
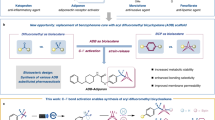
Abstract
We disclose a novel boron trifluoride induced C–H activation and difluoroboronation at room temperature, thus providing a straightforward gateway to a series of N,O-bidentate organic BF2 complexes. The scope of the method is demonstrated with 24 examples. All the synthesized compounds exhibit fluorescence and some of them have large Stokes shifts.
Graphical Abstract

Similar content being viewed by others


Introduction
Small organic fluorescent dyes are popular and have been attracting attention owing to their remarkable optical properties. Due to the characteristics associated with high fluorescence intensity and quantum yields, sharp absorption and fluorescence emission spectra, high photo- and chemical stability, organic difluoroboron (BF2) complexes have played increasingly important roles in many fields involving biological fluorophores, fluorescent indicators, photosensitizers, light-emitting materials, photodynamic therapy, laser dye and solar cells as well. Fluorescent materials with large Stokes shift play an important role in biological field [1,2,3,4,5,6,7,8]. Besides, fluorescence properties of organic difluoroboron complexes can be managed by changing the structure of the organic ligand. Since these advantages make organic difluoroboron complexes becomes a research hotspot.
At present, these organic difluoroboron complexes are classified into three categories: N,N-bidentate (Fig. 1a), O,O-bidentate (Fig. 1b) and N,O-bidentate (Fig. 1c) complexes [9]. After years of research, the synthetic strategies.for access to N,N-bidentate and O,O-bidentate organic difluoroboron complexes are gradually mature. Boradipyrromethene (BODIPY) (Fig. 1a), a typical fluorescent dye of N,N-bidentate complexes, as well as its derivatives are continuously explored [10,11,12,13,14,15,16,17,18,19,20,21]. Meanwhile, extensive research has also been carried on difluoroboron β-diketonate (Fig. 1b) [22,23,24]. However, N,O-bidentate organic BF2 complexes are seldom investigated [25,26,27,28], especially for the synthesis of such compounds.

Three types of organic difluoroboron BF2 complexes
In recent year, a new class of N,O-bidentate organic BF2 complexes are prepared by 2-phenylpyridine derivatives (Scheme 1a) [28]. The reaction involves a Cu(OAc)2 catalyzed bimetallic system for the efficient C-H activation of 2-phenylpyridines, but restricted substrates limit the structural diversity of N,O-bidentate organic BF2 complexes. Besides, other published approaches to build the N,O-bidentate organic BF2 motifs suffer from their disadvantages. For example, substrates of the reaction in Scheme 1b are limited to a few of non-commercial available compounds endowed with specific structures [29]. Another adverse factor is that noble metal catalysts are required for the complete transformation of substrates (Scheme 1c) [30]. Moreover, characterized by tedious experimental operations and high energy consumption, multi-step methods usually have their limitations (Scheme 1d) [31]. In addition, harsh conditions and low yield are drawbacks for further industrial production and commercial application (Scheme 1e) [26]. From the perspective of foundation and application, more effective and convenient strategies are required for the development of the N,O-bidentate organic BF2 complexes synthetic chemistry.

Different strategies to synthesize N,O-bidentate organic BF2 complexes
In this paper, we report the synthesis and fluorescence properties of a novel pyrazineboron complex. Boron trifluoride and potassium t-butoxide induce regioselective C–H activation and difluoroboronation at room temperature. N,O-bidentate complexes are obtained by the reaction and excellent fluorescence properties of the products are shown in further results. In comparison to what described in the literatures, our synthesis method is superior in catalyst-free system, low-cost process and step economy.
Results and discussions
Acetophenone 1a and 2-cyanopyrazine 2 were selected as model substrates to produce compound 4a (Figure of Table 1). A mixture of acetophenone 1a (0.20 mmol), 2-cyanopyrazine 2 (0.30 mmol), potassium t-butoxide (0.60 mmol) and boron trifluoride tetrahydrofuran (0.60 mmol) in THF (2.0 mL) was stirred in nitrogen atmosphere at room temperature for 24 h, at last providing compound 4a in 73% yield based on acetophenone (Table 1, entry 1). The structure of 4a was determined by X-ray crystallography (Scheme 2, 4a). The crystal data of compounds 4a, 4aa, 4ab and 5 are included in additional file 1, 2. Other alkali salts and/or organic base such as K2CO3, KOH and Et3N afforded the product in lower yields or no yield (entries 2–4). Higher temperature was a disadvantage to the reaction (entries 1, 5–7). If the temperature exceeds 100 ℃, no product will be obtained. The effect of solvent was found to be essential for the generation of the product (entries 1, 8–10).

