
Abstract
A novel series of 4-nitrophenylpiperazine derivatives (4a–m) was designed and synthesized as potential tyrosinase inhibitors. Comprehensive characterization using 1H-NMR, 13C-NMR, CNH, and IR techniques was performed for all target compounds. Subsequently, the derivatives were evaluated for their inhibitory activity against tyrosinase. Among them, compound 4l, featuring an indole moiety at the N−1 position of the piperazine ring, exhibited a significant tyrosinase inhibitory effect with an IC50 value of 72.55 μM. Enzyme kinetics analysis revealed that 4l displayed mixed inhibition of the tyrosinase enzymatic reaction. Molecular docking was carried out in the enzyme’s active site to further investigate the enzyme-inhibitor interactions. Based on the findings, compound 4l shows promise as a lead structure for the design of potent tyrosinase inhibitors. This study paves the way for the development of more effective tyrosinase inhibitors for potential applications in various fields.
Similar content being viewed by others
Introduction
Melanin is a critical biopolymer that significantly determines the color of mammalian skin, eyes, and hair. Beyond its pigmentation function, melanin serves as a protective shield, safeguarding skin cells from the harmful effects of UV radiation and eliminating reactive oxygen species, which are damaging byproducts of cellular processes [1]. Melanin is synthesized in the specialized organelles called melanosomes in melanocytes. However, the aberrant production and accumulation of melanin in the face and neck usually cause common pigmentary disorders, such as melasma and post-inflammatory hyperpigmentation, Parkinson’s, Ochronosis, and Alkaptonuria [2, 3].
Tyrosinases (TYRs, EC 1.14.18.1) are widely distributed enzymes found in various organisms, including bacteria, fungi, insects, plants, and mammals. They play a crucial role in regulating the synthetic pathway of melanin formation, a process involved in pigmentation and other essential biological functions. X-ray structures of tyrosinase showed that the enzyme from different origins is a type-3 metalloenzyme with a conserved catalytic domain comprising six histidine residues and two copper ions (CuA and CuB) [4, 5]. Tyrosinases are the key enzyme that catalyzes the conversion of the substrate tyrosine into the intermediate products l-dopa and o-quinone (dopaquinone), which is further oxidized into eumelanin and pheomelanin through other interrelated enzymatic and non-enzymatic reactions [6]. As a result, tyrosinase inhibition is a crucial strategy to control melanin synthesis; therefore, tyrosinase inhibitors have gained interest in therapies for skin disorders associated with abnormal pigmentation and dermo-cosmetic treatments [7].
Available tyrosinase inhibitors such as kojic acid, hydroquinone, and arbutin are anti-melanin agents, but they exhibit various adverse side effects, such as contact dermatitis, irritation, leukoderma, and hypochromic [8]. Therefore, it is of great interest for medical and cosmetic applications to synthesize novel inhibitors. Over the past few years, different tyrosinase inhibitors have been developed, including azole and thiazolidine, thiosemicarbazones, quinone, xanthate, and carboxylic acids [6].
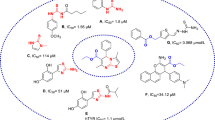
Tyrosine (compound A, Fig. 1) is the natural substrate of tyrosinase enzyme, and a variety of inhibitors mimic the chemical structure of tyrosinase’s natural substrate to hinder the oxidation process of tyrosinase [9]. Cinnamic acid, as an organic acid with low toxicity (compound B), presents naturally in plants and has been under investigation for a long time due to its therapeutic relevance [10, 11]. This α,β-unsaturated carbonyl structure demonstrated good tyrosinase inhibition. Also, variation of its structural properties exhibited a wide range of tyrosinase inhibitory potentials, from almost inactive, such as caffeic acid, and 3,4-dihydroxycinnamic acid, to highly potent inhibitors [12]. In the year 2019, cinnamamides, through the conversion of the carboxylic acid functional group into an amido group to increase lipophilicity, were designed. The structure–activity relationship (SAR) showed that N-methyl piperazine (compound C) had higher activity than morpholine, cyclopentamine, and cyclohexylamine derivatives. Analysis of tyrosinase activity and melanin content in B16F10 cells showed that compound C dose-dependently inhibited both cellular tyrosinase activity and melanin content [13]. Also, it was demonstrated that derivatives with heteroatom capable of H-bound interaction group on the aromatic ring of the scaffold generally showed high tyrosinase inhibitory activities [14]. Romagnoli et al. also developed cinnamic acid derivatives linked to aryl piperazines (compound D) in 2022. It was shown that the piperazine ring could provide the proper balance between flexibility and rigidity to correctly orientate the substituted moiety into the active site of tyrosinase. As reported by the previous study, the presence of electron-withdrawing substituent (–NO2) and electron-releasing substituent (–OCH3) capable of H-bound interaction with enzyme improves the inhibition [15]. In another study, a series of 1-(4-fluorobenzyl)piperazine (compound E) fragments were introduced as potent tyrosinase inhibitors with IC50 values in the range of 0.48–14.66 µM [16].

Potent inhibitors of mushroom tyrosinase from previous studies and newly designed compounds (4a–m)
In this study, we utilized the structural similarity between l-tyrosine and the previously reported potent derivatives by incorporating aryl substitutions linked to nitrophenyl piperazine. This approach was explored as a potential strategy for designing new tyrosinase inhibitors. After synthesizing all the compounds, their tyrosinase inhibition was evaluated using tyrosinase. The most potent derivative from this group was then selected for further investigation through kinetic and molecular docking studies to better understand its inhibitory properties and binding interactions with the enzyme.
