
Abstract
Background
Xanthine oxidase is an important enzyme of purine catabolism pathway and has been associated directly in pathogenesis of gout and indirectly in many pathological conditions like cancer, diabetes and metabolic syndrome. In this research rutin, a bioactive flavonoid was explored to determine the capability of itself and its derivatives to inhibit xanthine oxidase.
Objective
To develop new xanthine oxidase inhibitors from natural constituents along with antioxidant potential.
Method
In this report, we designed and synthesized rutin derivatives hybridized with hydrazines to form hydrazides and natural acids to form ester linkage with the help of molecular docking. The synthesized compounds were evaluated for their antioxidant and xanthine oxidase inhibitory potential.
Results
The enzyme kinetic studies performed on rutin derivatives showed a potential inhibitory effect on XO ability in competitive manner with IC50 value ranging from 04.708 to 19.377 µM and RU3a3 was revealed as most active derivative. Molecular simulation revealed that new rutin derivatives interacted with the amino acid residues PHE798, GLN1194, ARG912, GLN 767, ALA1078 and MET1038 positioned inside the binding site of XO. Results of antioxidant activity revealed that all the derivatives showed very good antioxidant potential.
Conclusion
Taking advantage of molecular docking, this hybridization of two natural constituent could lead to desirable xanthine oxidase inhibitors with improved activity.

Similar content being viewed by others
Introduction
Xanthine oxidase (XO) having molecular weight of around 300 kDa is oxidoreductase enzyme represented in the form of a homodimer. Both the monomers of XO are almost identical and each of them contains three domains namely (a) molybdopterin (Mo-pt) domain at the C-terminal having 4 redox centers where oxidation takes place (b) a flavin adenine dinucleotide (FAD) domain at the centre generally considered as binding site domain and (c) 2[Fe–S]/iron sulfur domain at the N-terminal [1,2,3]. The catalytic oxidation of XO is two substrates reaction on the xanthine and oxygen at the enzymatic centre. While xanthine undergoes oxidation reaction near to the Mo-pt center/substrate binding domain of XO, simultaneously substrate oxygen undergoes reduction at FAD center and electron transfer takes place leading to formation of superoxide anion (O2−) or hydrogen peroxide (H2O2) free radicals. [4,5,6,7,8]. This catalytic reaction results in formation uric acid as a final product and oxygen reactive species in form of free radicals. The excessive generation of uric acid leads to a condition like hyperuricemia which is a key factor in development of gout [1, 9], and uncontrolled amounts of reactive oxygen species causes many pathological conditions like cardiovascular disorders, inflammatory diseases and hypertensive disorders. Xanthine oxidase (XO; EC 1.17.3.2) has been considered as significantly potent drug target for the cure and management of pathological conditions prevailing due to high levels of uric acid in the blood stream. [10,11,12,13,14,15,16,17]. Considering the above fact, by inhibiting XO selectively could be better treatment plan for disorders caused by XO directly or indirectly including gout, inflammatory disease, oxidative damage and cancer [3, 18, 19]. Generally, XO inhibitors have been categorized into purine and non-purines inhibitors differentiated on the basis of their chemically derived skeleton structure. The first purine derived XO inhibitor discovered and approved by US FDA was Allopurinol as marketed drug for gout and hyperuricemia [20, 21]. Considering the life threatening side effects like Stevens–Johnsons syndrome caused by allopurinol use, scientists turned their interest into non-purine XO inhibitors and an immense accomplishment has been received in this direction with development of new drug Febuxostat [22,23,24,25]. This non-purine candidate produced minor and non-life threatening adverse effects in comparison to Allopurinol [26,27,28,29]. Extending our previous successful effort to achieve new xanthine oxidase inhibitors from natural sources, in this report we investigated and developed some new rutin derived xanthine oxidase inhibitor [30].
Rutin is a well characterized bioactive plant flavonoid having great therapeutic importance for the treatment of many disease like conditions including cytotoxicity, antioxidant activity, antibacterial property and anti-inflammatory action [31,32,33,34]. Due to these pharmacological activities rutin is explored widely and great success have been achieved in order to get drug like candidates.

Taking advantage of molecular docking techniques new compounds with potential drugability for the targeted enzyme might be achieved with a precise knowledge of mechanism of action. With the combined approach of molecular docking and synthetic chemistry, in this research we developed some new potential compounds against xanthine oxidase (Fig. 1).

