
Abstract
Pyrazole-bearing compounds are known for their diverse pharmacological effects including potent antileishmanial and antimalarial activities. Herein, some hydrazine-coupled pyrazoles were successfully synthesized and their structures were verified by employing elemental microanalysis, FTIR, and 1H NMR techniques. The in vitro antileishmanial and in vivo antimalarial activities of the synthesized pyrazole derivatives (9–15) were evaluated against Leishmania aethiopica clinical isolate and Plasmodium berghei infected mice, respectively. The result revealed that compound 13 displayed superior antipromastigote activity (IC50 = 0.018) that was 174- and 2.6-fold more active than the standard drugs miltefosine (IC50 = 3.130) and amphotericin B deoxycholate (IC50 = 0.047). The molecular docking study conducted on Lm-PTR1, complexed with Trimethoprim was acquired from the Protein Data Bank (PDB ID:2bfm), justified the better antileishmanial activity of compound 13. Furthermore, the target compounds 14 and 15 elicited better inhibition effects against Plasmodium berghei with 70.2% and 90.4% suppression, respectively. In conclusion, the hydrazine-coupled pyrazole derivatives may be considered potential pharmacophores for the preparation of safe and effective antileishmanial and antimalarial agents.
Similar content being viewed by others
Introduction
Leishmaniasis and malaria are communicable, devastating, and neglected tropical diseases (NTDs) affecting more than 500 million people worldwide. The causative agents of leishmaniasis (Leishmania strains) and malaria (Plasmodium strains) can be transmitted through the bite of sandflies and mosquitoes, respectively [1, 2]. Globally, nearly 350 million people are at risk of leishmaniasis. The overall prevalence of leishmaniasis in the world is 12 million [3]. For instance, visceral leishmaniasis (VL) was implicated in approximately 6% (1.4 million) of all disability-adjusted life-years (DALYs) caused by NTDs in 2015 [2], echoing the economic implication of leishmaniasis in low- and middle-income countries. In addition, nearly half of the global population is at risk of contracting malaria infection by the WHO in 2021. Moreover, 241 million cases and 627,000 deaths were occurred in 2020 slightly higher compared to 2019 [4]. Despite the global healthcare importance of leishmaniasis and malaria, there are only a few drugs often deployed to treat them in the clinical setting. Ironically, multiple reports revealed that the efficacy of the existing antileishmanial and antimalarial drugs is often conceded due to suboptimal treatment outcomes and the advent of drug-resistant Plasmodium falciparum [5,6,7,8,9,10,11] which is responsible for most of the mortality and morbidity associated with malaria [12,13,14]. Thus, the discovery of new antileishmanial and antimalarial agents with desirable therapeutic efficacy and tolerable side effects is highly demanded to treat infections caused by leishmaniasis and malaria [15,16,17].
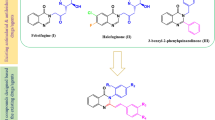
Pyrazoles are a class of bioactive compounds with diverse pharmacological effects such as anticancer [18, 19], antiviral [20, 21], antifungal [22], antibacterial [23], anti-inflammatory [24], antioxidant activities [25]. Moreover, hydrazine-coupled pyrazole derivatives are shown to have promising antimalarial and antileishmanial activities [26,27,28]. In this regard, Bekhit and coworkers [29] successfully synthesized different hydrazine-coupled pyrazole derivatives (I-VI) with potent antimalarial activities having high percent suppression (94.19 to 97.67%) against mice infected with Plasmodium berghei at a dose of 48.4 µM/kg per day. In addition, the compounds (Fig. 1) displayed desirable IC50 values (0.0364 to 0.0418 µM) against chloroquine-resistant RKL9 strains compared to chloroquine phosphate (IC50 = 0.1920 µM). Furthermore, they also exhibited superior antileishmanial activities with IC50 ranging from 0.0241 to 0.0341 µg/mL which was far better than the conventional drugs miltefosine (IC50 = 3.1921 µg/mL) and amphotericin B deoxycholate (IC50 = 0.0472 µg/mL). Interestingly, all the synthesized pyrazoles were safe and well tolerated by mice when treated with 300 mg/kg and 100 mg/kg through oral and parenterally administration, respectively. Inspired by the appealing pharmacological profiles of the aforementioned compounds, this study aimed to synthesize some hydrazine-coupled pyrazole derivatives by incorporating different moieties with potential effects on the solubility and interactions of target compounds with biological macromolecules. In addition, the antimalarial activities, antileishmanial activities and acute toxicities of the target compounds were assessed.

Design rationale for hydrazine-coupled pyrazole derivatives based on the existing dual-acting antimalarial and antileishmanial agents
Results and discussion
Chemistry
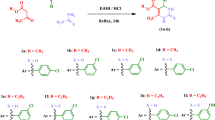
The target hydrazine-coupled pyrazole derivatives (9–15) were synthesized by employing nucleophilic addition–elimination reaction of intermediates (5–8) with different hydrazine derivatives (salicyl hydrazide, hydrazine hydrate, and phenyl hydrazine). In this study, the target compounds were prepared with good yields ranging from 61.64 to 95.5%. The percent yield of the synthesized compounds was comparable with previously synthesized hydrazine-coupled pyrazole derivatives [27]. The structures of the synthesized compounds were verified by employing different techniques such as elemental microanalysis, FTIR, and 1H NMR. For instance, the FTIR spectrum of compound 13 resonated the presence of a characteristic absorption band at 3292 cm−1 attributed to the NH group. It appeared at a relatively higher frequency due to the absence of amidic carbonyl moiety. Two medium bands corresponding to C = N asymmetric and symmetric stretching vibrations appeared at 1615 and 1593 cm−1, respectively. The presence of a band for the NH group and the absence of bands for C = O stretching vibrations around 1678 cm−1 verified the formation of compound 13. Moreover, the 1H NMR spectrum also reiterated the successful synthesis of the target compounds. In this regard, the peaks between δ 7.89–9.97 integrated for three protons were attributed to the overlapped doublet peaks of benzenesulfonamide-C3,5 and singlet peak of pyrazole-C5 hydrogens, respectively. Moreover, the presence of a single peak for the N = CH at δ 8.99, a singlet peak for NH at δ 10.25, and the absence of the singlet peak for the CHO group at δ 10.1 proved the synthesis of compound 13 beyond the reasonable doubt. The chemical structures of the synthesized compounds were verified using spectral and physical measurements. The elemental microanalysis, specific stretching and bending IR vibration frequencies, and the 1H NMR chemical shift data for each of the hydrazine-coupled pyrazoles (9–15) are presented in the experimental section. Overall, the protocols followed in the preparation of target compounds resulted in the formation of colored well-defined crystals characteristic of hydrazine-coupled pyrazole derivatives.