Results and discussion
Chemistry
The synthesis and mechanism of the compounds 4a–m were carried out during a one-pot stepwise synthesis (OPSS) that, were schematically described in Schemes 1 and 2 respectively. As shown in the mechanism of this reaction, in the first stage, DABCO attacked 4-fluoronitrobenzene as a nucleophile, which caused the formation of intermediate A. This intermediate has a quaternary amine in its structure and is under pressure due to its bicyclic structure. In the second stage, the cesium carbonate (strong mineral base) absorbs hydrogen of acid derivatives and the resulting carboxylates, as a nucleophile, attack the carbons adjacent to the ammonium group which are in the same position, causing the opening of the bicyclic ring and the release of nitrogen from the positive charge pressure, creating the final products 4a–m. The yield of the final products as mentioned in the spectral information section was obtained in the range of 75–88%, which indicates the cheap and fast synthesis chosen for this reaction. Finally, the structure of final products 4a–m was confirmed using NMR, IR spectroscopy, and elemental analysis.

Synthesis of 4-nitrophenyl piperazine derivatives 4a–m

Mechanism of 4-nitrophenyl piperazine derivatives 4a–m
Tyrosinase inhibitory activity assay
Tyrosinase inhibitory assay of all synthesized nitrophenyl piperazine derivatives 4a–m was performed using l-dopa as the substrate, and the results are summarized in Table 1.
Compound 4a with no substitute group was selected as the template compound exhibited IC50 = 174.71 μM. The presence of 2-Br (4b, IC50 > 200 μM) and 2,4-dichloro (4c, IC50 > 200 μM) as halogen substitution on the phenyl ring did not improve the potencies compared with 4a. Also, nitro substituent as a strong electron-withdrawing group at the para (4d) or meta (4e) position of the phenyl ring did not empower the potency vs 4a. Also, 2-chloro-4-nitrophenyl substitution (4f) appeared to deteriorate the inhibitory activities (IC50 > 200 µM). Incorporating 2,3-dimethoxy benzoate in compound 4h results in a slight increase in the activity compared to the rest of the halogen-substituted groups.
Next, the elongation of the linker was also evaluated, and it was revealed that compound 4i bearing benzyl moiety had less potency than the phenyl counterpart, 4a, with an IC50 value of 184.24 µM. Also, the effect of bound unsaturation with the same structure as cinnamic acid was examined. Such substitution in 4j did not alleviate the activity (IC50 > 200 µM).
To better extract the SARs, ring replacement was performed; noteworthy, 3-pyridine substitution (4k) significantly improved the potency compared with phenyl and benzyl counterparts with IC50 value of 82.68 µM. The best results in this set of compounds came back to 4l bearing indole moiety at R position with IC50 = 72.55 µM. In contrast, 4m bearing 5-nitrofuran reduced the activity compared to 4k and 4l; it appeared to have lower inhibitory activities than most compounds.
In summary, the presence of phenyl or benzyl at the R position did not show significant inhibition, and any substitutions on this ring were also unfavorable for tyrosinase inhibition. However, replacing benzyl with 3-pyridine or 2-indole led to a notable improvement in potency compared to the rest of the derivatives. These findings highlight the importance of specific substitutions at the R position in enhancing the inhibitory activity of the compounds against tyrosinase.
Inhibition mechanism
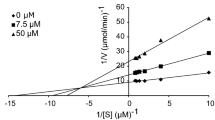
The most potent derivative, compound 4l, was studied for its enzyme inhibition mode using Lineweaver–Burk plot analysis. The results, depicted in Fig. 2 and summarized in Table 2, showed that the Km and Vmax values increased as the inhibitor concentration was raised. The Lineweaver–Burk plots for tyrosinase inhibition with various concentrations of 4l and l-dopa displayed straight lines intersecting the x-axis at similar points. These findings indicate that compound 4l acts as a mixed inhibitor against mushroom tyrosinase, affecting substrate binding and enzyme catalysis.

Lineweaver–Burk plot for the inhibition of mushroom tyrosinase catalyzed l-dopa oxidation by 4l
Molecular docking
The reliability of the applied docking protocol was assessed by re-docking of tropolone into the active site of the tyrosinase enzyme. The key characteristic of a good docking program is its ability to reproduce the experimental or crystallographic binding modes of ligands. To test this, a ligand is taken out of the X-ray structure of its protein–ligand complex and re-docked into its binding site. The docked binding mode is then compared with the experimental binding mode, and the RMSD is calculated; a prediction of a binding mode is considered successful if the RMSD is below a certain value (usually < 2.0 Å). Figure 3 shows the superimposed structures between the docked and the crystallographic tropolone over tyrosinase active site which its RMSD is in acceptable value within the cutoff limit (1.02 Å). This protocol was then similarly applied to all synthesized compounds (4a–4m).

Representation of the tyrosinase active site, the tropolone co-crystallized and the corresponding re-docked form are represented in green and cyan color, respectively
To gain insight into the inhibitory activity of compounds, molecular docking was performed to investigate its interaction pattern with the active site of tyrosinase. Analysis of the docked ligand poses showed that His61, His85, His259, His263 were the top residues producing the greatest number of interactions at the tyrosinase bi nuclear active site pocket. Table 3 shows docking binding score and the free binding energy calculation based on MM-GBSA calculation of the synthesized compounds (4a–4m). The obtained MM-GBSA energy are close to and correlated with the mentioned experimental results. Compounds Kojic acid and 4l with the highest inhibitory activity represent the MM-GBSA energy of − 80.6 and − 76.3 kcal mol−1, respectively, while compounds with lowest IC50 represent lower MM-GBSA energy (Table 3).