Design strategy for the development of rutin derivatives
Experimental
Chemicals and instrumentation
For this research, the analytical grade chemicals necessary for synthesis and antioxidant activity were purchased from Hi-media Laboratories. The in vitro evaluation of the human xanthine oxidase inhibitory activity was performed by measuring hydrogen peroxide (H2O2) production from oxidation of xanthine oxidase by the substrate xanthine, utilizing the human xanthine oxidase assay kit (Sigma USA). The progress of reaction was observed through thin layer chromatography (TLC) on 0.25 mm precoated silica gel plates purchased from Merck, reaction spots were envisaged in iodine compartment and UV. Melting points were measured using a Sonar melting point apparatus and uncorrected. 1H NMR and 13C NMR spectra were documented in DMSO and deuterated CDCl3 respectively on Bruker Avance II 400 NMR spectrometer at the frequency of 400 MHz using tetramethylsilane standard (downfield) moreover chemical shifts were expressed in ppm (δ) using the residual solvent line as internal standard. Infrared (IR) spectra were recorded on Perkin Elmer FTIR spectrophotometer by utilizing KBr pellets system.
Molecular docking
In silico docking studies was done with integrated Schrodinger software using Glide module for enzyme ligand docking [35].
Protocol followed for docking procedures
Preparation of protein
The 3D crystal structure of human xanthine oxidase co-crystalised with salicylic acid was retrieved from Protein Data Bank (PDB ID. 2E1Q). The targeted protein structure was further refined in the Protein Preparation Wizard to obtain the optimized and chemically accurate protein configuration. For that, the co-crystalised enzyme (XO) was retrieved directly from Protein data bank in maestro panel followed by removal of water molecules, addition of H atoms, addition of missing side chains and finally minimization was done to obtain the optimized structure.
Preparation of ligand
The 3D-structures of rutin derived compounds to be docked against XO were built in maestro building window. Ligand preparation was performed in Ligprep module.
Active site prediction
To predict the binding site/active site Site Map application of glide was utilized. Out of top three active site, the one having larger radius was selected. Validation of binding site was done by redocking the salicylic acid and RMSD value was observed. RMSD value of less than 0.2 validated the docking procedure and active site was defined for docking of new rutin analogs.
Glide docking
To carry out docking, Firstly the receptor grid generation tool was utilized to around the active/binding site of xanthine oxidase and glide docking with extra precision was used to visualize the interaction of protein and ligand. The top active ligand was selected for wet lab synthesis and evaluation of pharmacological activity.
Synthetic procedures
Procedures for synthesis of rutin derivatives (Scheme 1)
-
(A)
General procedure for synthesis of hydrazine derivatives RU3a (1– 4)
0.001 mol of rutin was taken in round bottom flask and dissolved in 50 ml of ethanol. Different hydrazines (0.001 mol) were added to the flask and reaction mixture was refluxed for 5–6 h at 40 °C. Completion of reaction was monitored by TLC. The product thus obtained was filtered and filtrate was concentrated to obtain the final product. The final product was recrystallised to obtain the pure compound.
-
(B)
General procedure for synthesis of anilline derivatives RU4b (1– 2)
0.001 mol of the intermediate obtained above was taken in round bottom flask and dissolved in 50 ml of ethanol. Different anillines (0.001 mol) were added to the flask and reaction mixture was refluxed for 8–10 h at 40 °C. Completion of reaction was monitored by TLC. The product thus obtained was filtered and filtrate was concentrated to obtain the final product. The final product was recrystallised to obtain the pure compound.
-
(C)
General procedure for synthesis of methylated rutin derivatives RU7c (1– 3)
Rutin was methylated by methyl sulphate in presence of potassium carbonate and dimethyl formamide by stirring along with reflux at 40 °C for 48 h to generate tetramethylated rutin. Acidolysis of above was done to obtain the intermediate compound (RUI) by refluxing it with HCl and 95% ethanol for 4 h. The intermediate compound (RUI) was then refluxed with different phenolic acid to obtain their ester derivatives.