Biological activity results
In vitro antileishmanial activity results
The viability of promastigotes can be estimated by microscopically counting live cells, measuring enzyme activities via the 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay, etc. The frequent use of these methods is hampered due to the lengthy and time-consuming procedures followed in the test. AlamarBlue® has been widely employed in vitro antileishmanial sensitivity assays as it is nontoxic for cells even during long incubation times, water-soluble, and highly stable in complete media. In the living cells, the non-fluorescent resazurin is metabolically reduced to a fluorescent active resorufin thereby changing its color from blue to red. The extent of fluorescence produced by each sample is strongly correlated with the number of living cells. In this study, the antipromastigote assays of the target compounds were carried out in triplicates and the results were presented as IC50 values. The result disclosed that all target compounds except compounds 11 (IC50 = 5.738 µg/mL) and 14 (IC50 = 3.263 µg/mL) had far better antipromastigote activity (Table 1) than miltefosine (IC50 = 3.13 µg/mL). However, all the hydrazine-coupled pyrazoles except compound 13, exhibited lower antipromastigote activity as compared to amphotericin B deoxycholate (IC50 = 0.047 µg/mL). Most of the test compounds lacking the C = O group adjacent to the C = N bond demonstrated superior antileishmanial activities. Exceptionally, compound 13 had the highest antipromastigote activity (IC50 = 0.018 µg/mL) which is about 174- and 2.6-fold more active than miltefosine and amphotericin B deoxycholate, respectively. This might be attributed to the strong H-bonding interactions between NH moiety and the residual amino acids in the active site of the enzyme. To validate this, compound 13 was fitted into the simulated active site of Lm-PTR1 and the result was consistent with the in vitro antileishmanial data. Moreover, compound 9 which contains bromine had the second-highest antipromastigote activity with an IC50 value of 0.084 µg/mL. On the contrary, compound 11 with a methyl substituent at the same position elicited far less antileishmanial activity than compound 9, highlighting the importance of electron-withdrawing substituent at the 4-position of the benzene ring attached to N-1 of the pyrazole ring for improved antipromastigote activity. Overall, the hydrazine-coupled pyrazole derivatives with the aforementioned attributes may serve as a potential scaffold for the preparation of potent antileishmanial agents.
In vivo antimalarial activity results
The four-day suppressive test was implemented to screen the antimalarial activity of the target compounds as it is effective in evaluating early infections and convenient to assess the prodrug effects of test compounds [30]. In this study, the in vivo antimalarial activity of the target compounds was investigated by using mice infected with Plasmodium berghei (achieve steady-state infection) at a dose of 48.4 µmol/kg per day. The results of the antimalarial activity screening of target compounds against the control groups were expressed based on mean parasitemia and percent inhibition of parasitemia (Table 2) which is known as a reliable parameter for in vivo antimalarial screening tests [31]. The treated with compounds 13, 14, and 15 (with no amidic carbonyl group placed at the 4-position of the pyrazole ring) demonstrated percent suppressions greater than 50% but suboptimal as compared to the positive control (which did not experience any death in the study period). While compounds 9, 10 and 11 containing amidic carbonyl group had less than 50% mean percent suppression compared to the positive control groups. Among the tested compounds, bis-[N,N-dimethylaminomethylene-4-(3-(4-methylphenyl)-4-(hydrazonomethylene)-1H-pyrazole-1-yl)benzenesulfonamide] (15) showed a desirable level of mean parasitemia (5.0 ± 1.4) and highest percent suppression (90.4%) against Plasmodium berghei infected mice but the mean survival time of the mice in the group was not appreciably different (7.7 ± 1.4) as compared to the negative control (7.0 ± 1.2). On the other hand, there was an increase in the mean survival time (9.8 ± 1.3) of mice treated with a half dose (24.2 µmol/mL) of compound 15, highlighting the potential dose-dependent toxicity associated with bis-structure due to cleavage of acid labile C = N bond and subsequent increase in concentration in the systemic circulation. 4-[3-(4-methylphenyl)-4-hydrazonomethylene-1H-pyrazol-1-yl]benzenesulfonamide (14) displayed the next better antimalarial activity (70.2%) that is complemented by the relatively lower mean parasitemia level (15.5 ± 4.1) and longer mean survival time of 9.0 ± 2.0 days. This could be attributed to the presence of a primary amine group adjacent to the C = N bond that can increase the strength of binding interaction with the active site of the receptor by hydrogen bonding. Moreover, the hydrophobic interaction imparted by the methylated phenyl group with the amino acid residues in the enzyme pocket may be beneficial for the observed antimalarial activity of compound 14. The compounds containing C = O bond (9–12) near C = N had relatively lower antimalarial activity due to the electron-withdrawing effect and subsequent decrease in hydrogen bonding interactions with the different functional groups reside in the enzyme pocket. In general, some hydrazine-coupled pyrazoles which possessed substituents or moieties that favor a desirable binding interaction with the enzyme active site demonstrated promising antimalarial activity and deserve further optimization and investigations.
Molecular docking study
A molecular simulation study was performed to justify the potent in vitro antipromastigote activity of compound 13, which has desirable fitting pattern in the LmPTR1 pocket (active site) characterized by lower binding free energy (− 9.8 kcal/mol). Inspection of the fitting pattern of compound 13 (Fig. 3) illustrated H-bonding interactions with both Arg287 and Tyr283 through its diethylaminomethylsulphonamide fragment. Additionally, strong hydrophobic interactions with His241 of the catalytic residue were observed. Furthermore, the 1-phenyl-3-tolylpyrazole scaffold of compound 13 substantially anchored within the side chains of Tyr283, Phe113, Gln186, Leu188, Leu226, and Val230 in the Lm-PTR1 pocket. The subsequent hydrophobic interactions were implicated in the desirable in vitro antipromastigote activity of different compounds. The finding was in line with prior reports on Lm-PTR1 [32, 33]. More importantly, the phenylhydrazonomethylene moiety displayed H-bonding interactions with co-factor NADPH and Tyr194 (three H-bonds). It also has strong π-π stacking modes with NADPH and Phe113. The aforementioned desirable interactions of hydrophilic and hydrophobic nature would firmly positioned compound 13 in the Lm-PTR1 pocket resulting in deactivation of PTR1 enzyme. Generally speaking, the in vitro antipromastigote activity and subsequent molecular docking results necessitated additional rigorous experimental protocols to confirm the broad-spectrum antipromastigote activity of compound 13 against various isolates of Leishmania parasite and different animal models.