The scope of acetophenone derivatives used for the synthesis of compounds 4a,b. aReaction conditions: 1 (0.20 mmol), 2 (0.30 mmol), KTB (0.60 mmol), THF (2.0 mL), BF3∙THF (0.60 mmol), N2, rt, 24 h. bIsolated yields
With the best conditions in hand, we sought to investigate the generality of the reaction (Scheme 2). The yields was discussed in terms of electronic effect and the steric effect of functional groups on the substrates. Firstly, experimental results showed that the electronic effect on the phenyl rings of 1 will affect the production of compound 4. Substitution of the electron-donating groups or weak electron-withdrawing groups at the para-position of the phenyl group would bring about products in yields of 50–70% (4a-4k). Substitution of electron-donating groups at the para-position of the phenyl group had little influence on the reaction yield. The strong electron-donating group -OMe substituted product (4b) could be obtained in good yield. When the para-position of the phenyl group was electron withdrawing group, the situation was just the opposite. Substrates bearing weak electron-withdrawing groups could still react to give products 4h-4k while the yield decreased in succession. However, the reaction was totally suppressed when strong electron-withdrawing groups such as − CF3, − NO2 and group -COOEt were introduced to the substrates. Secondly, the steric effect of substituents on the substrates was studied by employing methoxy- (4b, 4l, 4m) at the para, meta, and ortho positions of the phenyl group respectively. With the shift of the functional groups from the para to ortho positions, there was no obvious change in reaction yield. Moreover, substrates with two substituents at the 3,5 positions of phenyl (4o) or three substituents at the 2,4,6 positions of phenyl (4p) had little effect on the yield of the reaction. Some fused-ring and heterocyclic substrates were also tested, most of them providing corresponding compounds in good yields (45%-58%) (4q-4u). However, the yield of 4t decreased significantly (45%). In addition, the synthetic utility of our reaction was also examined by running the experiments on gram scale. The reactions of acetophenone 1a (9.0 mmol) with 2-cyanopyrazine 2 (13.5 mmol) in our system afforded the product 4a in 46% yield (Additional file 3: Section 2.2).
With the progress of building the N,O-bidentate organic BF2 motifs, several experiments were carried out for the investigation of the reaction mechanism (Scheme 3). In experiment shown in Scheme 3a, compound 5 was isolated instead of compound 3 or 3’ (Scheme 4) without the addition of boron trifluoride, indicating that C-H activation process was relevant to the use of boron trifluoride. When extra five equivalent free radical trapping agent (TEMPO) was added in standard conditions, the reaction proceeded effectively to afford 4a in 66% yield (Scheme 3b), so we thought no free radical process was involved in the reaction pathways.

Control Experiments

Proposed reaction mechanism for the generation of compound 4a
Based on the experimental results presented above, a possible reaction mechanism is proposed (Scheme 4). In the presence of base, acetophenone 1a is deprotonated to form intermediate (a). Reaction of 2-cyanopyrazine 2 with one equivalent boron trifluoride produces the intermediate (b). After nucleophilic attack, intermediate (a) reacts with intermediate (b) to generate intermediate (c). Intermediate (c) loses one molecule of HBF3− by intermolecular electron transfer, then affording intermediate 3. In solution, 3 is transformed into enol 3' via keto-enol tautomerization. Finally, it 3' immediately picks up another molecule of boron trifluoride to give target product 4a in the presence of alkaline.
Compounds with D − π − A structure usually possess excellent luminescence property. Boron heterocycle is highly electron-deficient while -OMe at para position of phenyl in compound 4b is an electron-donating group, which is favorable for the formation of the D − π − A system. The introduction of strong electron-donating group -OMe makes the compound 4b exhibit excellent fluorescence performance. Fluorescence quantum yield of the compound 4b was tested to be 79%. The fluorescent lifetime of it is 4.3 ns. Data of other compounds can be found in Additional file 3: Section 9.
The extension of conjugation system in the fluorescent compound can reduce the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), which leads to the red shift of emission wavelength. With that in mind, structural modification methods are discussed in order to obtain compounds with larger red shift. Fluorescence properties of compounds are usually enhanced by introducing fluorene. On the other hand, thiophene has distinctive electronic transmission capability [32, 33]. Therefore, it is common to introduce fluorene or thiophene ring to extend the conjugation system and improve the optical properties of compounds. Long-chain alkoxy group not only has stronger electron donating ability, but also can increase the solubility of compounds. According to the analysis, we designed and synthesized compounds 4aa, 4ab and 4ac. The structures of 4aa and 4ab were determined by X-ray crystallography (Fig. 2).