Synthesis of rutin derivatives
Spectral data
RU3a1 yield 69.6% Rf 0.6 [Mobile Phase for TLC—Methanol:Glacial acetic acid:Formic acid:Water (3:2.9:0.8:0.5)] M.pt. (231–232) IR (KBR pellets) cm−1 1) 3222 (O–H str., Ar), 1609 (C=N str.), 1501 (C=C str.), 1206 (O–CH3), 1128 (C=S Str.) 1H NMR (400 MHz, DMSO-d6) δ 7.81 (dd, J = 7.5, 1.5 Hz, 1H), 7.59 (d, J = 1.5 Hz, 1H), 6.82 (d, J = 7.5 Hz, 1H), 6.48 (dd, J = 15.0, 1.5 Hz, 2H), 6.28 (t, J = 7.0 Hz, 1H), 4.13 (t, J = 7.0 Hz, 1H), 3.89–3.81 (m, 3H), 3.71 (dd, J = 12.4, 6.9 Hz, 1H), 3.67–3.54 (m, 3H), 2.32 (dt, J = 12.4, 7.0 Hz, 1H), 2.28–2.16 (m, 2H), 2.06–2.04 (m, 1H), 1.97–1.92 (m, 2H), 1.74–1.66 (m, 2H). 13C NMR (100 MHz, Chloroform-d) δ 180.16, 163.73, 155.81, 154.70, 152.34, 148.70, 145.50, 133.79, 133.45, 120.73, 120.41, 115.79, 115.09, 102.38, 99.59, 99.00, 91.11, 80.48, 73.58, 73.26, 72.40, 71.83 (d, J = 10.5 Hz), 66.02, 40.22, 37.43, 28.26, 26.90. m/z found for C28H33N3O15S: 683 (M+) 687 (M + 1)+. Anal calcd for C28H33N3O15S: C, 52.91; H, 5.23; N, 6.61; O, 35.20; S, 5.04 Found: C, 52.93; H, 5.21; N, 6.60; O, 35.19; S, 5.06.
RU3a2 yield 72.5% Rf 0.7 [Mobile Phase for TLC—Methanol:Glacial acetic acid:Formic acid:Water (3:2.9:0.8:0.5)] M.pt. (255–257) IR (KBR pellets) cm−1) 3468 (O–H str., Ar), 1639 (C=N str.), 1596 (C=C str.), 1218 (O–CH3), 1150 (C=S Str.) 1H NMR (400 MHz, DMSO-d6) δ 7.78–7.60 (m, 3H), 7.49 (d, J = 1.5 Hz, 1H), 7.39–7.29 (m, 2H), 7.10–7.01 (m, 1H), 6.86 (d, J = 7.5 Hz, 1H), 6.52 (dd, J = 15.0, 1.5 Hz, 2H), 6.24 (t, J = 7.0 Hz, 1H), 4.04 (t, J = 7.0 Hz, 1H), 3.98–3.88 (m, 3H), 3.78 (dd, J = 12.4, 6.9 Hz, 1H), 3.68–3.64 (m, 3H), 2.28 (dt, J = 12.4, 7.0 Hz, 1H), 2.14–2.11 (m, 2H), 2.09–2.06 (m, 1H), 1.87–1.84 (m, 2H), 1.74–1.71 (m, 2H). 13C NMR (100 MHz, Chloroform-d) δ 174.93, 164.50, 160.96, 155.78, 150.30, 148.16, 145.55, 139.23, 130.44, 128.67, 124.46, 123.85, 123.09, 122.39, 121.81, 116.06, 115.83, 103.40, 99.09, 97.71, 95.05, 82.37, 73.06 (d, J = 19.1 Hz), 72.87 (d, J = 12.2 Hz), 72.47, 72.35, 71.92, 65.19, 41.10, 38.86, 29.40, 27.86. m/z found for C34H37N3O15S: 759 (M+) 760 (M + 1)+. Anal calcd for C34H37N3O15S: C, 53.75; H, 4.91; N, 5.53; O, 31.59; S, 4.22. Found: C, C, 53.77; H, 4.93; N, 5.56; O, 31.59; S, 4.24.
RUT3a3 yield 61% Rf 0.6 [Mobile Phase for TLC—Methanol:Glacial acetic acid:Formic acid:Water (3:2.9:0.8:0.5)] M.pt. (235–237) IR (KBR pellets) cm−1) 3475 (O–H str., Ar), 1641 (C=N str.), 1580 (C=C str.), 1220 (O–CH3), 1155 (C=S Str.) 1H NMR (400 MHz, DMSO-d6) δ 7.70 (dd, J = 7.5, 1.5 Hz, 1H), 7.56 (d, J = 1.5 Hz, 1H), 7.46–7.38 (m, 2H), 7.32–7.23 (m, 2H), 7.07–6.98 (m, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.35 (dd, J = 15.0, 1.5 Hz, 2H), 6.19 (t, J = 7.0 Hz, 1H), 4.09 (t, J = 7.0 Hz, 1H), 4.02–3.88 (m, 3H), 3.68 (dd, J = 12.4, 6.9 Hz, 1H), 3.66–3.54 (m, 3H), 2.33 (dt, J = 12.4, 7.0 Hz, 1H), 2.21–2.19 (m, 2H), 1.96–1.88 (m, 2H), 1.87–1.85 (m, 2H) (Additional file 1). 13C NMR (100 MHz, Chloroform-d) δ 164.50, 160.96, 155.78, 150.30, 148.16, 145.55, 143.60, 132.14, 129.50, 124.46, 122.39, 121.81, 121.19, 118.32, 116.06, 115.83, 104.75, 94.15, 93.97, 91.01, 83.98, 79.41 (d, J = 19.1 Hz), 78.77 (d, J = 12.2 Hz), 77.09, 73.82, 68.48, 42.85, 37.51, 23.82, 23.17. m/z found for C33H36N2O15: 700 (M+) 701 (M + 1)+. Anal calcd for C33H36N2O15: C, 56.57; H, 5.18; N, 4.00; O, 34.25. Found: C, 56.58; H, 5.20; N, 4.00; O, 34.27.