In vitro cytotoxicity results
The in vitro cytotoxicity of compound 13 was assessed against VERO cells at different concentrations. The result revealed that 117.79 µM of compound 13 was required to suppress the growth and proliferation of 50% of cells (CC50) which is 194-fold higher than the amount required to kill 50% of Leishmania aethiopica isolates (Table 3) reaffirming its selectivity [34]. Moreover, it exhibited marked selectivity and pronounced safety profile compared to the conventional drug miltefosine.
Acute toxicity results
The in vivo acute toxicity study of compound 13 in mice model revealed that it was devoid of any death at all dose (50, 100, 200, and 300 mg/kg) in the study period (14 days). In addition, physical and gross behavioral observations of the mice also suggested no visible symptoms of toxicity even at the highest dose of 300 mg/kg. Moreover, compound 13 was devoid of any inherent dose-dependent toxicity or death after intraperitoneal injection (≤ 140 mg/kg) within 24 h or the entire study period.
Experimental
Materials
Chemicals and reagents
Salicylhydrazide, phenyl hydrazine, 2-(4-bromophenyl)-1-(1-phenylethylidene)-hydrazine, 3-phenyl-1-(4-methylphenyl)-1H-pyrazole-4-carboxaldehyde, N,N-Dimethylaminomethylene-4-[3-(4-methylphenyl)-4-formyl-1H-pyrazol-1-yl]-benzenesulfonamide, HCl, ethyl acetate, hydrazine hydrate, acetonitrile, chloroform, ethanol, absolute methanol, AlamarBlue®, dimethyl sulfoxide (DMSO), acetic acid, phosphorous oxychloride (donated by Drug Discovery Center, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Alexandria University, Egypt), distilled water, iodine, Giemsa stain, Tween 80, 1% acacia gum, Roswell Park Memorial Institute 1640 (RPMI-1640) medium, heat-inactivated fetal calf serum (HIFCS) and penicillin–streptomycin solution were used in the study. The aforementioned chemicals and reagents were analytical grade and used without further purification.
Instruments and apparatuses
Melting points were determined in open capillaries using electro-thermal 9100 melting point apparatus at the Ethiopian Food and Drug Administration, Addis Ababa, Ethiopia, and were uncorrected. Elemental microanalysis was conducted using a Perkin Elmer 2400 elemental analyzer at the Microanalytical Unit, Faculty of Science, Cairo University, Egypt. The FTIR spectra in nujol were recorded with the SHIMADZU 8400SP FT-IR spectrophotometer (Shimadzu Corporation, Nakagyo-Ku, Kyoto, Japan) in the range of 4000 to 500 cm−1 at Ethiopian Pharmaceutical Manufacturing (EPHARM), Addis Ababa, Ethiopia, and nuclear magnetic resonance (NMR) spectral data were performed on Bruker Avance DMX400 FT-NMR spectrometer (Bruker, Billerica, MA, USA) using tetramethylsilane (TMS) as an internal standard in Department of Chemistry, Faculty of Science, AAU, Ethiopia. BIO-PLUS microscope was used to count parasites at the Department of Pharmaceutical Chemistry and Pharmacognosy, School of Pharmacy, AAU, Ethiopia. Enzyme-linked Immunosorbent assay (ELISA) plate was also used to determine the absorbance for samples in the antipromastigote assay at Ethiopian Health and Nutrition Research Institute, Addis Ababa, Ethiopia.
Experimental animals and strains
Swiss albino mice of either sex weighing 24–30 g and 6-week-old donated by the Ethiopian Health and Nutrition Institute, Addis Ababa, Ethiopia were used in the antimalarial activity and acute toxicity study of the target compounds. The mice were housed in cages, maintained in a standard pelleted diet, and acclimatized to the laboratory conditions (temperature of 23–25 ℃, relative humidity of 60–65, and a light/dark cycle of 12 h) for 7 days before each experiment. The rodent malaria parasite, Plasmodium berghei ANKA strain was obtained from Biomedical Laboratory at the Department of Biology, Faculty of Science, AAU, Ethiopia. The Leishmania aethiopica isolates used in the study were acquired from the Leishmania Diagnostic and Research Laboratory (LDRL), School of Medicine, AAU, Ethiopia.
Culture medium and conditions
A complete culture medium was prepared from RPMI-1640, 10% HIFCS, 1% penicillin–streptomycin, and 1% L-glutamine for the in vitro antipromastigote assay. The Leishmania aethiopica isolate was grown first on Novy-MacNeal-Nicolle (NNN) medium and then in tissue-culture flasks containing RPMI-1640 medium supplemented with 10% HIFCS and 1% 100 IU penicillin/mL-100 µg/mL streptomycin solution at 22 ℃ [35].
Reference drugs
Chloroquine phosphate (Ethiopian Pharmaceutical Manufacturing (EPHARM), Addis Ababa, Ethiopia) was used as a reference drug in the determination of the antimalarial activities of the target compounds. Amphotericin B deoxycholate (Fungizone®, ER Squibb, Middlesex, UK) and miltefosine/hexadecylphosphocholine (AG Scientific, San Diego, CA, USA) were employed as reference drugs in the in vitro antileishmanial activity testing of the synthesized compounds.
Methods
Synthesis of target compounds
Synthesis of the target compounds (9–15) was realized through the condensation of selected pyrazole aldehydes with NH2-bearing nucleophiles such as salicyl hydrazide, phenylhydrazine, and hydrazine hydrate [27] as depicted in Fig. 2. Details for the general reaction conditions and purification techniques employed in the synthesis of the target compounds are summarized below.

Schematic representation of the different reaction routes used in the hydrazine-coupled pyrazole derivatives
General procedure for the synthesis of reaction intermediates (4–8) °
To an ice-cold DMF (6.93 mL, 89.5 mmol), 2.42 g (15.8 mmol) phosphorous oxychloride was added dropwise with continuous stirring for 30 min. The mixture was further stirred for 45 min. Accordingly, the compounds (1–4) (2.25 g, 6.88 mmol) were added to the mixture and heated at 70 ℃ for 2 h. After cooling, each solution was poured into crushed ice (55 g) in water (100 mL). Finally, the corresponding mixture was boiled, cooled and the resulting precipitate was filtered, dried, and recrystallized from methanol to obtain pure products (5–8) [27]. As a representative of the synthesized reaction intermediates, the 1H NMR data of compound 4 is presented as follows.