Docking study of compound 4l showed that it formed deep interactions with the active site residues and di-copper ions which are surrounded by α3/α4 and α10/α11 helices over the active pocket of tyrosinase enzyme (Fig. 4a). Also, Fig. 4b shows that the 4l indole group orients toward the two-Copper ions through several hydrophobic pi-pi interactions with the imidazolidine rings of His85, His259 and His263, which coordinate to the di-copper core of the active site. Additionally, the indolyl NH group provides H-bond interaction with the backbone carbonyl group of Met280. Additionally, Trp227 and Phe264 are the remaining residues for stabilizing compound 4l over the active site pocket through hydrophobic interaction with the ester linker and the nitro group of the mentioned compound.

Representation of the compounds docking poses over the tyrosinase enzyme (a) close-up illustration and the non-boding interaction pattern of compound 4l over the tyrosinase active site (b)
Molecular dynamic (MD) simulation
In order to study the stability of the complex, the best induced fit docked pose of compound 4l was implemented as starting points for 150 ns MD simulation in order to predict the motion and the dynamic behavior of complexed systems at an atomistic level [17].
Root mean square deviation (RMSD) values are indicative of the conformational stability and perturbations of system. When RMSD values no longer follow a specific trend but fluctuate around a certain point it can be argued that the complex has reached equilibrium [18].
Figure 5 depicts the protein backbone RMSD values for the tyrosinase-4l complex over about 150 ns MD simulation time. It is obvious that the RMSD value of the tyrosinase complexed with compound 4l fluctuate through the first 80 ns and reaches to an equilibration for the rest of simulation time. Such observation indicated that the employed simulation time was enough to obtain an equilibrium structure over the simulation time.

RMSD plot of tyrosinase backbone in complex with compound 4l, throughout the 150 ns of the simulation time
The total contact diagram of MD procedure revealed that, the quantity of ligand-tyrosinase interaction decreased from 4 contacts at the beginning of the MD simulation to zero during the short period time and after that it raised and fluctuated between 2 and 6 contacts for the majority of the MD simulation time (Fig. 6a). Also, the mentioned phenomena can be tracked by Fig. 6b in which at a short period time of MD simulation time there are few ligand–residue interaction while after that time it increased (dark orange color) and stabilized for the rest of the MD simulation time.

The total tyrosinase residues contact (a) and the timeline representation of the interactions of compound 4l in each trajectory frame during the total MD simulation time (the more contact of ligand with residues, the darker shade of orange color) (b). The 3D representation of tyrosinase with compound 4l in two different binding pockets related to 0–6.4 ns (c) and 6.4 to the rest of simulation time (d) (Desmond v5.3)
Additionally, the visual inspection of the MD trajectories revealed that the MD simulation time divided in two sections; the first part with short duration was from 0 to 6.4 in which compound 4l located at the heart of the active site and oriented towards the di-copper catalytic active site through its indoline moiety (Fig. 6c). Otherwise the other one belongs to the significant amount of time last from 6.4 to 150 ns in which the ligand resided to the secondary binding pocket which was far from the bi-metal active site and which faced toward the final C-terminal helix of the tyrosinase enzyme (Fig. 5d). The new pose of compound 4l increased an average number of ligand-enzyme contacts from 0 to about 4–6 interactions (Fig. 6a).
Furthermore, the 2D interactions diagram of compound 4l-tyrosinase complex in with the enzyme are depicted in Fig. 7a shows that the aromatic property of the indoline ring and corresponding –NH group stabilized with Arg225 and Trp141 through the direct hydrogen bond and pi-cation interaction for about the majority of the MD simulation time (93% and 95% of the simulation time, respectively). Also, the carbonyl part of the ester group interacted through water mediated hydrogen bond by His390 for about 35% of MD simulation time. Also, the nitrogen atom belongs to the piperazine ring provided indirect H-bond interaction with Tyr140 for about 15% and finally the p-nitro phenyl group provided the occasionally salt-bridge, Pi-cation and pi–pi stacking interaction with Trp386 and Lys389 at the C-terminal domain of the enzyme. Previously, Tyr140 and the correspond environment introduced as allosteric binding site of tyrosinase enzyme [19]. Based on the RMSD plot it can be revealed that the mentioned ligand interactions caused decreasing the corresponding residue fluctuation at different domain of the enzyme (Fig. 7b).

2D interaction diagram of compound 4l-tyrosinase complex which is responsible for through the whole MD simulation time (a), RMSF plot of the tyrosinase residue in complexed with compound 4l over 150 ns MD simulation time (b) α-helical and ß-strand regions are highlighted in light pink and blue backgrounds, respectively
In summary, through MD simulation investigation, it is revealed that the resulted IFD posed of compound 4l is slightly stay at the bi-metal active site and stable at the allosteric secondary binding pocket which is far from the active site and is responsible for many important interactions. Based on enzyme kinetic studies, compound 4l exerted a mixed-type inhibition, binding ability of 4l with both the catalytic site and the allosteric sites confirmed its mixed-type inhibition of tyrosinase.
In silico prediction of pharmacokinetic properties
In order to predict the oral and gastrointestinal absorption of compound 4l and 4k as higher biological activated compound in this series and kojic acid as standard inhibitor of tyrosinase, the predicted aqueous solubility (Log S), the predicted apparent Caco-2 cell permeability as a model for the gut-blood barrier (non-active transport) (LogCaco-2) and the predicted % human intestinal absorption (%HIA) calculated with the aid of pkCSM web server (http://biosig.unimelb.edu.au/pkcsm).