RU4b1 yield 74.3% Rf 0.6 [Mobile Phase for TLC—Methanol:Glacial acetic acid:Formic acid:Water (3:2.9:0.8:0.5)] M.pt. (259–260) IR (KBR pellets) cm−1 1) 1725 (C=O str.), 1631 (C=N str.), 1603 (C=C str.), 1234 (O–CH3), 1268 (C–O str., ester) 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 1.5 Hz, 1H), 8.15 (dd, J = 7.5, 1.5 Hz, 1H), 7.69 (dd, J = 7.5, 1.5 Hz, 1H), 7.2 (d, J = 1.5 Hz, 1H), 7.40 (d, J = 7.5 Hz, 1H), 6.81 (d, J = 7.5 Hz, 1H), 6.47 (dd, J = 10.8, 1.5 Hz, 2H), 6.22 (t, J = 7.0 Hz, 1H), 4.11 (t, J = 7.0 Hz, 1H), 3.98–3.90 (m, 3H), 3.79 (dd, J = 12.4, 6.9 Hz, 1H), 3.71–3.61 (m, 3H), 2.42 (dt, J = 12.4, 7.0 Hz, 1H), 2.39– 2.31 (m, 2H), 2.29–2.28 (m, 1H), 1.87–1.77 (m, 2H). 13C NMR (100 MHz, Chloroform-d) δ 169.14, 168.95, 168.11, 166.86, 150.94, 144.52, 144.24, 142.37, 140.47, 131.18, 128.56, 125.41, 123.81, 122.54 (d, J = 14.8 Hz), 121.81, 113.64, 113.17, 106.71, 97.09, 96.89, 93.98, 82.37, 75.79 (d, J = 19.1 Hz), 73.17 (d, J = 12.2 Hz), 73.06, 72.69, 71.01, 65.19, 41.10, 38.86, 28.85, 27.44. m/z found for H33ClN2O17: 764 (M+) 766 (M + 2)+. Anal calcd for C33H33ClN2O17: C, 51.81; H, 4.35; Cl, 4.63; N, 3.66; O, 35.55. Found: C, 51.83; H, 4.36; Cl, 4.65; N, 3.64; O, 35.53.
RU4b2 yield 83.5% Rf 0.8 [Mobile Phase for TLC—Methanol:Glacial acetic acid:Formic acid:Water (3:2.9:0.8:0.5)] M.pt. (253–254) IR (KBR pellets) cm−1 1) 1785 (C=O str.), 1637 (C=N str.), 1561 (C=C str.), 1258 (O–CH3), 1234 (C–O str., ester) 1H NMR (400 MHz, DMSO-d6) δ 8.21–8.14 (m, 2H), 7.79 (dd, J = 7.5, 1.5 Hz, 1H), 7.59 (d, J = 1.5 Hz, 1H), 7.32–7.25 (m, 2H), 6.75 (d, J = 7.5 Hz, 1H), 6.44 (dd, J = 14.1, 1.5 Hz, 2H), 6.27 (t, J = 7.0 Hz, 1H), 4.15 (t, J = 7.0 Hz, 1H), 3.98–3.95 (m, 3H), 3.88 (dd, J = 12.4, 6.9 Hz, 1H), 3.67–3.55 (m, 3H), 2.22 (dt, J = 12.4, 7.0 Hz, 1H), 2.14–2.11 (m, 2H), 2.09–2.06 (m, 1H), 1.76–1.73 (m, 2H), 1.67–1.55 (m, 2H).
