
Abstract
The 3C-like protease (3CLpro), known as the main protease of SARS-COV, plays a vital role in the viral replication cycle and is a critical target for the development of SARS inhibitor. Comparative sequence analysis has shown that the 3CLpro of two coronaviruses, SARS-CoV-2 and SARS-CoV, show high structural similarity, and several common features are shared among the substrates of 3CLpro in different coronaviruses. The goal of this study is the development of validated QSAR models by CORAL software and Monte Carlo optimization to predict the inhibitory activity of 81 isatin and indole-based compounds against SARS CoV 3CLpro. The models were built using a newer objective function optimization of this software, known as the index of ideality correlation (IIC), which provides favorable results. The entire set of molecules was randomly divided into four sets including: active training, passive training, calibration and validation sets. The optimal descriptors were selected from the hybrid model by combining SMILES and hydrogen suppressed graph (HSG) based on the objective function. According to the model interpretation results, eight synthesized compounds were extracted and introduced from the ChEMBL database as good SARS CoV 3CLpro inhibitor. Also, the activity of the introduced molecules further was supported by docking studies using 3CLpro of both SARS-COV-1 and SARS-COV-2. Based on the results of ADMET and OPE study, compounds CHEMBL4458417 and CHEMBL4565907 both containing an indole scaffold with the positive values of drug-likeness and the highest drug-score can be introduced as selected leads.
Similar content being viewed by others

Introduction
In the end of February 2003, a novel human coronavirus was detected as the causative agent of the first major pandemic of the twenty-first century, severe acute respiratory syndrome (SARS). The first case of "atypical pneumonia" was declared in China and quickly and unexpectedly spread to 29 countries, especially in Asia and North America, alarming the World Health Organization (WHO). Within several months of the outbreak in 2003, the WHO reported that it had caused 916 deaths out of 8422 cases worldwide (10–15% case fatality rate) [1]. In early 2003, a new human coronavirus known as SARS coronavirus (SARS CoV) was recognized as the causative agent of SARS [2].
COVID-19 is the active pandemic which was first reported in late 2019 in Wuhan, China. In February 2020, SARS-COV-2 was announced as the causative agent. As of October 24th 2021, 243 million cases and over 4.9 million deaths have been reported. The 3C-like protease (3CLpro) enzyme or major protease (Mpro), is essential for the process of viral replication and infection, thereby making it an ideal target for antiviral therapy [1]. The coronavirus 3CLpro is a cysteine protease consisting of about 300 amino acids and containing three domains. Domains I (amino acids 8 to 99) and II (amino acids 100 to 183) consist of beta barrels that simulate the chymotrypsin and 3C proteinases. The binding site is located between the mentioned domains, and about 16 residues join domains I and II to residues 200 to 300 as the C-terminal domain III. The proteolytic activity of 3CLpro has been performed by this third five helices domain [3]. The 3CLpro enzymes show a highly conserved structure among known coronavirus species, and several common characteristics are shared among different coronavirus 3CLpro substrates [4]. Comparative sequence analysis has shown that the 3CLpros of the three coronaviruses of SARS-CoV-2, SARS-CoV, and MERS-CoV are very similar in structure and conservatism [5]. These findings indicate that 3CLpro could be used as a homologous target for the development of anti-coronavirus drugs that can inhibit the proliferation of various coronaviruses [4].
Based on various studies, a combination of nucleoside analogues such as ribavirin can be used for the treatment of SARS along with corticosteroids such as methylprednisolone and hydrocortisone [6,7,8,9]. Since the beginning of the COVID-19 pandemic different options for the treatment of this disease have been used including monoclonal antibodies, protease inhibitors, corticosteroids, convalescent plasma and so on. However, the definitive efficacy of these drugs has not been proven.
Previous research has revealed that isatin and its derivatives have a broad range of anti-bacterial and anti-viral activities such as anti-HIV [10, 11], anti-rhinovirus [12] and against mycobacterium tuberculosis [13]. The derivatized isatin scaffold may be a good candidate for the SARS CoV 3CLpro inhibitor because both proteases (human SARS CoV and rhinovirus) are cysteine proteases and are structurally similar in the active site [14].
In 2005, Chen et al. investigate that N-substituted isatin derivatives with anti-rhinovirus activity may also have anti-SARS activity. Therefore, based on these compounds, they synthesized new isatin derivatives and evaluated their inhibition activities against SARS CoV 3CLpro. The IC50 values showed that the mentioned isatin derivatives could inhibit SARS CoV 3CLpro in the low micro molar range (0.95–17.50 µM) [15]. Using the results of the previous study, Zhou et al. designed and synthesized a series of N-substituted 5-carboxamide-isatin compounds and evaluated their activities. They introduced some compounds as SARS CoV 3CLpro inhibitors which the most potent compound showed an IC50 of 0.37 µM [2]. In 2014 Liu et al. in order to improve the inhibitory activity of isatin derivatives against SARS CoV 3CLpro, investigated a replacement of the carboxamide group using a series of substituted sulfonamide groups in isatin. Optimization of 5-sulfonyl isatin derivatives led to the discovery of a new compound with the strongest potency (IC50 = 1.04 µM) [16].