According to Table 4, the predicted solubility of all compounds are in favorable value. In this all compound exhibited high HIA in which their value is higher than 30%.
Conclusion
In conclusion, this study designed, synthesized, and screened nitrophenyl piperazine derivatives with aryl substitutions against tyrosinase. Among them, compound 4l containing an indole derivative exhibited the highest potency with an IC50 value of 72.55 µM. The SAR analysis revealed that replacing the benzyl or phenyl ring with indole or pyridine moiety significantly improved the potencies of the compounds. Kinetic studies of the most potent derivative indicated mixed inhibition against tyrosinase. Molecular docking studies provided insights into the key interactions, including hydrogen bonding, π–π interactions, and salt bridge interactions, between tyrosinase and compound 4l. These findings suggest that nitrophenylpiperazine derivatives hold promise as potential anti-tyrosinase agents for applications in medicine, agriculture, and the food industry.
Materials and methods
Synthesis
General synthesis procedure of 4-nitrophenyl piperazine derivatives 4a–m
Synthesis of the compounds 4a–m. There is a conventional reaction between DABCO and 4-fluoronitrobenzene led to the formation of A mixture of 4-fluoronitrobenzene (compound 1, 1 mmol), DABCO (compound 2, 1.2 mmol), benzoic acid derivatives (compound 3, 1.2 mmol), and Cs2CO3 (1.3 mmol) in DMF (5 ml) was stirred at 80 °C for 4 h. A stopwatch monitored the time of the reaction. The progress of the reaction was monitored by thin-layer chromatography. On completion of the reaction, the reaction mixture was drowned in water. The precipitated product was filtered and dried.
Spectral information
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl benzoate (4a)
White solid; isolated yield: 75%, M.p: 176–178 °C; IR (ν, cm−1): 1729, 1552, 1450, 1348. 1H NMR (500 MHz, DMSO-d6) δ 8.00 (dd, J = 32.8, 8.6 Hz, 4H), 7.66 (t, J = 7.5 Hz, 1H), 7.53 (t, J = 7.7 Hz, 2H), 7.01 (d, J = 8.9 Hz, 2H), 4.43 (t, J = 5.7 Hz, 2H), 3.44 (t, J = 5.0 Hz, 4H), 2.77 (t, J = 5.7 Hz, 2H), 2.62 (t, J = 5.0 Hz, 4H). 13C NMR (125 MHz, DMSO-d6) δ 166.13, 155.17, 137.31, 133.78, 130.23, 129.60, 129.56, 129.23, 126.13, 113.05, 112.99, 62.74, 56.34, 52.83, 46.82. Anal. Calcd. for C19H21N3O4 (355.39): C, 64.21; H, 5.96; N, 11.82. Found: C, 64.11; H, 5.84; N, 11.65 (Additional file 1).
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl 2-bromobenzoate (4b)
White solid; isolated yield: 83%, M.p: 185–187 °C; IR (ν, cm−1): 1726, 1549, 1354, 1293, 656. 1H NMR (500 MHz, DMSO-d6) δ 8.03 (d, J = 8.9 Hz, 2H), 7.75 (t, J = 7.6 Hz, 2H), 7.49 (p, J = 7.6 Hz, 2H), 7.00 (d, J = 9.0 Hz, 2H), 4.43 (t, J = 5.7 Hz, 2H), 3.43 (t, J = 5.0 Hz, 4H), 2.74 (t, J = 5.7 Hz, 2H), 2.60 (t, J = 5.0 Hz, 4H). 13C NMR (125 MHz, DMSO-d6) δ 166.15, 155.15, 137.31, 134.32, 133.49, 133.07, 131.23, 128.30, 126.12, 120.54, 113.04, 62.98, 56.20, 52.75, 46.82. Anal. Calcd. for C19H20BrN3O4 (434.28): C, 52.55; H, 4.64; N, 9.68. Found: C, 52.37; H, 4.85; N, 9.53.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl 2,4-dichlorobenzoate (4c)
White solid; isolated yield: 87%, M.p: 196–198 °C; IR (ν, cm−1): 1741, 1549, 1428, 1347, 1185, 773. 1H NMR (400 MHz, DMSO-d6) δ 8.07–8.00 (m, 2H), 7.84 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 2.0 Hz, 1H), 7.57 (dd, J = 8.4, 2.1 Hz, 1H), 7.06–6.97 (m, 2H), 4.44 (t, J = 5.6 Hz, 2H), 3.43 (t, J = 5.1 Hz, 4H), 2.74 (t, J = 5.6 Hz, 2H), 2.60 (t, J = 5.1 Hz, 4H). 13C NMR (100 MHz, DMSO-d6) δ 164.64, 155.16, 137.62, 137.28, 133.61, 132.93, 130.84, 129.42, 128.18, 126.15, 113.05, 63.10, 56.15, 52.72, 46.79. Anal. Calcd. for C19H19Cl2N3O4: C, 53.79; H, 4.51; N, 9.90. Found: C, 53.61; H, 4.72; N, 9.80.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl 4-nitrobenzoate (4d)