13C NMR (100 MHz, Chloroform-d) δ 173.89, 164.58, 163.50, 158.34, 152.36, 151.92, 148.16, 146.53, 145.55, 128.56, 125.27, 124.36, 122.39, 121.81, 116.06, 115.83, 108.81, 93.06, 97.81, 90.53, 82.19, 73.80 (d, J = 19.1 Hz), 72.67 (d, J = 12.2 Hz), 72.36, 72.12, 71.08, 64.86, 42.81, 36.15, 28.55, 26.98. m/z found for C33H34N2O17:730 (M+) 731 (M + 1)+. Anal calcd for C33H34N2O17: C, 54.25; H, 4.69; N, 3.83; O, 37.23. Found: C, 54.27; H, 4.70; N, 3.85; O, 37.25.
RU7C1 yield 83.5% Rf 0.8 [Mobile Phase for TLC—Methanol:Glacial acetic acid:Formic acid:Water (3:2.9:0.8:0.5)] M.pt. (189–190) IR (KBR pellets) cm−1 1) 1715 (C=O str.), 1627 (C=N str.), 1607 (C=C str.), 1234 (O–CH3), 11,944 (C–O str., ester) 1H NMR (400 MHz, DMSO-d6) δ 9.11 (d, J = 1.5 Hz, 1H), 8.77–8.70 (m, 1H), 8.14 (dt, J = 7.5, 1.5 Hz, 1H), 7.92 (dd, J = 7.5, 1.5 Hz, 1H), 7.68 (d, J = 1.5 Hz, 1H), 7.51 (t, J = 7.5 Hz, 1H), 6.93–6.83 (m, 2H), 6.23 (d, J = 1.5 Hz, 1H), 3.92 (s, 3H), 3.83 (d, J = 0.9 Hz, 6H), 3.76 (s, 3H). 13C NMR (100 MHz, Chloroform-d) δ 174.99, 164.48, 164.18, 160.33, 157.96, 156.60, 153.53, 151.74, 150.80, 149.32, 138.25, 128.95, 123.72, 123.22, 122.87, 122.65, 113.70, 112.82, 107.81, 95.68, 93.25, 56.20, 55.88 (d, J = 2.6 Hz), 55.62. m/z found for C25H21NO8:463 (M+) 464 (M + 1)+. Anal calcd for C25H21NO8: C, 64.79; H, 4.57; N, 3.02; O, 27.62. Found: C, 64.80; H, 4.58; N, 3.00; O, 27.60.
RU7C2 yield 62.5% Rf 0.6 [Mobile Phase for TLC—Methanol:Glacial acetic acid:Formic acid:Water (3:2.9:0.8:0.5)] M.pt. (186–188) IR (KBR pellets) cm−1 1) 1764 (C=O str.), 1619 (C=N str.), 1595 (C=C str.), 1277 (O–CH3), 1214 (C–O str., ester) 1H NMR (400 MHz, DMSO-d6) δ 7.91 (ddd, J = 7.5, 6.5, 1.5 Hz, 2H), 7.67 (d, J = 1.5 Hz, 1H), 7.47 (td, J = 7.5, 1.5 Hz, 1H), 7.09 (td, J = 7.5, 1.5 Hz, 1H), 6.97–6.88 (m, 2H), 6.86 (d, J = 1.5 Hz, 1H), 6.28 (d, J = 1.5 Hz, 1H), 3.97 (s, 3H), 3.80 (d, J = 0.7 Hz, 6H), 3.67 (s, 3H). 13C NMR (100 MHz, Chloroform-d) δ 171.85, 168.95, 167.67, 165.22, 158.95, 157.67, 148.53, 146.92, 133.72, 131.16, 128.84, 124.78, 124.78, 123.22, 122.87, 116.52, 113.70, 108.53, 104.92, 92.81, 90.38, 53.06, 52.81, 52.76 (d, J = 2.6 Hz), 51.65. m/z found for C26H22O9:478 (M+) 479 (M + 1)+. Anal calcd for C26H22O9: C, 65.27; H, 4.63; O, 30.10. Found: C, 65.27; H, 4.63; O, 30.10.