Quantitative structure–activity relationship (QSAR) is one of the critical computational techniques for ligand-based drug design, which can statistically show the correlation between the structural and bioactive properties of compounds [17]. Molecular docking is a computational technique for predicting the optimal interaction of two molecules that creates a binding model, typically a small ligand with a protein receptor [18], most commonly used in drug discovery [19]. CORAL is a new software for developing the reliable and predictive QSAR/QSPR models based on SMILES or quasi-SMILES of materials and Monte Carlo optimization [17, 20].
The main goal of this study is to create the simple and reliable QSAR models by CORAL software to predict the inhibitory activity of 81 isatin and indole-based compounds against SARS CoV 3CLpro. In addition, the effect of using the index of ideality correlation (IIC) as the objective function for modeling in CORAL software has been investigated [21]. Moreover, the results from Monte Carlo optimization-based QSAR modeling with the further addition of molecular docking studies applied for pharmacologically important endpoints. SMILES notation-based optimal descriptors, defined as molecular fragments, identified as main contributors to the increase/decrease of biological activity, which are used further to search compounds from the ChEMBL database with targeted activity based on computer calculation, are presented. Here, molecular docking was applied as an additional method to validate the calculated activity of proposed compounds as novel SARS CoV 3CLpro inhibitors.
Data and methods
Dataset
In this study 81 isatin and indole-based SARS 3CLpro inhibitors were gathered from literature [2, 15, 16, 22,23,24,25]. The number isatin based compounds were 41 and the rest were indole-based compounds. The IC50 (µM) values for inhibitors were converted into their pIC50 (− logIC50). Table 1 shows the structure of the molecules along with their pIC50 (range between 4.08 and 7.77). BIOVIA Draw 2020 was used to draw the molecular structures of the compounds and convert them into SMILES symbols. The dataset divided the active training (≈25%), passive training (≈20%), calibration (≈20%), and validation (≈35%) sets randomly. To construct the QSAR models based on Monte Carlo optimization, four separate random partitions were performed.
Descriptors
There are three categories of optimal descriptors in CORAL software, including SMILES-based, graph-based and a combination of SMILES with molecular graph descriptors as hybrid descriptors. The optimal descriptors used in this research to construct the QSAR model are a combination of hydrogen suppression graph (HSG) and SMILES descriptors. The below equation indicates the optimal type of molecular descriptors for QSAR modeling for pIC50 of isatin and indole-based compounds as SARS 3CLpro inhibitors:
where, Sk, SSk and SSk are one, two and three-character SMILES features, respectively. BOND represents a global SMILES descriptor that demonstrate the presence/absence of various bonds including double ( =), triple (#), and stereochemical (@) bonds. The NOSE indicates the presence/absence of nitrogen, oxygen, sulfur, and phosphorus atoms in the SMILES symbol of molecules. HALO is the presence/absence of halogen in the structure of molecules. HARD is the combination of BOND, NOSP, and HALO in the structure of compounds. Cmax, Nmax, and O max show the maximum number of rings (the range 0–9), the maximum number of nitrogen atoms, and the maximum number of oxygen atoms in the molecular structure, respectively. In addition, C5 and C6 indicate the presence of five- and six-membered rings in the molecular structures, respectively. The CW(x) represents the correlation weight of a SMILES feature or an HSG invariant.
The following equation indicates the correlation between the sum of correlation weights (DCW) of the optimal descriptors and pIC50 of the compounds:
a is the intercept point and b is the slope of the line obtained by the least-squares method. DCW (Descriptors of Correlation Weights) is the sum of correlation weights for the optimal descriptor derived from HSG and SMILES and calculated by Monte Carlo optimization. The T* and N* indicate the optimal threshold value and the number of Monte Carlo optimization cycles, respectively.
A flowchart of a Monte Carlo optimization cycle is presented by Sokolovic et al. [26]. At first cycle, the CW(x) of features is randomly generated and then optimized based on the proposed objective function. There are different objective functions to obtain a reliable QSAR model in CORAL software. TF0, TF1 are two objective functions that we used here to obtain correlation weights for attributes and compare the extracted models based on each of them [27, 28].
The RATRN and RPTRN denote the correlation coefficients between the experimental and predicted pIC50 for the active training and passive training sets, respectively and, c and c’ represent empirical values which are generally constant.