Crystal structures of compounds 4aa and 4ab with all non-hydrogen atoms shown as 50% probability ellipsoids
As shown in Fig. 3, the UV–vis and fluorescence spectra of representative N,O-bidentate organic BF2 complexes in dichloromethane were tested. The absorption and emission maxima of these BF2 complexes vary from 433 to 510 nm, and 472 nm to 615 nm respectively. Compounds 4a and 4ac exhibit good fluorescence properties (Table 2). Data of other compounds can be found in Additional file 3: Section 9.

Absorption spectra and emission spectra of 4a, 4aa, 4ab and 4ac in dichloromethane at a concentration of 210–5 mol∙L−1
The extension of conjugation system in the fluorescent compound effectively makes compounds 4aa and 4ab red shifts. The solution-state fluorescence spectra of 4aa and 4ab exhibited larger Stokes shifts. The large Stokes shifts of fluorescent materials has the advantages of low background interference, small light damage to biological samples, strong sample penetrability and high detection sensitivity [34,35,36]. These compounds have potential for biological imaging.
Conclusions
In summary, we have developed the diversity-oriented efficient one-pot synthesis of a series of a novel pyrazineboron complexes. Boron trifluoride and potassium t-butoxide induce C–H activation and difluoroboronation at room temperature. These compounds show excellent photophysical properties, including high fluorescence quantum yields in solution, large Stokes shifts and excellent stability. Further structural modification was carried out to improve the fluorescent properties of the products.
Experimental
A mixture of acetophenone 1a (0.20 mmol), 2-cyanopyrazine 2 (0.30 mmol), potassium t-butoxide (0.60 mmol) and boron trifluoride tetrahydrofuran (0.60 mmol) in THF (2.0 mL) was stirred in nitrogen atmosphere at room temperature for 24 h. After reaction, 10 mL water was added and the reaction mixture was exacted with dichloromethane (3×40 mL). Filtered through a pad of silica gel, and concentrated under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/dichloromethane (10:3 to absolute dichloromethane v/v) to afford the products 4a-4u. 4aa, 4ab and 4ac were obtained by the same method.
Availability of data and materials
The data underlying this study are available in the published article and its online supplementary material. The Supporting Information is available free of charge via the Internet at 2217152, 2217156, 2217158 and 2217159 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 44 1223 336033.
References
Gorman A, Killoran J, O’Shea C, Kenna T, Gallagher WM, O’Shea DF. In vitro demonstration of the heavy-atom effect for photodynamic therapy. J Am Chem Soc. 2004;126:10619–31.
Yogo T, Urano Y, Ishitsuka Y, Maniwa F, Nagano T. Highly efficient and photostable photosensitizer based on BODIPY chromophore. J Am Chem Soc. 2005;127:12162–3.
Oleynik P, Ishihara Y, Cosa G. Design and synthesis of a BODIPY-α-tocopherol adduct for use as an Off/On fluorescent antioxidant indicator. J Am Chem Soc. 2007;129:1842–3.
Lovell JF, Liu TWB, Chen J, Zheng G. Activatable photosensitizers for imaging and therapy. Chem Rev. 2010;110:2839–57.
Ozlem S, Akkaya EU. Thinking outside the silicon box: molecular and logic as an additional layer of selectivity in singlet oxygen generation for photodynamic therapy. J Am Chem Soc. 2009;131:48–9.