RU7C3 yield 71% Rf 0.7 [Mobile Phase for TLC—Methanol:Glacial acetic acid:Formic acid:Water (3:2.9:0.8:0.5)] M.pt. (165–166) IR (KBR pellets) cm−1 1) 1710 (C=O str.), 1637 (C=N str.), 1596 (C=C str.), 1258 (O–CH3), 1194 (C–O str., ester) 1H NMR (400 MHz, DMSO-d6) δ 7.98 (dd, J = 7.5, 1.5 Hz, 1H), 7.76 (d, J = 1.5 Hz, 1H), 7.30–7.20 (m, 5H), 6.91–6.86 (m, 2H), 6.23 (d, J = 1.5 Hz, 1H), 3.93 (s, 3H), 3.88 (d, J = 0.9 Hz, 6H), 3.69 (s, 3H), 2.93–2.84 (m, 2H), 2.73 (td, J = 7.0, 0.8 Hz, 2H). 13C NMR (100 MHz, Chloroform-d) δ 175.20, 170.26, 164.48, 160.33, 157.96, 156.95, 150.80, 149.32, 139.89, 128.47–128.31 (m), 126.14, 123.22, 122.87, 113.70, 112.82, 107.81, 99.41, 98.77, 53.17, 53.06 (d, J = 2.6 Hz), 52.69, 51.86, 34.56, 30.26. m/z found for C28H24O8:488 (M+) 489 (M + 1)+. Anal calcd for C28H24O8: C, 68.85; H, 4.95; O, 26.20. Found: C, 68.87; H, 4.90; O, 26.20.
Evaluation of biological activity
In vitro evaluation of xanthine oxidase inhibitory activity
The method opted to evaluate the inhibitory potential of rutin derivatives was a modified protocol of Sigma, done by UV-spectrophotometric method by using xanthine oxidase activity assay kit purchased from sigma (MAK078, sigma-aldrich.co, USA). The colorimetric product obtained in the form of hydrogen peroxide generated during the oxidation of XO was determined by a coupled enzyme technique, measured at 570 nm in a 96-well plate, using the plate reader EPOCH™ “MICROPLATE READER (BIOTEK).one unit of XO is defined as the amount of enzyme that catalyzes the oxidation of xanthine substrate, yielding 1.0 µmol of uric acid and hydrogen peroxide per minute at 25 °C. Reagents used were 44 µL of xanthine oxidase assay buffer, 2 µl xanthine substrate solution and 2 µl of Xanthine Oxidase enzyme solution. All the solutions mentioned above were mixed to prepare reaction mixture. The different concentrations of synthesized derivatives having final volume 50 µl were prepared in dimethyl sulfoxide (DMSO) and added to 96 well plate. To each well 50 µl of reaction mix was added and mixed well. After 2–3 min initial measurement was taken. The plates were incubated at 25 °C taking measurements at every 5 min. Allopurinol served as positive control. Absorbance at different time intervals was noted for further statistical analysis.
In vitro evaluation of antioxidant activity by DPPH method
The antioxidant potential of rutin derivatives was performed by DPPH method evaluated in the form of IC50 estimated using the ELISA plate reader EPOCH™ “MICROPLATE READER (BIOTEK). This method opted for evaluation of free radical scavenging activity of DPPH was based on modified procedure described by Dhiman et al. [36]. The tested compounds were prepared in methanolic solution and reacted with methanolic solution of DPPH at 37 °C. The reaction mixture was prepared in 96-well plate by adding 50 µL of sample, 50 µl of methanol and 50 µl of DPPH solution prepared in 0.1 mM methanol. The mechanism of action of DPPH assay was based on the fact that DPPH radical get reduced during its reaction with an antioxidant compound and results in changes of color (from deep violet to light yellow). The absorbance was read at 517 nm for 30 min at an interval of 5 min of using ELISA microplate reader. The mixture of methanol (5.0 ml) and tested compounds (0.2 ml) serve as blank. Ascorbic acid served as positive control.
Hydrogen peroxide scavenging (H2O2) assay
To compare and best evaluate the antioxidant potential of newly synthesized rutin derivatives, hydrogen peroxide assay was performed by the method described by Patel et al. [37] with some modifications. The solution of H2O2 (100 mM) was prepared via adding up different concentrations of synthesized derivatives ranging from 5 to 80 μg/ml to H2O2 solution (2 ml), prepared in 20 mM phosphate buffer of pH 7.4. Finally, the absorbance of H2O2 was measured at 230 nm after incubating for 10 min next to a blank reading of phosphate buffer without H2O2. For every measurement, a fresh reading of blank was taken to carry out background correction. For control sample containing H2O2 was scanned for absorbance at 230 nm. Results calculated as percentage of hydrogen peroxide inhibition was estimated by the formula [(Ab–At)/A0] × 100, where Ab is the absorbance of the control and At is the absorbance of compounds/standard taken as l-ascorbic acid (5–80 μg/ml) are shown in Table 5.