The IICCAL for calibration (CAL) set is obtained according to the following equation:
The RCAL indicates the correlation coefficient for the calibration set. MAECAL (Mean Absolute Error for calibration set) is calculated based on Eqs 6 to 8:
The ‘k’ is the index (1, 2... N) and the experimental k and predicted k are related to the pIC50. The CWs for each attribute of Split 1 is provided as an example in Additional file 1: Table S1, total number of attributes is 383.
QSAR model Validation
There are various criteria for evaluating the predictive ability of QSAR models, such as internal validation, external validation, and Y-scrambling. In this study, some standard statistical criteria were used to check the validity of the QSAR models, such as coefficient of determination (R2), concordance correlation coefficient (CCC), Q2, Q2F1, Q2F2, Q2F3, standard error of estimation (s), mean absolute error (MAE), r2m and new Y-scrambling criteria (\({\text{C}}_{{\text{R}}_{\text{P}}^{2}}\)) [29,30,31,32]. In addition, the IIC of models was used to improve the predictability of the models [33, 34].
Applicability domain
The range of compounds for which a QSAR model can make reliable predictions is defined based on the applicability domain (AD) of model as the Organization of Economic Co-operation and Development (OECD) principle 3. Here, the AD is calculated based on the distribution of SMILES features in the training and calibration sets and is defined as “\({\text{Defect}}_{{\text{A}}_{\text{K}}}\)”[17].
where PTRN(Fk) and PCAL(Fk) represent the probabilities of kth feature (Fk) in the training and calibration set, respectively; NTRN(Fk) and NCAL(Fk) denote the frequency of kth feature (Fk) in the training and calibration set, respectively.
According to the SMILES of molecules, the molecule is included in AD if:
where \({\overline{\text{Defect}} }_{\text{TRN}}\) is the average \({\text{Defect}}_{\text{molecule}}\) in the training set.
The interpretation of QSAR models
CORAL software provides a simple approach to interpret QSAR models. Three categories of features can be extracted with numerical data of correlation weights in several Monte Carlo optimization cycles: (I) features with a positive correlation weight in all runs that increase the endpoint; (II) features with a negative correlation weight in all runs that decrease the endpoint; and also (III) features with both negative and positive correlation weight in different optimization runs, these features have an undefined role and not be classified as an increasing/decreasing promoters of the endpoint [35].
Molecular docking study
Molecular docking method as a common virtual screening technique can help to find the most favorable ligand binding mode in protein for computer-aided drug discovery [36,37,38]. The X-ray crystallographic structures of SARS-COV-2 3CLpro were obtained from the Protein Data Bank (PDB: 6XHO) based on a good experimental resolution (1.45 Å), R-value free (0.239), and R-value work (0.211). The native ligand in active site of this protein was ethyl (4R)-4-({N-[(4-methoxy-1H-indol-2-yl)carbonyl]-L-leucyl}amino)-5-[(3S)-2-oxopyrrolidin-3-yl]pentanoate (Query on V34), thus we use this pdb code for molecular docking of indole derivatives. The selected receptor for molecular docking simulation was the x-ray structure of SARS-COV-1 (PDB ID: 1UK4) based on a good experimental resolution (2.5 Å), R-value free (0.231), and R-value work (0.213). The native ligand in active site of this protein was 5-mer peptide. 6XHO and1 UK4 structures consist of a dimer composed of two identical sequences. The side chain A was chosen for molecular docking and the side chain B was removed. The protein structure was prepared using adding hydrogens removing water molecules and native ligands. Then, the Kollmann charges were assigned to the receptor. All compounds were sketched using the by ChemOffice15 (PerkinElmer Inc.), and assigned gasteiger charges and energy optimization of ligands using the steepest descent algorithm carried out by Open Babel [39]. The docking studies were done with the Smina program. Smina is a version of AutoDock Vina with a modified scoring function that is particularly optimized to offer high-throughput scoring (http://smina.sf.net) [40].
The grid parameter file is according to the grid box that comprised 20 × 20 × 20 points with 1 Å space and was centered on the active site of SARS-COV-2 3CLpro (x = 9.412, y = 1.383, and z = 8.836). The grid parameter file is according to the grid box that comprised 14 × 14 × 14 points with 1 Å space and was centered on the active site of SARS-COV-1 (x = 66.036, y = 3.288, and z = 5.254).
The X-ray crystallographic structures of SARS-COV-1, SARS-COV-2 3CLpro were obtained from the Protein Data Bank (PDB: 1UK and 6XHO). The structures of compounds were drawn by BIOVIA Discovery Studio Visualizer 2021. The calculation of energy optimization was done using the steepest descent method. Smina was performed with default settings for three proteins and 9 best conformations of ligand were introduced (Additional file 1: Table S4). The computational docking approach was evaluated based on the root-mean-square deviation (RMSD) value from re-docking the co-crystalized native ligand back into the active pocket site of the receptor [41].