Gómez-Durán CFA, García-Moreno I, Costela A, Martin V, Sastre R, Bañuelos J, López Arbeloa F, López Arbeloa I, Peña-Cabrera E. 8-PropargylaminoBODIPY: unprecedented blue-emitting pyrromethene dye. Synthesis, photophysics and laser properties. Chem Commun. 2010;46:5103–5.
Rousseau TCA, Bura T, Ulrich G, Ziessel R, Roncali J. BODIPY derivatives as donor materials for bulk heterojunction solar cells. Chem Commun. 2009;13:1673–5.
Chang MC, Chantzis A, Jacqueminb D, Otten E. Boron difluorides with formazanate ligands: redox-switchable fluorescent dyes with large stokes shifts. Dalton Trans. 2016;45:9477–84.
Mellerupab SK, Wang S. Boron-based stimuli responsive materials. Chem Soc Rev. 2019;48:3537–49.
Patil NG, Basutkar NB, Ambade AV. Visible light-triggereddisruption of micelles of an amphiphilic block copolymer with BODIPY at the junction. Chem Commun. 2015;51:17708–11.
Guo S, Zhang H, Huang L, Guo Z, Xiong G, Zhao J. Porous material-immobilized iodo-Bodipy as efficient photocatalyst for photoredox catalytic organic reaction to prepare pyrrolo[2,1-a]isoquinoline. Chem Commun. 2013;49:8689–91.
Wang S, Lan H, Xiao S, Tan R, Lu Y. Highly fluorescent non-conventional boron difluoride-based π organogel with gelation-assisted piezochromism. Chem Asian J. 2018;12:198–202.
Wang X, Liu Q, Yan H, Liu Z, Yao M, Zhang Q, Gong S, He W. Piezochromic luminescent behaviors of two new benzothiazole-enamido boron difluoride complexes: intra- and inter-molecular effects induced by hydrostatic compression. Chem Commun. 2015;51:7497–500.
Barbon SM, Bender SD, Groom H, Luyt LG, Gilroy JB. Copper-assisted azide–alkyne cycloaddition chemistry as a tool for the production of emissive boron difluoride 3-cyanoformazanates. Org Chem Front. 2017;4:178–90.
Zhou B, Guo M, Pan Q, Zhou M, Xu L, Rao Y, Wang K, Yin B, Zhou J, Song J. Rhodium-catalyzed annulation of pyrrole substituted BODIPYs with alkynes to access π-extended polycyclic heteroaromatic molecules and NIR absorption. Org Chem Front. 2021;8:868–75.
Sun Y, Yuan H, Di L, Zhou Z, Gai L, Xiao X, He W, Lu H. Non-symmetric thieno[3,2-b]thiophene-fused BODIPYs: synthesis, spectroscopic properties and providing a functional strategy for NIR probes. Org Chem Front. 2019;6:3961–8.
Blázquez-Moraleja A, Maierhofer L, Mann E, Prieto-Montero R, Oliden-Sánchez A, Celada L, Martínez-Martínez V, Chiara M, Luis CJ. Acetoxymethyl-BODIPY dyes: a universal platform for the fluorescent labeling of nucleophiles. Org Chem Front. 2022;9:5774–89.
Li H, Lv F, Guo X, Wu Q, Wu H, Tang B, Yu C, Wang H, Jiao L, Hao E. Direct C-H alkoxylation of BODIPY dyes via cation radical accelerated oxidative nucleophilic hydrogen substitution: a new route to building blocks for functionalized BODIPYs. Chem Commun. 2021;57:1647–50.
Hou CL, Yao Y, Wang D, Zhang J, Zhang JL. Orthogonally arranged tripyrrin-BODIPY conjugates with an “edge to plane” mode. Org Chem Front. 2019;6:2266–74.
Di L, Yang J, Tang W, Gai L, Zhou Z, Lu H. Nonsymmetric benzo[a]fused and thiophene/thieno[3,2-b]thiophene[b]fused BODIPYs: synthesis and photophysical properties. J Org Chem. 2021;86:601–8.
Nakano T, Sumida A, Naka K. Synthesis and characterization of boron difluoride complexes bearing π-expanded pyridine ligands as organic fluorochromes. J Org Chem. 2021;86:5690–701.
Wang JX, Yu YS, Niu LY, Zou B, Wang K, Yang QZ. Difluoroboron β-diketonate based thermometer with temperature-dependent emission wavelength. Chem Commun. 2020;56:6269–72.
Sugiura S, Kobayashi Y, Yasuda N, Maeda H. Multiply aryl-substituted dipyrrolyldiketone boron complexes exhibiting anion-responsive emissive properties. Chem Commun. 2019;55:8242–5.