ADMET studies
The pharmacokinetic and pharmacological parameters of newly synthesized compounds were predicted with the help of Schrodinger suite. In-silico ADMET-related properties were computed using Qikprop application of Schrodinger software (Table 1). QikProp program generates set of physicochemically significant descriptors which further evaluates ADMET properties. The whole ADME-compliance score-drug-likeness parameter is used to predict the pharmacokinetic profiles of the ligands. This parameter determines the number of property descriptors calculated via QikProp which fall outside from the optimum range of values for 95% of noted drugs. Initially, all compound structures were neutralized before operated through Qikprop. The neutralizing step is crucial, as QikProp is unable to neutralize ligands in normal mode. Qikprop predicts both pharmacokinetically significant properties and physicochemically significant descriptors. It application run in normal mode which predicted IC50 value for blockage of HERG K + channels (log HERG), predicted apparent Caco-2 cell permeability in nm/s (QPPCaco), brain/blood partition coefficient (QPlogBB), predicted skin permeability (QPlogKp), prediction of binding to human serum albumin (QPlogKhsa) and predicted apparent Madin–Darby Canine Kidney (MDCK) cell permeability in nm/s (QPPMDCK). Solubility of drug was predicted as octanol/water partition coefficient (QPlogPo/w). Aqueous solubility of compound defined in terms of log S (S in mol dm−3) is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid.
Result and discussion
Molecular docking
To rationalize the structure activity relationship observed in this research and to foreknow the potential interaction of the synthesized compounds with XO, molecular simulation studies were carried out using Schrödinger suite (Schrödinger Release 2018-2, Schrödinger, LLC, New York, NY, 2018).The crystal structure of xanthine oxidase with PDB code 2E1Q was adopted for the docking calculations. Based on the docking score and binding energy calculation, top ranking derivatives were established and compared with the IC50 calculated from in vitro activity (Table 2). Important interactions were depicted as hydrophobic regions, hydrogen bonding, polar interactions and pi–pi bonding visualized in the active pocket of xanthine oxidase revealed through Site map application of Schrodinger suite. The derivatives having better docking scores than rutin were kept for further synthetic procedures and the remaining were discarded. To observe the binding interaction in detail, 3D poses of two most active compounds RU3a3 and RU3a1 were visualized and compared with native rutin and standard drug Allopurinol. The residues of binding pocket involved in the interaction were reported as GLN 1194, ARG912, MET1038, GLN1040, PHE798 and SER1080. Similar binding cavity was observed by Li et al. during the docking analysis of newly synthesized non-purine XO inhibitors [38].
Visual inspection of 3D poses of RU3a3 displayed a compact arrangement of polar and hydrophobic residues around the ligand forming a narrow passage in XO binding cavity with a docking score/binding score of − 13.244 and binding energy − 91.242 kJ/mol. An interesting pi–pi bonding was observed between benzene ring of phenyl hydrazine and hydrophobic residue PHE 798 of active site (Figs. 1, 2, 3). Along with this a strong hydrogen bonding was observed between OH group of rutinoside and polar residue GLN 1194 and negatively charged ARG 912 (Fig. 4). Similarly ARG 912 was found essential in the study of Shen et al. during the comparison of curcumin derivatives with quercetin and leuteolin [39]. Another hydrogen bonding was visualized between Chromene moiety and the residues of active site namely GLY 795 ad GLN585. Other hydrophobic amino acid residues closely placed within the cavity were observed as PHE 798, VAL1200, ALA1198, TYR 592, MET 1038 and ILE1229.

3D pose of RU3a3 inside the binding pocket

2D pose of RU3a3 inside the binding pocket

3D pose of RU3a3 showing hydrogen bonding (yellow) with GLN1194, ARG 912, GLY795, GLN 585 and π–π bonding (blue) with PHE798
On the other hand, during the visualization of RU3a1 the hydrogen bond was observed with OH group of phenyl ring and hydrophobic residue MET 1038 (Figs. 5, 6). Another hydrogen bond was found similar to RU3a3 between OH group of rutinoside and polar residue GLN1194 (Fig. 7). One more hydrogen bonding was observed between one of the OH group of dihydroxyphenyl ring and GLY1039. One more interaction was observed with the surrounding residue GLN 767 which forms a hydrogen bond with MOS 1328 (molybdenum metal ion) forming a closed channel to prevent the entry of substrate in the binding site. Other residues surrounding the ligand were observed as ARG 912, HIE 579, GLU 1261, ALA 1189 and ILE1198. When the 3D poses of these two compounds were compared with the native rutin structure, GLN 1194 forms 2 H-bonds, one with the C=O group of rutin and another with OH group of rutinoside (Fig. 8). The amino acid residues GLU1261 and GLN 1194 were found to be interacted similarly in the study of verbascoside by Wan et al. [40]. Beside this one H-bond was formed between OH group of chromene ring and MET1038. No pi–pi interaction was in the native structure rutin. In case of Allopurinol, the active site residues surrounding ligand were almost similar and placed near to MOS 1328. The hydrogen bond was observed between purine ring of allopurinol and GLN1194 (Fig. 9).