Results and discussion
QSAR models
To build the reliable QSAR models, two objective functions were used: objective function without IIC (TF0) and with IIC (TF1). The range of finding the optimal threshold value (T) and the number of epochs (N) were 1–3 and 1–15, respectively. The QSAR models to predict the inhibitory activity against SARS 3CLpro for four splits were built based on TF1 are given below:
Split 1:
Split 2:
Split 3:
Split 4:
where \({\text{R}}_{\text{ATRN}}^{2}\), \({\text{R}}_{\text{PTRN}}^{2}\) R2CAL, and R2VAL are coefficient of determination for active training, passive training, calibration, and validation set, respectively. \({\text{ n}}_{\text{ATRN}}, {{\text{ n}}_{\text{PTRN}},\text{ n}}_{\text{CAL}}\), and \({\text{n}}_{\text{VAL}}\) indicate the number of molecules in the training, calibration, and validation set, respectively.
Table 2 indicates the statistical criteria of QSAR models for predicting of pIC50 isatin and indole derivatives based on TF0 and TF1 for each split. Regarding the QSAR models, the models developed based on IIC (TF1) are more predictive than the models developed using TF1. Therefore, it can be stated that the QSAR models built with the modified objective function TF1 using IIC are more reliable and robust than the models built by the objective function TF0. Thus, the QSAR model built for split 3 with TF1 was selected as the best model because the coefficient of determination (R2) was the highest for the validation set of this model.
Y-randomization test (Y-test) was done by CORAL software to confirm the non-chance correlation of developed QSAR models. After ten repetitions of new random models were developed and the values of average value of R2 were found below 0.1 (see Additional file 1: Table S2). These values confirm that the correlation between pIC50 and molecular attributes is not based on chance correlation. Moreover, for the Y-randomization test, the value of CR2p for all models was more than 0.8 (Table 2).
Additional file 1: Table S3 shows the SMILES symbol of isatin and indole derivatives, the set of each compound, the observed and calculated pIC50 of four models, and AD in four splits using TF1. The average \({\overline{\text{Defect}} }_{\text{TRN}}\) for Split 1 to 4 of constructed models based of TF0 are 5.91, 3.19, 5.18, and 5.05, respectively. So, compounds fall into AD if DefectSMILES < 11.82, 6.38, 10.36, and 10.10, for split 1 to 4 respectively. The percentages of data set in the AD of models were 82, 82, 83, and 88 for splits 1–4, respectively. This revealed that the four prediction models were capable of predicting more than 80% of the new data (Additional file 1: Table S3).
Figure 1 displays the plots of the calculated versus observed pIC50 of SARS 3CLpro inhibitors for four models developed based on TF1. It also shows that there is good agreement between the observed and experimental pIC50.

The graphical representation of the observed versus prediccted pIC50 for split 1 to 4
Mechanistic interpretation
Mechanistic interpretation as the fifth OECD principle of QSAR modeling states that the molecular features responsible for increased or decreased activity should be investigated whenever possible. The interpretation of the model can help to design and identify new isatin- and indole-based derivatives. The list of structural features extracted from the best QSAR model (split 3) for three independent probes is shown in Table 3. A short description of these descriptors is presented in the comments column of Table 3 which shows the structural features of increasing or decreasing pIC50 of isatin and indole derivatives. The identified promotors in the increase of pIC50 include the presence of nitrogen with double bond, presence of nitrogen with oxygen, presence of oxygen with double bond, presence of at least one ring, combination of aliphatic oxygen with double bond, presence of oxygen with double bond and branching and presence of aromatic carbon in first ring. The promoters of decrease of SARS 3CLpro inhibitory activity of isatin and indole derivatives are the presence of nitrogen with sulfur, presence of consecutive aliphatic carbon with aliphatic nitrogen with branching, presence of aromatic carbon with branching in fourth ring and presence of aliphatic carbon with branching in fourth ring.
Based on the favorable structural features and using the most active molecules among the 81 inhibitors which were gathered from literature, some compounds synthesized in various studies were extracted from ChEMBL database. In the ChEMBL database, newly synthesized compounds can be extracted with percentage similarity with desired compound, so we entered the ligand with the highest activity into ChEMBL and extracted some similar compounds from this database. The inhibitory activity (pIC50) of selected structures was calculated using best QSAR model (Split 3). Finally, eight most active compounds (isatin and indole scaffolds with most pIC50) were selected and introduced which are listed in Table 4. The predicted pIC50 range for the extracted compounds based on average prediction of four models was between 7.35 and 8.30. The AD analysis of these compounds based on the Split 3 model (the best model) shows that they fall into AD except for CHEMBL3103276.