1-(bromophenyl)-3- phenyl-1H-pyrazole-4-carboxaldehyde (4)
IR (Nujol) (cm−1): 1672 (C = O) and 1598 (C = N). 1H NMR (CDCl3/CCl4) ppm: 7.41–7.45 (t, 1H, phenyl-C4 H), 7.53–7.56 (t, 2H, phenyl-C3,5 H), 7.65 (d, 2H, J = 8.4 Hz, p-bromophenyl-C3,5 H), 7.78–7.82 (m, 4H, p-bromophenyl-C2,6 H & phenyl-C2,6 H), 8.59 (s, 1H, pyrazol-C5 H), and 10.10 (s, 1H, CHO) (Additional file 1: Figure S1). Anal. calcd. for C16H11BrN2O: C, 58.74; H, 3.39; N, 8.56; O, 4.89; Br, 24.42. Found: C, 58.74; H, 3.39; N, 8.56; O, 4.89; Br, 24.42.
General procedure for the synthesis of the target compounds (9–15)
A mixture of pyrazole aldehyde (5–8) and stoichiometric amounts of NH2-containing nucleophiles (salicyl hydrazide, hydrazine hydrate, or phenyl hydrazine) were dissolved in ethanol (20 mL) and subsequently acidified with two drops of HCl. The solution was heated under reflux at 70 ℃ for 4 to 6 h with continuous stirring. After cooling, the resulting white, the light-yellow, or yellow precipitate was filtered, washed with a large volume of ethanol, dried, and recrystallized from either ethanol, acetonitrile, methanol/ethyl acetate (v/v 2:1), or acetonitrile/ chloroform (v/v 3:1) to get well-defined crystals of target compounds (9–15) [27].
Nʹ-[(1-(4-bromophenyl)-3-phenyl-1H-pyrazol-4-yl) methylene] salicylhydrazide (9)
IR (Nujol) (cm−1): 3334 (OH), 3242 (NH), 1642, (C = O), 1611 and 1598 (C = N). 1H NMR (CDCl3/DMSO-d6) ppm: 6.79–6.85 (t, 1H, phenyl-C4 H), 6.90 (d, 1H, J = 8.3 Hz, hydroxyphenyl-C4 H), 7.27–7.49 (m, 4H, phenyl-C3,5 H & C2,6 H), 7.57–7.67 (2d, 2H each, J = 8.5 Hz each, p-bromophenyl-C3,5 H & C2,6 H), 7.85 (d, 3H, J = 7.8 Hz, hydroxyphenyl-C3,5 H & C6 H), 8.56 (s, 1H, pyrazol-C5 H), 8.76 (s, 1H, CH = N), 11.67 (s, 1H, NH), and 12.2 (s, 1H, OH) (Additional file 2: Figure S2). Anal. calcd. for C23H17BrN4O2: C, 59.88; H, 5.39; N, 12.15; O, 6.94; Br, 17.32. Found: C, 59.88; H, 5.39; N, 12.15; O, 6.94; Br, 17.32. Yield: 84.17%.
Nʹ-[(3-(4-chlorophenyl)-1-((4-N,N-dimethylaminomethylenesulfonamido)phenyl)-1H-pyrazol-4-yl) methylene] salicylhydrazide (10)
IR (Nujol) (cm−1): 3296 (NH), 1691 (C = O), 1620, 1593 (C = N), 1339 and 1148 (SO2). 1H NMR (DMSO-d6) ppm: 2.98, 3.15 (2 s, 2CH3, N(CH3)2), 6.98 (m, 2H, hydroxyphenyl-C3,5 H), 7.4- 7.5 (t, 1H, hydroxyphenyl-C4 H), 7.65 (d, 2H, J = 8.4 Hz, p-chlorophenyl-C3,5 H), 7.8–7.9 (2d, 2H, J = 8.4 Hz, p-chlorophenyl-C2,6 H & J = 7.9 Hz, hydroxyphenyl-C6 H), 7.92 (d, 2H, J = 8.7 Hz, benzenesulfonamide-C3,5 H), 8.20 (d, 2H, J = 8.7 Hz, benzenesulfonamide-C2,6 H), 8.28 (s, 1H, pyrazol-C5 H), 8.57 (s, 1H, SO2N = CH), 9.16 (s, 1H, CH = N), 11.83 (s, 1H, NH), and 12.00 (s, 1H, OH) (Additional file 3: Figure S3). Anal. calcd. for C26H23ClN6O4S: C, 55.92; H, 3.94; N, 15.65; O, 11.92; S, 5.97; Cl, 6.60. Found: C, 55.92; H, 3.94; N, 15.65; O, 11.92; S, 5.97; Cl, 6.60. Yield: 66.15%.
Nʹ-[(3-phenyl-1-(4-methylphenyl)-1H-pyrazol-4-yl)methylene] salicylhydrazide (11)
IR (Nujol) (cm−1): 3262 (OH), 3150 (NH), 1634 (C = O), 1615 and 1598 (C = N). 1H NMR (CDCl3/CCl4) ppm: 2.4 (s, 3H, p-tolyl-CH3), 6.80–6.86 (t, 1H, hydroxyphenyl-C4 H), 7.0 (d, 1H, J = 8.3 Hz, phenyl-C4 H), 7.26–7.35 (m, 4H, phenyl-C3,5 H & C2,6 H), 7.53–7.67 (2d, 2H each, J = 8.5 Hz each, p-tolyl-C3,5 H & C2,6 H), 7.83 (d, 3H, J = 7.8, hydroxyphenyl-C3,5 H & C6 H), 8.54 (s, 1H, pyrazol-C5 H), 8.72 (s, 1H, CH = N), 11.61 (s, 1H, NH), and 12.00 (s, 1H, OH) (Additional file 4: Figure S4). Anal. calcd. for C24H20N4O2: C, 72.71; H, 5.08; N, 14.13; O, 8.07. Found: C, 72.71; H, 5.08; N, 14.13; O, 8.07. Yield: 83.03%.
