine overproduction in Corynebacterium glutamicum–methanethiol and dimethyldisulfide as reduced sulfur sources. J Microbiol Biotechnol. 2010;20:1196–203.
Chen Z, Xia Y, Liu H, Liu H, Xun L. The mechanisms of thiosulfate toxicity against Saccharomyces cerevisiae. Antioxidants (Basel). 2021;10(5):646.
San Millan A, MacLean RC. Fitness costs of plasmids: a limit to plasmid transmission. Microbiol Spectr. 2017. https://doi.org/10.1128/microbiolspec.MTBP-0016-2017.
Lee SY, Kim HU. Systems strategies for developing industrial microbial strains. Nat Biotechnol. 2015;33:1061–72.
Zhang G, Wang W, Deng A, Sun Z, Zhang Y, Liang Y, Che Y, Wen T. A mimicking-of-DNA-methylation-patterns pipeline for overcoming the restriction barrier of bacteria. PLoS Genet. 2012;8: e1002987.
Hayashi K, Morooka N, Yamamoto Y, Fujita K, Isono K, Choi S, Ohtsubo E, Baba T, Wanner BL, Mori H, Horiuchi T. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol Syst Biol. 2006;2006(2):0007.
Bertani G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol. 1951;62:293–300.
Liu S, Xiao H, Zhang F, Lu Z, Zhang Y, Deng A, Li Z, Yang C, Wen T. A seamless and iterative DNA assembly method named PS-Brick and its assisted metabolic engineering for threonine and 1-propanol production. Biotechnol Biofuels. 2019;12:180.
Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol. 2015; 81:2506–2514.
Liu S, Liang Y, Liu Q, Tao T, Lai S, Chen N, Wen T. Development of a two-stage feeding strategy based on the kind and level of feeding nutrients for improving fed-batch production of L-threonine by Escherichia coli. Appl Microbiol Biotechnol. 2013;97:573–83.
Shang X, Chai X, Lu X, Li Y, Zhang Y, Wang G, Zhang C, Liu S, Zhang Y, Ma J, Wen T. Native promoters of Corynebacterium glutamicum and its application in L-lysine production. Biotechnol Lett. 2018;40:383–91.
Becker SA, Feist AM, Mo ML, Hannum G, Palsson B, Herrgard MJ. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat Protoc. 2007;2:727–38.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8.
Acknowledgements
We thank Dr. Guoxia Liu for assistance with Real-Time Reverse Transcription-PCR.
Funding
This work was supported by the National Natural Science Foundation of China (31800073 and 32171469), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (Y2022035), and the Innovation Academy for Green Manufacture, Chinese Academy of Sciences (IAGM-2019-A02).
Author information
Authors and Affiliations
Contributions
TW, SL, and ZL conceived and designed the study. ZL, QL, and JS performed the experiments and all data analysis with assistance from JS, ML, YZ, and AD. ZL wrote the manuscript with the revision from TW and SL. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Plasmids used in this study. Table S2. Primers used in this study. Figure S1. The expression of wild-type and mutated MetA was detected by SDS-PAGE in MET1. Lane 0, MET1/pBR322. Lane 1, MET1/pBR322-metA. Lane 2, MET1/pBR322-metAI124L. Lane 3, MET1/pBR322-metAI229Y. Lane 4, MET1/pBR322-metAI124L, I229Y. Lane 5, MET1/pBR322-metAR27C, I296S, P298L. Lane 6, MET1/pBR322-metAR27C, I296S, P298L, I124L. Lane 7, MET1/pBR322-metAR27C, I296S, P298L, I229Y. Lane 8, MET1/pBR322-metAR27C, I296S, P298L, I124L, I229Y. Figure S2. l-methionine relative titer and OD600 of MET4, MET5, MET6, and MET7. Data are presented by mean values with the standard deviation from three biological replicates. One-way analysis of variance was used to determine significant differences. Figure S3. Fed-batch fermentation process curve in a 5 L bioreactor. With ammonium thiosulfate feeding during the fed-batch cultivation of the MET17 strain. Data are presented as the mean values with the standard deviation from two replicates.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Z., Liu, Q., Sun, J. et al. Multivariate modular metabolic engineering for enhanced l-methionine biosynthesis in Escherichia coli. Biotechnol Biofuels 16, 101 (2023). https://doi.org/10.1186/s13068-023-02347-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-023-02347-7