Light brown solid; isolated yield: 87%, M.p: 172–174 °C; IR (ν, cm−1): 1736, 1556, 1343, 1210. 1H NMR (400 MHz, DMSO-d6) δ 8.41–8.29 (m, 2H), 8.25–8.13 (m, 2H), 8.12–7.98 (m, 2H), 7.01 (d, J = 9.4 Hz, 2H), 4.49 (t, J = 5.6 Hz, 2H), 3.44 (t, J = 5.1 Hz, 4H), 2.79 (t, J = 5.6 Hz, 2H), 2.63 (t, J = 5.1 Hz, 4H). 13C NMR (100 MHz, DMSO-d6) 164.72, 155.14, 150.67, 137.27, 135.61, 131.09, 126.15, 124.38, 113.03, 63.41, 56.19, 52.77, 46.78. Anal. Calcd for C19H20N4O6: C, 57.00; H, 5.04; N, 13.99. Found: C, 56.81; H, 5.19; N, 13.83.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl 3-nitrobenzoate (4e)
Light brown solid; isolated yield: 84%, M.p: 181–183 °C; IR (ν, cm−1): 1731, 1558, 1348, 1224. 1H NMR (400 MHz, DMSO-d6) δ 8.62 (t, J = 2.0 Hz, 1H), 8.50 (ddd, J = 8.3, 2.4, 1.2 Hz, 1H), 8.37 (dt, J = 7.8, 1.3 Hz, 1H), 8.11–7.95 (m, 2H), 7.85 (t, J = 8.0 Hz, 1H), 7.10–6.90 (m, 2H), 4.50 (t, J = 5.7 Hz, 2H), 3.44 (t, J = 5.1 Hz, 4H), 2.80 (t, J = 5.7 Hz, 2H), 2.63 (t, J = 5.1 Hz, 4H). 13C NMR (100 MHz, DMSO-d6) δ 164.59, 155.51, 150.53, 137.39, 135.27, 131.79, 129.15, 128.35, 126.58, 123.32, 113.35, 63.03, 55.95, 52.61, 46.95. Anal. Calcd for C19H20N4O6: C, 57.00; H, 5.04; N, 13.99. Found: C, 56.84; H, 5.26; N, 14.14.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl 2-chloro-4-nitrobenzoate (4f)
Light brown solid; isolated yield: 88%, M.p: 203–205 °C; IR (ν, cm−1): 1736, 1559, 1358, 1236, 756. 1H NMR (500 MHz, DMSO-d6) δ 8.39 (d, J = 2.0 Hz, 1H), 8.32–8.26 (m, 1H), 8.07–8.01 (m, 3H), 7.07–6.99 (m, 2H), 4.50 (t, J = 5.7 Hz, 2H), 3.44 (t, J = 4.9 Hz, 4H), 2.76 (t, J = 5.7 Hz, 2H), 2.61 (t, J = 4.9 Hz, 4H). 13C NMR (125 MHz, DMSO-d6) δ 164.48, 155.18, 149.77, 139.08, 137.32, 136.60, 132.31, 126.14, 125.92, 122.97, 113.08, 63.55, 56.10, 52.69, 46.82. Anal. Calcd for C19H19ClN4O6 (434.83): C, 52.48; H, 4.40; N, 12.88. Found: C, 52.31; H, 4.57; N, 12.72.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl 2-(4-methoxyphenyl)acetate (4g)
White solid; isolated yield: 77%, M.p: 207–209 °C; IR (ν, cm−1): 1734, 1555, 1357, 1286. 1H NMR (500 MHz, DMSO-d6) δ 8.05 (d, J = 9.4 Hz, 2H), 7.19 (d, J = 8.6 Hz, 2H), 7.00 (d, J = 9.4 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 4.17 (t, J = 5.6 Hz, 2H), 3.72 (s, 3H), 3.59 (s, 2H), 3.38 (t, J = 5.1 Hz, 4H), 2.58 (t, J = 5.7 Hz, 2H), 2.53–2.47 (m, 4H). 13C NMR (125 MHz, DMSO-d6) δ 171.77, 158.62, 155.15, 137.31, 130.81, 126.74, 126.14, 114.18, 113.01, 62.09, 56.26, 55.47, 52.75, 46.75, 40.02. Anal. Calcd. for C21H25N3O5 (399.44): C, 63.14; H, 6.31; N, 10.52. Found: C, 63.01; H, 6.20; N, 10.36.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl 6-formyl-2,3-dimethoxybenzoate (4h)
White solid; isolated yield: 78%, M.p: 176–178 °C; IR (ν, cm−1): 1723, 1545, 1347, 1258, 1151. 1H NMR (500 MHz, DMSO-d6) δ 9.85 (s, 1H), 8.04 (d, J = 9.2 Hz, 2H), 7.78 (d, J = 8.5 Hz, 1H), 7.36 (d, J = 8.5 Hz, 1H), 7.02 (d, J = 9.5 Hz, 2H), 4.43 (t, J = 5.6 Hz, 2H), 3.96 (s, 3H), 3.77 (s, 3H), 3.43 (t, J = 5.0 Hz, 4H), 2.72 (t, J = 5.7 Hz, 2H), 2.57 (t, J = 5.0 Hz, 4H). 13C NMR (125 MHz, DMSO-d6) δ 190.58, 166.17, 157.94, 155.17, 145.88, 137.30, 130.92, 128.70, 126.40, 126.13, 113.90, 113.06, 62.99, 61.76, 56.94, 56.00, 52.69, 46.74.