Nʹ-[(1-((4-(N,N-dimethylaminomethylenesulfonamido)-phenyl)-3-(4-methylphenyl)-1H-pyrazol-4-yl) methylene]-salicylhydrazide (12)
IR (Nujol) (cm−1): 3292 (NH), 1635 (C = O), 1622 and 1593 (C = N), 1345 and 1148 (SO2). 1H NMR (DMSO-d6) ppm: 2.4 (s, 3H, p-tolyl-CH3), 2.95,3.19 (2 s, 2CH3, N(CH3)2), 6.9–7.0 (m, 2H, hydroxyphenyl-C3,5 H), 7.37 (d, 2H, J = 7.9 Hz, p-tolyl-C3,5 H), 7.43–7.48 (t, 1H, hydroxyphenyl-C4 H), 7.66 (d, 2H, J = 7.9 Hz, p-tolyl-C2,6 H), 7.86 (d, 1H, J = 7.6 Hz, hydroxyphenyl-C6 H), 7.92 (d, 2H, J = 8.7 Hz, benzenesulfonamide-C3,5 H),8.20 (d, 2H, J = 8.7 Hz, benzenesulfonamide-C2,6 H), 8.28 (s, 1H, pyrazol-C5 H), 8.57 (s, 1H, SO2N = CH), 9.14 (s, 1H, CH = N), 11.85 (s, 1H, NH), and 12.0 (s, 1H, OH) (Additional file 5: Figure S5). Anal. calcd. for C27H26N6O4S: C, 61.12, H, 4.94; N, 15.84; O, 12.06; S, 6.04. Found: C, 60.45, H, 3.71; N, 16.27; O, 12.39; S, 6.03. Yield: 95.5%.
N,N-dimethylaminomethylene-4-[(3-(4-methylphenyl)-4-(phenylhydrazonomethylene))-1H-pyrazol-1-yl]benzenesulfonamide (13)
IR (Nujol) (cm−1): 3292 (NH), 1615 and 1593 (C = N), 1340 and 1148 (SO2). 1H NMR (DMSO-d6) ppm: 2.40 (s, 3H, p-tolyl-CH3), 2.95,3.19 (2 s, 2CH3, N(CH3)2), 6.76 (t, 1H, phenyl-C4 H), 7.03 (d, 2H, J = 7.8 Hz, phenyl-C3,5 H), 7.21 (d, 2H, J = 7.8 Hz, phenyl-C2,6 H), 7.36 (d, 2H, J = 8.0 Hz, p-tolyl-C3,5 H), 7.65 (d, 2H, J = 8.0 Hz, p-tolyl-C2,6 H), 7.89–7.97 (d, 2H, J = 8.8 Hz, benzenesulfonamide-C3,5 H & pyrazol-C5 H), 8.15 (d, 2H, J = 8.8 Hz, benzenesulfonamid-C2,6 H), 8.27 (s, 1H, SO2N = CH), 8.99 (s, 1H, CH = N), and 10.25 (s, 1H, NH) (Additional file 6: Figure S6). Anal. calcd. for C26H26N6O2S: C, 64.18; H, 5.38; N, 17.27; O, 6.58; S, 6.59. Found: C, 64.18; H, 4.68; N, 17.27; O, 6.59; S, 6.59. Yield: 61.64%.
4-[3-(4-methylphenyl)-4-hydrazonomethylene-1H-pyrazol-1-yl] benzenesulfonamide (14)
IR (Nujol, cm−1): 3350 and 3234 (NH2), 1625 and 1596 (C = N), 1340 and 1154 (SO2). 1H NMR (DMSO-d6): 2.4 (s, 3H, p-tolyl CH3), 7.38 (d, 2H, J = 8.0 Hz, p-tolyl C3,5 H), 7.49 (s, 2H, CH = N-NH2), 7.65 (d, 2H, J = 8.0 Hz, p-tolyl C2,6 H), 7.97, 8.23 (2d, 4H, J = 8.7 Hz, benzenesulfonamide-C3,5 H & C2,6 H), 8.69 (s, 1H, pyrazole-C5 H), and 9.27 (s, 1H, CH = N) (Additional file 7: Figure S7). Anal. calcd. for C20H22N6O2S: C, 58.52; H, 5.40; N, 20.47; O, 7.79; S, 7.81. Found: C, 57.45; H, 4.82; N, 19.7; O, 9.0; S, 9.02. Yield: 75.8%.
Bis-[N,N-dimethylaminomethylene-4-(3-(4-methylphenyl)-4-(hydrazonomethylene)-1H-pyrazole-1-yl) benzenesulfonamide] (15)
IR (Nujol) (cm−1): 1626 (C = N), 1340 and 1152 (SO2). 1H NMR (DMSO-d6): 2.4 (s, 3H, p-tolyl CH3), 2.95, 3.2 (2 s, 2CH3, N(CH3)2), 7.36, 7.64 (2d, 2H each, J = 8.0 Hz each, p-tolyl C3,5 H & C2,6 H), 7.92, 8.17 (2d, 2H each, J = 8.7 Hz, benzenesulfonamide-C3,5 H & C2,6 H), 8.26 (s, 1H, pyrazol-C5 H), 8.67 (s, 1H, SO2N = CH), and 9.23 (s, 1H, CH = N) (Additional file 8: Figure S8). Anal. calcd. for C40H40N10O4S2: C, 60.90, H, 5.12; N, 17.75; O, 8.11; S, 8.13. Found: C, 60.90; H, 5.11; N, 17.75; O, 8.11; S, 8.13. Yield: 67.63%.
Preparation of stock and working solutions
The entire test compound was dissolved in DMSO to a final concentration of 1 mg/mL. The final DMSO concentration was adjusted not to exceed v/v 1% to avoid its inhibitory effect against parasite proliferation or change in morphology. Six different concentrations of test compounds, ranging from 10 µg/mL to 0.04 µg/mL, were prepared by three-fold serial dilutions. Amphotericin B deoxycholate (with a concentration ranging from 0.5 µg/mL to 0.002 µg/mL) and miltefosine (with a concentration ranging from 40 µg/mL to 0.16 µg/mL) were used as a positive control and DMSO (v/v 1%) in complete media was used as a negative control to compare the antileishmanial activities of test compounds [36]. All the prepared drugs were stored at − 20 ℃ until used in the experiment.
In vivo antimalarial activity test
The in vivo antimalarial activity of the synthesized compounds was determined using the four-day standard suppressive test [37] using Swiss albino mice donated by the Ethiopian Health and Nutrition Institute, Addis Ababa, Ethiopia. Briefly, Swiss albino mice were infected with Plasmodium berghei ANKA strain (0.2 mL of 2 × 107 parasitized erythrocytes) through intraperitoneal injection. After 2 h of infection, the mice were weighed and randomly divided into nine groups of five mice per cage. Groups 1–7 received each test compound (48.4 µg/mL) dissolved in a vehicle containing 7% Tween 80, 3% ethanol, and water. Group 8 was treated with chloroquine phosphate (25 mg/kg) suspended in 7% Tween 80, 3% ethanol, and water which served as a positive control. Group 9 received 2 mL/100 g of the negative control (vehicle 7% Tween 80, 3% ethanol, and water). On day four (96 h post-infection), a blood smear was taken from the tail of each mouse, fixed with absolute ethanol, and stained with Giemsa stain. Then, the level of parasitemia was determined microscopically by counting four fields of approximately 100 erythrocytes per field. Finally, the antimalarial effects of control groups and test compounds were expressed in terms of blood parasitemia, percent suppression and mean survival time of mice [38].