Molecular docking analysis
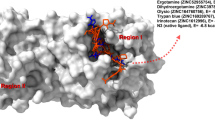
First, we perform a re-docking of the V34 ligand with the SARS-COV-2 3CLpro and 5-mer peptide with SARS-COV-1 receptors; this is done to validate the molecular docking protocol and also to get insight into the reference active amino acid residues involved in interactions inside the SARS-COV-2 3CLpro and SARS-COV-1 protein pocket (PDB code: 6XHO and 1UK4). Figure 2 displays 3D and 2D visualizations of the re-docking pathways of V34 inside the COVID-2 3CLpro and 5-mer peptide inside the SARS-COV-1 protein pockets with − 8.07 and − 9.4 kcal/mol, respectively. Figures indicate that the re-dock V34 located in the active site of SARS-COV-2 3CLpro interacts with the THR26, HIS41, PHE140, CYS145, HIS164, MET165, GLU166, PRO168, HIS172, GLN189, THR190, and ALA191. Also, the re-dock 5-mer peptide located in the active site of SARS-COV-1 interacts with the HIS41, PHE140, GLY143, SER144, and GLU166. These interactions were hydrophobic and hydrogen bonds. The root-mean-square deviation (RMSD) values were 0.14 and 1.1 Å for native and re-docked ligands of V34 and 5-mer peptide, respectively; which are lower than the tolerable marginal value of 2 Å (Additional file 1: Fig. S1).

V34 interaction patterns with active residues in the SARS-COV-2 3CLpro pocket (A), 5-mer peptide interaction patterns with active residues in the SARS-COV-1 pocket (B)
Figure 3a and b shows that the compound 12 and 53 were placed into the binding pocket of SARS-COV-1 3CLpro by representing three-dimensional diagram. Two‐dimensional diagram of compound 12 and 53 interactions was presented in Fig. 4a and b the compounds formed some important interactions with binding site residues of SARS-COV-1 3CLpro. As the molecular docking results are shown in Fig. 3a, the compound 12 formed two hydrogen bond interactions with SER144 and CYS145 the binding site of SARS-COV-1 3CLpro. Also, it has two hydrophobic interactions with HIS41 and MET49. Moreover, ALA46, CYS44, THR45, THR25, ASN142, GLY143, HIS163, PHE140, LEU141 and GLU166 have van der Walls interaction with the protein. Figure 3b shows various interactions of compound 57 with HIS41, MET49 and MET165, along with some hydrophobic interactions. In addition, the complex formed hydrogen bond interactions with residues SER144, THR26, CYS145, GLY143 and GLN189. LEU141, PHE140, HIS163, LEU27, THR25, ASN142, GLU166, THR190, ALA191, TYR54, ARG188, LEU167 and PRO168 had van der Walls interaction with the protein.

Three-dimensional diagram of compound 12(A) and 53(B) into the binding pocket of SARS-COV-1 3CLp

Two‐dimensional diagram of compound 12 (A) and 53 (B) interactions with binding site residues of SARS-COV-1 3CLpro
Comparing the molecular docking results of re-docked native ligands and compounds 12 and 53 as the most activist compounds; we can notice that all compounds 12 and 53 interacted with the majority of active residues in the COV-2 3CLpro and SARS-COV-1 pockets with which native ligands interacted.
Molecular docking results agree with some promoters regarding the increase in pIC50 in QSAR models; for instance, compounds 12 and 53 contain oxygen with double bonds, at least one ring, and branching, all of which interact with amino acids residues in protein active sites via hydrogen bonds and hydrophobic interactions.
Hexachlorophene was used as a SARS 3CLpro standard inhibitor (IC50 = 5 µM) according to Liu et al. [42]. We docked Hexachlorophene into the active site of 6XHO. The best binding mode of the Hexachlorophene in the binding site of SARS-COV-1 3CLpro (pdb: 6XHO) was − 8.05 kcal/mol.
Eight extracted compounds from CHEMBL based on scaffold of isatin or indole were docked into 1UK4 and 6XHO as well. Two and three‐dimensional diagrams of the interaction of the eight ligands from CHEMBLE with their receptors are presented in Additional file 1: Fig. S2. Molecular docking analysis shows that these ligands with the majority of active residues in the COV-2 3CLpro and SARS-COV-1 pockets with which native ligands interacted. As before we mentioned it for the activist compounds 12 and 53. It confirmed that indole and isatin are important cores in interaction with targets. As can be seen in Table 4, all eight compounds had higher binding energy compared to the most active compounds in data set and hexachlorophene. The results present a very good correlation between results obtained from Monte Carlo optimization modeling and molecular docking studies.
ADMET results
In silico ADMET (absorption, distribution, metabolism, excretion, and toxicity) screening of compounds can reduce the cost and time associated with the in vitro assay and/or in vivo experiments [43]. AdmetSAR online database was used to predict ADMET properties of extracted isatin- and indole-based compounds [44]. As ADMET properties are shown in Table 5, all eight compounds showed positive results for human intestinal absorption. Furthermore, it is necessary to check whether the proposed molecules are non-toxic because it plays an important role in the selection of drugs. Ames test was negative for all compounds except CHEMBL4443007 and based on acute oral toxicity all compounds were classified as non-toxic.