Anal. Calcd. for C22H25N3O7 (394.42): C, 59.59; H, 5.68; N, 9.48. Found: C, 59.41; H, 5.89; N, 9.32.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl 2-phenylacetate (4i)
White solid; isolated yield: 84%, M.p: 178–180 °C; IR (ν, cm−1): 1736, 1548, 1357, 1199. 1H NMR (500 MHz, DMSO-d6) δ 8.06 (d, J = 9.4 Hz, 2H), 7.30 (ddd, J = 18.2, 12.9, 7.4 Hz, 5H), 7.01 (d, J = 9.1 Hz, 2H), 4.19 (t, J = 5.7 Hz, 2H), 3.68 (s, 2H), 3.39 (t, J = 5.0 Hz, 4H), 2.59 (t, J = 5.7 Hz, 2H), 2.51 (d, J = 4.8 Hz, 4H). 13C NMR (125 MHz, DMSO-d6) δ 171.50, 155.15, 137.31, 134.86, 129.78, 128.78, 127.25, 126.16, 113.05, 62.13, 56.26, 52.75, 46.77, 40.91. Anal. Calcd. for C20H23N3O4 (369.41): C, 65.03; H, 6.28; N, 11.37. Found: C, 64.88; H, 6.45; N, 11.20.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl cinnamate (4j)
White solid; isolated yield: 77%, M.p: 185–187 °C; IR (ν, cm−1): 1720, 1546, 1357, 1288. 1H NMR (500 MHz, DMSO-d6) δ 8.04 (d, J = 8.7 Hz, 2H), 7.79–7.59 (m, 3H), 7.42 (d, J = 5.7 Hz, 3H), 7.01 (d, J = 9.0 Hz, 2H), 6.66 (d, J = 15.9 Hz, 1H), 4.31 (t, J = 5.8 Hz, 2H), 3.44 (t, J = 5.0 Hz, 4H), 2.68 (t, J = 5.8 Hz, 2H), 2.59 (t, J = 4.9 Hz, 4H). 13C NMR (125 MHz, DMSO-d6) δ 166.64, 155.17, 145.11, 137.31, 134.43, 130.97, 129.38, 128.86, 126.14, 118.47, 113.05, 61.89, 56.44, 52.85, 46.77. Anal. Calcd for C21H23N3O4 (381.43): C, 66.13; H, 6.08; N, 11.02. Found: C, 65.98; H, 6.23; N, 11.19.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl nicotinate (4k)
White solid; isolated yield: 77%, M.p: 168–170 °C; IR (ν, cm−1): 1723, 1552, 1353. 1H NMR (500 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.82 (d, J = 4.8 Hz, 1H), 8.29 (d, J = 8.0 Hz, 1H), 8.03 (d, J = 9.1 Hz, 2H), 7.57 (dd, J = 8.0, 4.9 Hz, 1H), 7.01 (d, J = 9.0 Hz, 2H), 4.46 (t, J = 5.6 Hz, 2H), 3.44 (t, J = 4.9 Hz, 4H), 2.78 (t, J = 5.6 Hz, 2H), 2.63 (t, J = 5.0 Hz, 4H). 13C NMR (125 MHz, DMSO-d6) δ 165.13, 155.16, 154.15, 150.45, 137.30, 126.17, 126.13, 124.40, 113.04, 63.07, 56.22, 52.78, 46.82. Anal. Calcd for C18H20N4O4 (356.38): C, 60.66; H, 5.66; N, 15.72. Found: C, 60.51; H, 5.81; N, 15.58.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl 1H-indole-2-carboxylate (4l)
White solid; isolated yield: 81%, M.p: 192–194 °C; IR (ν, cm−1): 3360, 1729, 1553, 1350, 1266. 1H NMR (500 MHz, DMSO-d6) δ 11.10 (s, 1H), 8.03 (d, J = 8.8 Hz, 2H), 7.46 (d, J = 7.8 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 6.98 (dd, J = 14.7, 8.5 Hz, 3H), 6.90 (t, J = 7.4 Hz, 1H), 6.53 (s, 1H), 4.45 (t, J = 5.7 Hz, 2H), 3.41 (d, J = 7.1 Hz, 4H), 2.63 (t, J = 5.0 Hz, 2H), 2.53 (t, J = 4.9 Hz, 4H). 13C NMR (125 MHz, DMSO-d6) δ 164.92, 155.10, 140.48, 137.37, 136.01, 128.50, 126.11, 121.21, 120.86, 118.69, 113.09, 112.34, 101.37, 62.47, 56.44, 52.87, 46.58. Anal. Calcd. for C21H22N4O4 (394.42): C, 63.95; H, 5.62; N, 14.20. Found: C, 63.81; H, 5.77; N, 14.01.
2-(4-(4-Nitrophenyl)piperazin-1-yl)ethyl 5-nitrofuran-2-carboxylate (4m)
Light brown solid; isolated yield: 85%, M.p: 173–175 °C; IR (ν, cm−1): 1728, 1560, 1358, 1274. 1H NMR (499 MHz, DMSO-d6) δ 8.05 (d, J = 9.1 Hz, 2H), 7.79 (d, J = 3.9 Hz, 1H), 7.60 (d, J = 3.9 Hz, 1H), 7.03 (d, J = 8.9 Hz, 2H), 4.49 (t, J = 5.6 Hz, 2H), 3.44 (t, J = 4.9 Hz, 4H), 2.75 (t, J = 5.7 Hz, 2H), 2.62 (t, J = 5.0 Hz, 4H). 13C NMR (126 MHz, DMSO-d6) δ 157.17, 155.17, 144.51, 137.31, 126.15, 120.47, 113.52, 113.07, 63.49, 56.12, 52.75, 46.78. Anal. Calcd for C17H18N4O7 (390.35): C, 52.31; H, 4.65; N, 14.35. Found: C, 52.17; H, 4.46; N, 14.19.