In vitro antileishmanial activity test
The antileishmanial activity of the test compounds was evaluated in vitro against isolates of Leishmania aethiopica promastigotes. 100 µL of the parasite suspension containing 3 × 106 promastigotes per milliliter was added to 96-well plates. The six different concentrations of test compounds and reference drugs were then added in triplicate. DMSO in complete RPMI media was used as a negative control to compare the antileishmanial activities of the test compounds. After 24 h incubation, 20 µL of fluorochrome AlamarBlue® (12.5 mg resazurin dissolved in 100 mL of distilled water) was added to each well. Then, the absorbance of each well was measured after 4 h of incubation at wavelengths of 492 and 630 nm using an ELISA reader for the quantitative determination of viable cells. Finally, the IC50 values were computed from sigmoidal dose–response curves using GraphPad Prism 5.0 software (GraphPad Software, Inc., San Diego, CA, USA) [35].
Molecular docking study
AutoDock Vina was used to perform the molecular simulation study [36] for compound 13 which elicited pronounced antileishmanial activity against Leishmania aethiopica promastigotes. The 3D structure of Lm-PTR1 complexed with Trimethoprim was obtained from the Protein Data Bank (PDB ID:2bfm), forming by chain A of the Lm-PTR1 heterodimer employed in the modeling study. Compound 13 was sketched in a manner that minimize energy and the protein was formulated via the Discovery studio suite (V5.1). The Python script (prepare receptor4.py) provided by the MGLTools package (version 1.5.4) was captured to convert protein files to PDBQT format for docking using AutoDock Vina (version 1.1.2). The efficiency of the search algorithm pertained to its default setting. The grid box docking dimensions were − 5.589 Å × 41.846 Å × 68.229 Å, with a spacing of 1 Å to deal with all the possible conformations of the docked molecule. Pymol was used to generate the graphical representations depicted in Fig. 3.

The docking pose of compound 13 as yellow sticks in the binding site of Lm-PTR1(PDB code:2bfm). NDP-0 is referred to as NADPH co-factor
In vitro cytotoxicity test
The cytotoxicity of compound 13 with promising antileishmanial activity was investigated in vitro against African green monkey kidney cells (VERO cells) [39]. In a 96-well plate, VERO cells (1 × 105 cells per well) were incubated in triplicates with different concentrations of compound 13 (0–100 µM) for 72 h at 37 ℃ incubator with 95% humidity and 5% CO2 and the cell viability was determined by measuring the optical density (OD) of formazan. The CC50 value (the concentration of compound 13 required to kill 50% of fibroblast cells) was and compared with the standard drug, miltefosine. The CC50 and IC50 values were computed using the formula: Growth inhibition (%) = \((ODcontrol-ODtest/ODcontrol) \times 100\). In addition, the selectivity indices (SI) were computed using the formula SI = CC50/IC50. IC50 refers to the concentration of compound 13 required to kill 50% of Leishmania aethiopica).
In vivo acute toxicity test
The oral acute toxicity of compound 13 with promising antileishmanial activity was investigated using male Swiss albino mice (~ 20 g each). The mice were weighed and divided into five groups of six mice per cage. After fasting them overnight, groups 1–4 received the test compound suspended in 1% acacia gum was administered orally at doses of 50, 100, 200, and 300 mg/kg [37]. Group 5 (served as a control) was treated with the solvent (1% acacia gum) at a maximum dose of 1 mL/100 gm of body weight [40]. In a separate experiment, compound 13 was administered with intraperitoneal injection at a dose of 40, 80, 120, and 140 mg/kg [41]. Then, each mouse was observed for gross acute toxicity signs like sedation, lacrimation, hair erection, blinking, sleep, coma, death, etc. Follow-up continued for 14 days with special attention for the first 24 h.
Ethical consideration
The protocols that involved experimental animals were assessed and approved by the Institutional Ethics Review Committee, School of Pharmacy, Addis Ababa University. The mice used in the study were donated by Ethiopian Health and Nutrition Institute (EHNI), Addis Ababa, Ethiopia and informed consent was obtained prior to use. Mouse work, such as injection with parasites or extracts, and euthanasia was implemented under general inhalation anesthesia induced with isoflurane (2%) to minimize animal suffering. Mice were euthanized by cervical dislocation at 30 days after parasite infection. In addition, the study finding was reported following the Animal Research Reporting of in vivo Experiments (ARRIVE) guidelines [42] and animals were handled according to the Guide for the Care and Use of Laboratory Animals (https://olaw.nih.gov/sites/default/files/Guide-for-the-Care-and-Use-of-Laboratory-Animals.pdf).
Statistical analysis
The % suppression and % parasitemia in the antimalarial activity testing were computed using the formulas given below and the resulting data along with the mean survival time was analyzed using Microsoft office excel 2007. The variation in the values of suppressive test was assessed by using one-way analysis of variance (ANOVA) and p < 0.05 is considered as stastically significant.
\(\mathrm{Parasitemia }(\mathrm{\%})=\frac{\mathrm{Number \,of \,infected \,red \,blood \,cells}}{\mathrm{Number \,of \,total \,red \,blood \,cells}}\times 100\)
\(\mathrm{Suppression }(\mathrm{\%})=\frac{\mathrm{Parasitemia \,in \,the \,untreated \,group }-\mathrm{Parasitemia \,in \,the \,treated \,group}}{\mathrm{Parasitemia \,in \,untreated \,group}}\times 100\)
Conclusion
In this study, some hydrazine-coupled pyrazole derivatives were synthesized and screened for their potential antimalarial and antileishmanial activity. Compound 13 displayed promising antileishmanial activity against Leishmania aethiopica isolate which is 174- and 2.6-fold more active than miltefosine and amphotericin B deoxycholate, respectively. The docking study also supported the desirable in vitro antileishmanial activity of compound 13. Moreover, compounds 14 and 15 displayed promising suppression of parasitemia in mice infected with the Plasmodium berghei ANKA strain. In general, the hydrazine-coupled pyrazole derivatives demonstrated desirable dual antileishmanial and antimalarial effects, suggesting the importance of the target compounds as potential pharmacophores in the development of safe and efficacious antileishmanial and antimalarial agents.