The Osiris Property Explorer (OPE) tool was used to assess the fragment-based drug-likeness of the extracted compounds [45, 46]. A positive value (0.1–10) indicates that the compound mainly contains fragments that are often found in commercial drugs. Also, using this program, the overall drug scores were evaluated that combines drug-likeness, ClogP, ClogS, molecular weight, and toxicity risk factors in one single value where the frequency of occurrence of each fragment is determined within the collection of approved drugs and within Fluka non-medicinal chemicals.
Finally, based on the results of the OPE study, compounds CHEMBL4458417 and CHEMBL4565907 both containing an indole scaffold with the positive values of drug-likeness and the highest drug-score can be introduced as selected leads.
Conclusion
Four simple, predictive, and reliable QSAR models were developed for the pIC50 values of 81 isatin and indole derivatives that inhibit SARS 3CLpro using Monte Carlo with the index of ideality of correlation (IIC) as the objective function. The statistical parameters of the models were suitable with high predictive power (\({R}_{Val}^{2}\) = 0.81–0.92, and MAE = 0.31–0.40). The four proposed models were satisfactory for predicting new isatin and indole derivatives as candidates for SARS 3CLpro inhibitors and can be used for pre-synthesis evaluation of new isatin and indole derivatives. A mechanistic interpretation of the models was done by examining the correlation weights of the different extracted molecular features extracted in several Monte Carlo optimization runs. These features were used to extract eight new and more active isatin and indole derivatives from the ChEMBL database. The activity of new compounds was further verified by molecular docking studies. The activity of the new compounds was further confirmed by molecular docking studies. The binding energy of these molecules with residues of active site were in correlation with calculated pIC50. Finally, the compounds CHEMBL4458417 and CHEMBL4565907 both containing an indole scaffold with the positive values of drug-likeness and the highest drug-score were introduced as selected leads.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem. 2016;59(14):6595–628.
Zhou L, Liu Y, Zhang W, Wei P, Huang C, Pei J, Yuan Y, Lai L. Isatin compounds as noncovalent SARS coronavirus 3C-like protease inhibitors. J Med Chem. 2006;49(12):3440–3.
Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–7.
Liu Y, Liang C, Xin L, Ren X, Tian L, Ju X, Li H, Wang Y, Zhao Q, Liu H, et al. The development of Coronavirus 3C-Like protease (3CL(pro)) inhibitors from 2010 to 2020. Eur J Med Chem. 2020;206: 112711.
Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, Hu Y, Tao Z-W, Tian J-H, Pei Y-Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–9.
Chu CK, Gadthula S, Chen X, Choo H, Olgen S, Barnard DL, Sidwell RW. Antiviral activity of nucleoside analogues against SARS-coronavirus (SARS-CoV). Antiviral Chem Chemother. 2006;17(5):285–9.
Morgenstern B, Michaelis M, Baer PC, Doerr HW, Cinatl J Jr. Ribavirin and interferon-beta synergistically inhibit SARS-associated coronavirus replication in animal and human cell lines. Biochem Biophys Res Commun. 2005;326(4):905–8.
Koren G, King S, Knowles S, Phillips E. Ribavirin in the treatment of SARS: a new trick for an old drug? CMAJ. 2003;168(10):1289–92.
Tai DY. Pharmacologic treatment of SARS: current knowledge and recommendations. Ann Acad Med Singap. 2007;36(6):438.
Corona A, Meleddu R, Esposito F, Distinto S, Bianco G, Masaoka T, Maccioni E, Menendez-Arias L, Alcaro S, Le Grice SF, et al. Ribonuclease H/DNA polymerase HIV-1 reverse transcriptase dual inhibitor: mechanistic studies on the allosteric mode of action of isatin-based compound RMNC6. PLoS ONE. 2016;11(1): e0147225.
Meleddu R, Distinto S, Corona A, Tramontano E, Bianco G, Melis C, Cottiglia F, Maccioni E. Isatin thiazoline hybrids as dual inhibitors of HIV-1 reverse transcriptase. J Enzyme Inhib Med Chem. 2017;32(1):130–6.
Webber SE, Tikhe J, Worland ST, Fuhrman SA, Hendrickson TF, Matthews DA, Love RA, Patick AK, Meador JW, Ferre RA. Design, synthesis, and evaluation of nonpeptidic inhibitors of human rhinovirus 3C protease. J Med Chem. 1996;39(26):5072–82.
Gao F, Yang H, Lu T, Chen Z, Ma L, Xu Z, Schaffer P, Lu G. Design, synthesis and anti-mycobacterial activity evaluation of benzofuran-isatin hybrids. Eur J Med Chem. 2018;159:277–81.
Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LL, Guan Y, Rozanov M, Spaan WJ, Gorbalenya AE. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol. 2003;331(5):991–1004.
Chen L-R, Wang Y-C, Lin YW, Chou S-Y, Chen S-F, Liu LT, Wu Y-T, Kuo C-J, Chen TS-S, Juang S-H. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg Med Chem Lett. 2005;15(12):3058–62.
Liu W, Zhu H-M, Niu G-J, Shi E-Z, Chen J, Sun B, Chen W-Q, Zhou H-G, Yang C. Synthesis, modification and docking studies of 5-sulfonyl isatin derivatives as SARS-CoV 3C-like protease inhibitors. Bioorg Med Chem. 2014;22(1):292–302.
Javidfar M, Ahmadi S. QSAR modelling of larvicidal phytocompounds against Aedes aegypti using index of ideality of correlation. SAR QSAR Environ Res. 2020;31(10):717–39.
Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384–421.
Lin X, Li X, Lin X. A review on applications of computational methods in drug screening and design. Molecules. 2020. https://doi.org/10.3390/molecules25061375.
Hamzehali H, Lotfi S, Ahmadi S, Kumar P. Quantitative structure–activity relationship modeling for predication of inhibition potencies of imatinib derivatives using SMILES attributes. Sci Rep. 2022;12(1):21708.
Lotfi S, Ahmadi S, Kumar P. A hybrid descriptor based QSPR model to predict the thermal decomposition temperature of imidazolium ionic liquids using Monte Carlo approach. J Mol Liq. 2021;338: 116465.
Hoffman RL, Kania RS, Brothers MA, Davies JF, Ferre RA, Gajiwala KS, He M, Hogan RJ, Kozminski K, Li LY. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J Med Chem. 2020;63(21):12725–47.
Zhang J, Pettersson HI, Huitema C, Niu C, Yin J, James MN, Eltis LD, Vederas JC. Design, synthesis, and evaluation of inhibitors for severe acute respiratory syndrome 3C-like protease based on phthalhydrazide ketones or heteroaromatic esters. J Med Chem. 2007;50(8):1850–64.
Ghosh AK, Gong G, Grum-Tokars V, Mulhearn DC, Baker SC, Coughlin M, Prabhakar BS, Sleeman K, Johnson ME, Mesecar AD. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg Med Chem Lett. 2008;18(20):5684–8.
Thanigaimalai P, Konno S, Yamamoto T, Koiwai Y, Taguchi A, Takayama K, Yakushiji F, Akaji K, Chen S-E, Naser-Tavakolian A. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: design, synthesis, biological evaluation, and docking studies. Eur J Med Chem. 2013;68:372–84.
Sokolović D, Stanković V, Toskić D, Lilić L, Ranković G, Ranković J, Nedin-Ranković G, Veselinović AM. Monte Carlo-based QSAR modeling of dimeric pyridinium compounds and drug design of new potent acetylcholine esterase inhibitors for potential therapy of myasthenia gravis. Struct Chem. 2016;27:1511–9.
Ahmadi S. Mathematical modeling of cytotoxicity of metal oxide nanoparticles using the index of ideality correlation criteria. Chemosphere. 2020;242: 125192.
Toropova AP, Toropov AA. Use of the index of ideality of correlation to improve models of eco-toxicity. Environ Sci Pollut Res. 2018;25(31):31771–5.
Aher R, Roy K. Exploring the structural requirements in multiple chemical scaffolds for the selective inhibition of Plasmodium falciparum calcium-dependent protein kinase-1 (Pf CDPK-1) by 3D-pharmacophore modelling, and docking studies. SAR QSAR Environ Res. 2017;28(5):390–414.
Shi LM, Fang H, Tong W, Wu J, Perkins R, Blair RM, Branham WS, Dial SL, Moland CL, Sheehan DM. QSAR models using a large diverse set of estrogens. J Chem Inf Comput Sci. 2001;41(1):186–95.
Schuurmann G, Ebert R-U, Chen J, Wang B, Kuhne R. External validation and prediction employing the predictive squared correlation coefficient Test set activity mean vs training set activity mean. J Chem Inf Model. 2008;48(11):2140–5.
Daoui O, Elkhattabi S, Chtita S, Elkhalabi R, Zgou H, Benjelloun AT. QSAR, molecular docking and ADMET properties in silico studies of novel 4, 5, 6, 7-tetrahydrobenzo [D]-thiazol-2-Yl derivatives derived from dimedone as potent anti-tumor agents through inhibition of C-Met receptor tyrosine kinase. Heliyon. 2021;7(7): e07463.
Kumar P, Kumar A. CORAL: QSAR models of CB1 cannabinoid receptor inhibitors based on local and global SMILES attributes with the index of ideality of correlation and the correlation contradiction index. Chemom Intell Lab Syst. 2020;200: 103982.