Tyrosinase inhibitory assay
The mushroom tyrosinase activity assays for the synthetic compounds were conducted using a modified procedure from a previous study. In summary, a 96-well microplate was utilized, and 160 μl of phosphate buffer (50 mM, pH 6.8), 10 μl of mushroom tyrosinase (500 U/ml, dissolved in PBS), and 10 μl of the test compound (dissolved in DMSO) were added to each well. Then, 20 μl of l-dopa (7 mM, dissolved in PBS) was added to initiate the enzymatic reaction. The change in absorbance at 475 nm was continuously monitored using a spectrophotometer. DMSO without test compounds was included as a control, while kojic acid was used as the positive control. The IC50 value, representing the compound’s inhibitory potency, was determined from dose–response curves of percentage inhibition. All experiments were performed in duplicate, and kojic acid served as the standard control for comparison [20].
Determination of the inhibition type
The most potent derivative was selected for kinetic analysis, and its inhibitory activity was assessed at inhibitor concentrations of 0, 10, 40, and 100 μM and the substrate (l-dopa) concentrations ranged from 0.5 to 2.8 mM according to the previously reported procedures [21].
Molecular docking
In order to find out the interaction’s mode of designed molecules over tyrosinase enzyme, Maestro Molecular Modeling platform (version10.5) by Schrödinger, LLC was performed [22]. The X-ray crystal structure of the receptor (PDB ID: 2Y9X) (in complex with tropolone) was obtained from the PDB database [23]. As tyrosinase is reported to have the catalytic active site in H subunit, all the docking studies were performed on H subnit. In addition, prosthetic group and co-factors are not directly involved in tyrosinase inhibition, so they totally removed before docking investigation. Water molecules and co-crystallized ligands were removed from the enzyme’s crystallographic structures. The 2D structures of all synthesized compounds were drawn in Marvin 15.10.12.0 program (http://www.chemaxon.com) [24] and converted into pdb file. The Protein Preparation Wizard [17] and the LigPrep [25] module was used to prepare protein and ligand structure properly. The missing side chains of the proteins were filled using the Prime tool and missing residues were updated.
The accurate side‑chain, backbone conformational changes or both during ligand binding at the active site of tyrosinase enzyme were predicted by IFD method using Glide software (Schrödinger LLC 2018, USA) [26]. The tropolone binding site was used to generate the grid for IFD calculation. The maximum 20 poses with receptor and ligand van der Waals radii of 0.7 and 0.5, respectively considered. Residues within 5 Å of the tropolone at the active site were refined followed by side-chain optimization. Structures whose Prime energy is more than 30 kcal/mol are eliminated based on extra precious Glide docking.
Molecular dynamic simulation
The X-ray crystallographic structure of tyrosinase (www.rcsb.org) and the structure of the compound with the best tyrosinase inhibition activity used. Molecular simulation was performed using the Desmond v5.3 (Schrödinger 2018‐4 suite) [27].
In order to build the system for MD simulation, the protein–ligand complexe was solvated with SPC explicit water molecules and placed in the center of an orthorhombic box of appropriate size in the periodic boundary condition. Sufficient counter‐ions and a 0.15 M solution of NaCl were also utilized to neutralize the system and to simulate the real cellular ionic concentrations, respectively. The MD protocol involved minimization, pre-production, and finally production MD simulation steps. In the minimization procedure, the entire system was allowed to relax for 2500 steps by the steepest descent approach. Then the temperature of the system was raised from 0 to 300 K with a small force constant on the enzyme in order to restrict any drastic changes. MD simulations were performed via NPT (constant number of atoms, constant pressure i.e., 1.01325 bar and constant temperature i.e. 300 K) ensemble. The Nose–Hoover chain method was used as the default thermostat with 1.0 ps interval and Martyna–Tobias–Klein as the default barostat with 2.0 ps interval by applying isotropic coupling style. Long‐range electrostatic forces were calculated based on particle‐mesh‐based Ewald approach with the he cut‐off radius for columbic forces set to 9.0 Å. Finally, the system subjected to produce MD simulations for 150 ns for each protein–ligand complex. During the simulation every 1000 ps of the actual frame was stored. The dynamic behavior and structural changes of the systems were analyzed by the calculation of the root mean square deviation (RMSD) and RMSF.
Prime MM-GBSA
The ligand binding energies (ΔGBind) were calculated using molecular mechanics/generalized born surface area (MM-GBSA) modules (Schrödinger LLC 2018) based on the following equation;
where ΔGBind is the calculated relative free energy which includes both ligand and receptor strain energy. EComplex is the MM-GBSA energy of the minimized complex, and ELigand is the MM-GBSA energy of the ligand after removing it from the complex and allowing it to relax. EReceptor is the MM-GBSA energy of relaxed protein after separating it from the ligand.
Prediction of pharmacokinetic properties
Prediction of the molecular properties of the synthesized compounds were performed using the online servers as pkCSM (http://biosig.unimelb.edu.au/pkcsm/).
Availability of data and materials
The datasets used or analyzed during the current study are available from the corresponding authors.
References
Solano F. Melanins: skin pigments and much more—types, structural models, biological functions, and formation routes. New J Sci. 2014;2014: 498276.
Liberti D, et al. A melanin-related phenolic polymer with potent photoprotective and antioxidant activities for dermo-cosmetic applications. Antioxidants. 2020;9(4):270.
Wang W, et al. Kojic acid showed consistent inhibitory activity on tyrosinase from mushroom and in cultured B16F10 cells compared with arbutins. Antioxidants. 2022;11(3):502.
Chang T-S. An updated review of tyrosinase inhibitors. Int J Mol Sci. 2009;10(6):2440–75.