Wang X, Sun Y, Wang G, Li J, Li X, Zhang K. TADF-Type organic afterglow. Angew Chem Int Ed. 2021;60:17138–47.
Balijapalli U, Manickam S, Thirumoorthy K, Sundaramurthy KN, Sathiyanarayanan KI. (Tetrahydrodibenzo[a, i]phenanthridin-5-yl)phenol as a fluorescent probe for the detection of aniline. J Org Chem. 2019;84:11513–23.
Kubota Y, Hara H, Tanaka S, Funabiki K, Matsui M. Synthesis and fluorescence properties of novel pyrazine-boron complexes bearing a β-iminoketone ligand. Org Lett. 2011;13:6544–7.
Tan G, Schrader ML, Daniliuc C, Strieth-Kalthoff F, Glorius F. C-H activation based copper-catalyzed one-shot synthesis of N, O-bidentate organic difluoroboron complexes. Angew Chem Int Ed. 2020;59:21541–5.
Wong CL, Poon CT, Yam VWW. Photochromic dithienylethene-containing boron(III) ketoiminates: modulation of photo-responsive behavior through variation of intramolecular motion. Chem Eur J. 2016;22:12931–40.
Kubota Y, Tsukamoto M, Ohnishi K, Jin J, Funabikia K, Matsui M. Synthesis and fluorescence properties of novel squarylium–boron complexes. Org Chem Front. 2017;4:1522–7.
Dohe J, Koßmann J, Müller TJJ. Diversity-oriented four-component synthesis of solid state luminescent difluoro oxazaborinines. Dyes Pigments. 2018;157:198–217.
Yang D, Liu P, Bai T, Kong J. N, N-Dimethyl-substituted boron ketoiminates for multicolor fluorescent initiators and polymers. Macromolecules. 2020;53:3339–48.
Skotheim TA, Reynolds JR. Handbook of Conducting Polymers. New York: CRC Pr I Llc; 1986.
Salaneck WR. Conjugated Polymer and related materials. Oxford: Oxford University Press; 1993.
Yu C, Fang X, Wu Q, Jiao L, Sun L, Li Z, So P, Wong WY, Hao E. A family of BODIPY-like highly fluorescent and unsymmetrical bis(BF2) pyrrolyl−acylhydrazone chromophores: BOAPY. Org Lett. 2020;22:4588–92.
Yu C, Huang Z, Wang X, Miao W, Wu Q, Wong WY, Hao E, Xiao Y, Jiao L. A family of highly fluorescent and unsymmetric bis(BF2) chromophore containing both pyrrole and N-heteroarene derivatives: BOPPY. Org Lett. 2018;20:4462–4.
Shandev PP, de Jong F, Veys K, Huang J, Santhini PV, Verhaeghe D, Van Meervelt L, Escudero D, Van der Auweraer M, Dehaen W. BOPAHY: a doubly chelated highly fluorescent pyrrole-acyl hydrazone–BF2 chromophore. Chem Commun. 2020;56:5791–4.
Acknowledgements
Not applicable.
Funding
This research was funded by the National Natural Science Foundation of China (Grant No. 21371171) and the Natural Science Foundation of Fujian Province (Grant No. 2020J01114). There is no role of the funding body in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
JG: conceptualization, methodology, data curation and original draft; WF: investigation, validation and formal analysis; ZL: software, interpretation of data; DH: project administration, funding acquisition, resources, supervision, review & editing. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Checkcifs of the compounds 4a, 4aa, 4ab and 5.
Additional file 2.
Cifs of the compounds 4a, 4aa, 4ab and 5.
Additional file 3.
Supporting document showing the 1H NMR, 13C NMR, IR spectra and photophysical data of each compound studied in this paper.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guang, J., Fan, W., Liu, Z. et al. Synthesis of N,O-bidentate organic difluoroboron complexes and their photophysical studies. BMC Chemistry 17, 53 (2023). https://doi.org/10.1186/s13065-023-00974-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-023-00974-7