Availability of data and materials
The datasets that are not included in this article can be shared with the corresponding author upon request.
References
Asmare G. Willingness to accept malaria vaccine among caregivers of under-5 children in Southwest Ethiopia: a community based cross-sectional study. Malaria J. 2022;21(1):146.
Akao Y, Canan S, Cao Y, Condroski K, Engkvist O, Itono S, Kaki R, Kimura C, Kogej T, Nagaoka K, et al. Collaborative virtual screening to elaborate an imidazo[1,2-a]pyridine hit series for visceral leishmaniasis. RSC Med Chem. 2021;12(3):384–93.
Prinsloo IF, Zuma NH, Aucamp J, N’Da DD. Synthesis and in vitro antileishmanial efficacy of novel quinazolinone derivatives. Chem Biol Drug Design. 2021;97(2):383–98.
Chan AHY, Fathoni I, Ho TCS, Saliba KJ, Leeper FJ. Thiamine analogues as inhibitors of pyruvate dehydrogenase and discovery of a thiamine analogue with non-thiamine related antiplasmodial activity. RSC Med Chem. 2022;13:817–21.
Srivastava S, Mishra J, Gupta AK, Singh A, Shankar P, Singh S. Laboratory confirmed miltefosine resistant cases of visceral leishmaniasis from India. Parasit Vectors. 2017;10(1):49.
Mondelaers A, Sanchez-Cañete MP, Hendrickx S, Eberhardt E, Garcia-Hernandez R, Lachaud L, Cotton J, Sanders M, Cuypers B, Imamura H, et al. Genomic and molecular characterization of Miltefosine resistance in Leishmania infantum strains with either natural or acquired resistance through experimental selection of intracellular amastigotes. PLoS ONE. 2016;11(4):e0154101.
Ponte-Sucre A, Gamarro F, Dujardin J-C, Barrett MP, López-Vélez R, García-Hernández R, Pountain AW, Mwenechanya R, Papadopoulou B. Drug resistance and treatment failure in leishmaniasis: a 21st century challenge. PLoS Negl Trop Dis. 2017;11(12):e0006052.
Faral-Tello P, Greif G, Satragno D, Basmadjián Y, Robello C. Leishmania infantum isolates exhibit high infectivity and reduced susceptibility to amphotericin B. RSC Med Chem. 2020;11(8):913–8.
Ikeda M, Kaneko M, Tachibana S-I, Balikagala B, Sakurai-Yatsushiro M, Yatsushiro S, Takahashi N, Yamauchi M, Sekihara M, Hashimoto M. Artemisinin-resistant Plasmodium falciparum with high survival rates, Uganda, 2014–2016. Emerg Infect Dis. 2018;24(4):718.
Uwimana A, Umulisa N, Venkatesan M, Svigel SS, Zhou Z, Munyaneza T, Habimana RM, Rucogoza A, Moriarty LF, Sandford R. Association of Plasmodium falciparum kelch13 R561H genotypes with delayed parasite clearance in Rwanda: an open-label, single-arm, multicentre, therapeutic efficacy study. Lancet Infect Dis. 2021;21(8):1120–8.
Rudrapal M, Chetia D, Prakash A. Synthesis, antimalarial-, and antibacterial activity evaluation of some new 4-aminoquinoline derivatives. Med Chem Res. 2013;22:3703–11.
Rudrapal M, Chetia D, Bhattacharya S. Development of phytomedicines as novel antimalarial lead molecules: progress towards successful antimalarial drug discovery. In: Rudrapal M, editor. Drug repurposing-advances, scopes and opportunities in drug discovery. London: IntechOpen; 2022. https://doi.org/10.5772/intechopen.108729
Rudrapal M, Washmin Banu Z, Chetia D. Newer series of trioxane derivatives as potent antimalarial agents. Med Chem Res. 2018;27(2):653–68.
Rudrapal M, Chetia D, Singh V. Novel series of 1, 2, 4-trioxane derivatives as antimalarial agents. J Enz Inhib Med Chem. 2017;32(1):1159–73.
Rudrapal M, Chetia D. Plant flavonoids as potential source of future antimalarial leads. Sys Rev Pharm. 2017;8(1):13–8.
Sharma D, Chetia D, Rudrapal M. Design, synthesis and antimalarial activity of some new 2-Hydroxy-1,4-naphthoquinone-4-hydroxyaniline hybrid mannich bases. Asian J Chem. 2015;28(4):782–8.
Kalita J, Chetia D, Rudrapal M. Design, synthesis, antimalarial activity and docking study of 7-chloro-4-(2-(substituted benzylidene) hydrazineyl) quinolines. Med Chem. 2020;16(7):928–37.
Nitulescu GM, Matei L, Aldea IM, Draghici C, Olaru OT, Bleotu C. Ultrasound-assisted synthesis and anticancer evaluation of new pyrazole derivatives as cell cycle inhibitors. Arab J Chem. 2019;12(6):816–24.
Farag PS, AboulMagd AM, Hemdan MM, Hassaballah AI. Annulated pyrazole derivatives as a novel class of urokinase (uPA) inhibitors: green synthesis, anticancer activity, DNA-damage evaluation, and molecular modelling study. Bioorg Chem. 2023;130:106231.
Tantawy AS, Nasr MN, El-Sayed MA, Tawfik SS. Synthesis and antiviral activity of new 3-methyl-1, 5-diphenyl-1 H-pyrazole derivatives. Med Chem Res. 2012;21:4139–49.
Morsy AR, Ramadan SK, Elsafty MM. Synthesis and antiviral activity of some pyrrolonyl substituted heterocycles as additives to enhance inactivated Newcastle disease vaccine. Med Chem Res. 2020;29:979–88.
Ahmed W, Yan X, Hu D, Adnan M, Tang R-Y, Cui Z-N. Synthesis and fungicidal activity of novel pyrazole derivatives containing 5-Phenyl-2-Furan. Bioorg Med Chem. 2019;27(19):115048.
Patil SV, Suryavanshi MB, Nagargoje DR, Kokate SV. Synthesis and antimicrobial evaluation of some new pyrazole derivatives containing thiazole scaffolds. Chem Proc. 2021;8(1):46.
Yan R, Huang X, Deng X, Song M. Synthesis and activity evaluation of some pyrazole-pyrazoline derivatives as dual anti-inflammatory and antimicrobial agents. Polycycl Aromat Compd. 2022;42(8):5006–19.
Dhonnar SL, Jagdale BS, Adole VA, Sadgir NV. PEG-mediated synthesis, antibacterial, antifungal and antioxidant studies of some new 1,3,5-trisubstituted 2-pyrazolines. Mol Divers. 2022. https://doi.org/10.1007/s11030-022-10562-x.