Shu P, et al. Six natural phenylethanoid glycosides: total synthesis, antioxidant and tyrosinase inhibitory activities. ChemistrySelect. 2020;5(35):10817–20.
Zolghadri S, et al. A comprehensive review on tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2019;34(1):279–309.
Peng Z, et al. A systematic review of synthetic tyrosinase inhibitors and their structure–activity relationship. Crit Rev Food Sci Nutr. 2022;62(15):4053–94.
Pillaiyar T, Manickam M, Namasivayam V. Skin whitening agents: medicinal chemistry perspective of tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2017;32(1):403–25.
Sanginabadi F, et al. Exploring the potential of ω3 derivatives as tyrosinase inhibitors: a comprehensive study combining experimental, computational, and biological approaches. ChemistrySelect. 2023;8(22): e202300373.
De P, Baltas M, Bedos-Belval F. Cinnamic acid derivatives as anticancer agents—a review. Curr Med Chem. 2011;18(11):1672–703.
Sova M. Antioxidant and antimicrobial activities of cinnamic acid derivatives. Mini Rev Med Chem. 2012;12(8):749–67.
Iwai K, et al. In vitro antioxidative effects and tyrosinase inhibitory activities of seven hydroxycinnamoyl derivatives in green coffee beans. J Agric Food Chem. 2004;52(15):4893–8.
Ullah S, et al. Tyrosinase inhibition and anti-melanin generation effect of cinnamamide analogues. Bioorg Chem. 2019;87:43–55.
Nazir Y, et al. Hydroxyl substituted benzoic acid/cinnamic acid derivatives: tyrosinase inhibitory kinetics, anti-melanogenic activity and molecular docking studies. Bioorg Med Chem Lett. 2020;30(1): 126722.
Romagnoli R, et al. Cinnamic acid derivatives linked to arylpiperazines as novel potent inhibitors of tyrosinase activity and melanin synthesis. Eur J Med Chem. 2022;231: 114147.
Ielo L, et al. Exploiting the 1-(4-fluorobenzyl)piperazine fragment for the development of novel tyrosinase inhibitors as anti-melanogenic agents: design, synthesis, structural insights and biological profile. Eur J Med Chem. 2019;178:380–9.
Schrödinger release 2018-4: Schrödinger release 2018–4 protein preparation wizard; Epik, Schrödinger, LLC, New York, NY, 2016; impact, Schrödinger, LLC, New York, NY, 2016; Prime, Schrödinger, LLC, New York, NY, 2019.
Schrödinger release 2018-4: LigPrep, Schrödinger, LLC, New York, NY, 2018.
Jung HJ, et al. (E)-1-(Furan-2-yl)-(substituted phenyl) prop-2-en-1-one derivatives as tyrosinase inhibitors and melanogenesis inhibition: an in vitro and in silico study. Molecules. 2020;25(22):5460.
Divar M, et al. Benzyl-triazole derivatives of hydrazinecarbothiamide derivatives as potent tyrosinase inhibitors: synthesis, biological evaluation, structure–activity relationship and docking study. ChemistrySelect. 2023;8(8): e202203382.
Najafi Z, et al. Synthesis and molecular modeling of new 2-benzylidenethiobarbituric acid derivatives as potent tyrosinase inhibitors agents. J Chin Chem Soc. 2022;69(4):692–702.
Schrödinger suite 2018, induced fit docking protocol; glide version 5.5, S., LLC, New York, NY, 2009; Prime version 2.1, Schrödinger, LLC, New York, NY, 2009.
Ismaya WT, Rozeboom HJ, Weijn A, Mes JJ, Fusetti F, Wichers HJ, Dijkstra BW. Crystal structure of Agaricus bisporus mushroom tyrosinase: identity of the tetramer subunits and interaction with tropolone. Biochemistry. 2011;50:5477.
ChemAxon Marvin was used for drawing, displaying and characterizing Chemical structures, substructures and reactions, Marvin 15.10.12.0 (version number), 2015, ChemAxon. http://www.chemaxon.com.
Schrödinger LLC. Schrödinger release 2018-4: LigPrep. Schrödinger LLC; 2018. [Google Scholar].
Schrödinger release 2018-4: induced fit docking protocol; glide, Schrödinger, LLC, New York, NY, 2018; Prime, Schrödinger, LLC, New York, NY, 2018.
Schrödinger release 2018-4: Desmond molecular dynamics system, D. E. Shaw research, New York, NY, 2018. Maestro-Desmond interoperability tools, Schrödinger, New York, NY, 2018.
Acknowledgements
This publication was supported by the Research Council of Tehran University of Medical Sciences, Tehran, Iran.
Funding
Tehran University of Medical Sciences: 1402-2-104-67632.
Author information
Authors and Affiliations
Contributions
Mehdi Asadi and Fahime Fayazi synthesized compounds. Homa Azizian conducted the molecular modeling; docking study, MD, predictive ADME and contributed to the preparation of the manuscript computational part, Aida Iraji contributed to the preparation of the manuscript. Reyhaneh and Mannan Hajimahmoodi performed the biological assay. Mohammad Mahdavi and Bagher Larijani contributed to the design and characterization of compounds as well as the preparation of the manuscript. Massoud Amanlou supervised all phases of the study.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
The Supplementary Information file contains NMR spectra.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Asadi, M., Fayazi, F., Iraji, A. et al. Nitrophenylpiperazine derivatives as novel tyrosinase inhibitors: design, synthesis, and in silico evaluations. BMC Chemistry 18, 67 (2024). https://doi.org/10.1186/s13065-024-01167-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-024-01167-6