Toropov AA, Toropova AP, Marzo M, Benfenati E. Use of the index of ideality of correlation to improve aquatic solubility model. J Mol Graph Model. 2020;96: 107525.
Ahmadi S, Mardinia F, Azimi N, Qomi M, Balali E. Prediction of chalcone derivative cytotoxicity activity against MCF-7 human breast cancer cell by Monte Carlo method. J Mol Struct. 2019;1181:305–11.
Mahapatra MK, Karuppasamy M. Fundamental considerations in drug design. In: Computer Aided Drug Design (CADD): from ligand-based methods to structure-based approaches. Elsevier; 2022: 17–55.
Daoui O, Elkhattabi S, Chtita S. Rational design of novel pyridine-based drugs candidates for lymphoma therapy. J Mol Struct. 2022;1270: 133964.
Daoui O, Nour H, Abchir O, Elkhattabi S, Bakhouch M, Chtita S. A computer-aided drug design approach to explore novel type II inhibitors of c-Met receptor tyrosine kinase for cancer therapy: QSAR, molecular docking, ADMET and molecular dynamics simulations. J Biomol Struct Dyn. 2022. https://doi.org/10.1080/07391102.2022.2124456.
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3(1):1–14.
Koes DR, Baumgartner MP, Camacho CJ. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model. 2013;53(8):1893–904.
Daoui O, Elkhattabi S, Chtita S. Rational identification of small molecules derived from 9, 10-dihydrophenanthrene as potential inhibitors of 3CLpro enzyme for COVID-19 therapy: a computer-aided drug design approach. Struct Chem. 2022;33(5):1667–90.
Liu Y-C, Huang V, Chao T-C, Hsiao C-D, Lin A, Chang M-F, Chow L-P. Screening of drugs by FRET analysis identifies inhibitors of SARS-CoV 3CL protease. Biochem Biophys Res Commun. 2005;333(1):194–9.
Alam S, Khan F. Virtual screening, docking, ADMET and system pharmacology studies on Garcinia caged Xanthone derivatives for anticancer activity. Sci Rep. 2018;8(1):1–16.
Matin MM, Hasan MS, Uzzaman M, Bhuiyan MMH, Kibria SM, Hossain ME, Roshid MH. Synthesis, spectroscopic characterization, molecular docking, and ADMET studies of mannopyranoside esters as antimicrobial agents. J Mol Struct. 2020;1222: 128821.
Almasirad A, Mousavi Z, Tajik M, Assarzadeh MJ, Shafiee A. Synthesis, analgesic and anti-inflammatory activities of new methyl-imidazolyl-1, 3, 4-oxadiazoles and 1, 2, 4-triazoles. Daru J Pharm Sci. 2014;22(1):1–8.
Faizi M, Jahani R, Ebadi SA, Tabatabai SA, Rezaee E, Lotfaliei M, Amini M, Almasirad A. Novel 4-thiazolidinone derivatives as agonists of benzodiazepine receptors: design, synthesis and pharmacological evaluation. EXCLI J. 2017;16:52.
Acknowledgements
The authors would like to express their deepest gratitude to Dr. Alla P. Toropova and Dr. Andrey A. Toropov for providing the CORAL software.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
S.A. conceived of the presented idea. N.S. performed drawing of structures and developed the models and wrote original draft. S.A. performed model validation and interpretation. F.S. contributed to molecular docking. A.A. performed in silico ADMET screening of compounds. All authors discussed the results and commented on the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file1: Table S1.
CWs for each attribute of Split 1. Table S2. The results of Y-randomization test for all splits constructed based on TF1. Table S3. SMILES notations of isatin and indole derivatives, the compound set, their experimental, predicted pIC50, and applicability domain in four splits using TF1. Table S4. The affinity of nine conformations docked into SARS-COV-1 3CLpro (PDB: 1UK4 and 6XHO) for compounds 12 and 53. Figure S1. 3D superposition of original (black) and re-docked (yellow) (A) V34 ligand in the 6XHO (RMSD=0.14 Å), (A) 5-mer peptide ligand in the 1UK4 (RMSD=1.1 Å). Figure S2. Two and three‐dimensional diagram of (A) CHEMBL4524939 (B) CHEMBL4458417 (C) CHEMBL4452760 (D) CHEMBL4565907 (E) CHEMBL4443007 interactions with binding site residues of SARS-COV-1 3CLpro (6XHO) and (F) CHEMBL383761 (G) CHEMBL210543 (H) CHEMBL3103276interactions with binding site residues of SARS-COV-1 (1UK4).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Soleymani, N., Ahmadi, S., Shiri, F. et al. QSAR and molecular docking studies of isatin and indole derivatives as SARS 3CLpro inhibitors. BMC Chemistry 17, 32 (2023). https://doi.org/10.1186/s13065-023-00947-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-023-00947-w