Ibezim A, Ofokansi MN, Ndukwe X, Chiama CS, Obi BC, Isiogugu ON, Ikechukwu PE, Onwuka AM, Ihim SA, Asegbeloyin JN. Evaluation of anti-malarial potency of new pyrazole-hydrazine coupled to Schiff base derivatives. Malaria J. 2022;21(1):1–9.
Bekhit AA, Hymete A, Asfaw H, Bekhit AEDA. Synthesis and biological evaluation of some pyrazole derivatives as anti-malarial agents. Arch Pharm. 2012;345(2):147–54.
Bekhit AA, Nasralla SN, Bekhit SA. Bekhit AE-DA: novel dual acting antimalarial antileishmanial agents derived from pyrazole moiety. Biointerface Res Appl Chem. 2022;12:6225–33.
Bekhit AA, Hassan AM. Abd El Razik HA, El-Miligy MM, El-Agroudy EJ, Bekhit AE-DA: New heterocyclic hybrids of pyrazole and its bioisosteres: design, synthesis and biological evaluation as dual acting antimalarial-antileishmanial agents. Eur J Med Chem. 2015;94:30–44.
Waako P, Gumede B, Smith P, Folb P. The in vitro and in vivo antimalarial activity of Cardiospermum halicacabum L. and Momordica foetida Schumch. Et Thonn. J Ethnopharmacol. 2005;99(1):137–43.
Trigg PI, Kondrachine AV. The current global malaria situation. In: Malaria: parasite biology, pathogenesis and protection. Washington, DC: ASM Press; 1998; 11–22.
Eldehna WM, Almahli H, Ibrahim TM, Fares M, Al-Warhi T, Boeckler FM, Bekhit AA, Abdel-Aziz HA. Synthesis, in vitro biological evaluation and in silico studies of certain arylnicotinic acids conjugated with aryl (thio) semicarbazides as a novel class of anti-leishmanial agents. Eur J Med Chem. 2019;179:335–46.
Bekhit AA, Saudi MN, Hassan AMM, Fahmy SM, Ibrahim TM, Ghareeb D, El-Seidy AM, Nasralla SN. Bekhit AE-DA: Synthesis, in silico experiments and biological evaluation of 1,3,4-trisubstituted pyrazole derivatives as antimalarial agents. Eur J Med Chem. 2019;163:353–66.
Mohamed MAA, Kadry AM, Bekhit SA, Abourehab MAS, Amagase K, Ibrahim TM, El-Saghier AMM, Bekhit AA. Spiro heterocycles bearing piperidine moiety as potential scaffold for antileishmanial activity: synthesis, biological evaluation, and in silico studies. J Enz Inhib Med Chem. 2023;38(1):330–42.
Birhan YS, Bekhit AA, Hymete A. Synthesis and antileishmanial evaluation of some 2,3-disubstituted-4(3H)-quinazolinone derivatives. Org Med Chem Lett. 2014;4(1):10.
Seifu GW, Birhan YS, Beshay BY, Hymete A, Bekhit AA. Synthesis, antimalarial, antileishmanial evaluation, and molecular docking study of some 3-aryl-2-styryl substituted-4(3H)-quinazolinone derivatives. BMC Chem. 2022;16(1):107.
Birhan YS, Bekhit AA, Hymete A. In vivo antimalarial evaluation of some 2,3-disubstituted-4(3H)-quinazolinone derivatives. BMC Res Notes. 2015;8(1):589.
Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193(4254):673–5.
Tonelli M, Gabriele E, Piazza F, Basilico N, Parapini S, Tasso B, Loddo R, Sparatore F, Sparatore A. Benzimidazole derivatives endowed with potent antileishmanial activity. J Enz Inhib Med Chem. 2018;33(1):210–26.
Lorke D. A new approach to practical acute toxicity testing. Arch Toxicol. 1983;54(4):275–87.
Adnan Ahmed B, Tizita H, Ariaya H. Evaluation of some 1H-pyrazole derivatives as a dual acting antimalarial and anti-leishmanial agents. Pak J Pharm Sci. 2014;27(6):1767–73.
Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, et al. The ARRIVE guidelines 2.0 Updated guidelines for reporting animal research. PLoS Biol. 2020;18(7):e3000410.
Acknowledgements
We appreciate Prof. Wondimagegn Mammo for running the NMR data. We would like to recognize the Alexandria University, Department of Pharmaceutical Chemistry for the elemental microanalysis and providing some chemicals. AAU is also acknowledged for financially supporting this research work.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
AAB and AH designed the study. HGB and YSB prepared the hydrazine-coupled pyrazoles, carried out the bioactivity testing, and wrote the manuscript. BYB performed the molecular simulation study and wrote the manuscript. HJH performed the toxicity study. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The protocols that involved experimental animals were assessed and approved by the Institutional Ethics Review Committee, School of Pharmacy, AAU. The mice used in the study were acquired from Ethiopian Health and Nutrition Institute (EHNI) and informed consent was obtained prior to use. In addition, all the methods were implemented by strictly following relevant guidelines and regulations. Moreover, the study is reported following the Animal Research Reporting of in vivo Experiments (ARRIVE) guidelines (https://arriveguidelines.org/arrive-guidelines/experimental-animals) and animals were handled according to the Guide for the Care and Use of Laboratory Animals (https://olaw.nih.gov/sites/default/files/Guide-for-the-Care-and-Use-of-Laboratory-Animals.pdf).
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
1H NMR spectrum of compound 4 in CDCl3.
Additional file 2: Figure S2.
1H NMR spectrum of compound 9 in CDCl3.
Additional file 3: Figure S3.
1H NMR spectrum of compound 10 in DMSO-d6.
Additional file 4: Figure S4.
1H NMR spectrum of compound 11 in CDCl3/DMSO-d6.
Additional file 5: Figure S5.
1H NMR spectrum of compound 12 in DMSO-d6.
Additional file 6: Figure S6.
1H NMR spectrum of compound 13 in DMSO-d6.
Additional file 7: Figure S7.
1H NMR spectrum of compound 14 in DMSO-d6.
Additional file 8: Figure S8.
1H NMR spectrum of compound 15 in DMSO-d6.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Berhe, H.G., Birhan, Y.S., Beshay, B.Y. et al. Synthesis, antileishmanial, antimalarial evaluation and molecular docking study of some hydrazine-coupled pyrazole derivatives. BMC Chemistry 18, 9 (2024). https://doi.org/10.1186/s13065-023-01111-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-023-01111-0