3D pose of RU3a1 inside the binding pocket

2D pose of RU3a1 inside the binding pocket

3D pose of RU3a1 showing hydrogen bonding with GLN 1194, MET1038 and GLY 1039

3D pose of rutin showing hydrogen bonding with GLN 1194 and MET1038

3D pose of allopurinol showing hydrogen bonding with GLN 1194
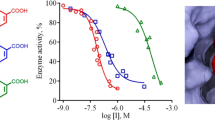
In-vitro xanthine oxidase inhibitory activity
In order to monitor the efficacy of different synthesized rutin derivatives, xanthine oxidase inhibitory activity was determined using xanthine oxidase activity assay kit purchased from Sigma-aldrich Co. Allopurinol (positive control) reported to inhibit xanthine oxidase was also screened under identical conditions for comparison. The inhibition ratios revealed the xanthine oxidase inhibitory activity of the synthesized rutin derivatives and the results were summarized in Table 3. As expected, these rutin derivatives exhibited remarkable activity comparable to the positive control. Based on the in vitro activity; it was observed that hydrazine (RU3a1–RU3a3) and anilline analogues (RU4b1–RU4b2) were considerably more effective than ester derivatives (RU7c1–RU7c3). All the compounds of hydrazine series (RU3a1–RU3a3) were effective with IC50-values ranging from 04.870 to 09.924 µM. Rutin hybridized with phenyl hydrazine demonstrated highest activity against xanthine oxidase. While thisemicarbazide and phenylthiosemicarbazide derivatives of rutin showed a slight decrease in activity indicating the role of sulfur group in diminishing the inhibition and NH–NH2 group in enhancing the activity of targeted enzyme. Surprisingly, substitution of NH–NH2 with NH2 group leads to decrease of inhibitory activity. Ester derivatives of rutin synthesized after the hydrolysis of rutin exhibited a weaker inhibition than the positive control Allopurinol.
The results of in vitro activity showed 80% similarity with the results of molecular docking with a few exceptions. In concordance with the screening and output of molecular docking RU3a3 comes out to be most active rutin derivative showing very good interaction with xanthine oxidase at molecular level. Elimination of rutinoside from rutin to synthesize ester derivatives results in a loss of potency with a threefold decrease of inhibitory potential.
Structure activity relationship (SAR)
Few interesting notions about the relationship of activity and structures of synthesized compounds emerged from the present research (Fig. 10): (A) Rutinoside moiety seems to be important for the activity, as deletion of this leads to loss of activity could be seen from xanthine oxidase inhibitory activity Table 3. Which shows RU3a3 (Having rutinoside group) exhibited highest activity with an IC50 value 04.870 µM among all the compounds and RU7c1 showed lowest activity and fivefold decrease of activity with an IC50 value 19.377 µM. (B) Hydrazine derivatives were found to be more effective than the aniline derivatives revealing the importance of NH–NH2 group. But substitution of sulfur group along with hydrazines decreases the activity as in RU3a3 and RU3a2 and substitution of phenyl group along with sulfur improves the activity (RU3a1). (C) Substitution with ester group leads to a decrease of inhibitory activity.

Structure activity relationship (SAR) of synthesized compounds
Enzyme kinetic analysis for XO-inhibitory activity
To determine the XO-inhibitory mechanisms of newly synthesized derivatives, we carried out kinetic studies of most active compound RU3a3 using Graph pad prism software. Firstly Michaelis–Menten curve was plotted for the enzyme activity at different concentrations of RU3a3 against different concentration of substrate (xanthine) Fig. 11.

Lineweaver–Burk plot for RU3a3 against different concentrations
Then double reciprocal plot (Lineweaver–Burk) analysis was done in the presence (0.25, 0.5, and 1.0 µM) and absence of RU3a3 from in vitro data generated during the oxidation of xanthine in presence of xanthine oxidase (Fig. 12). The x- and y axis intercepts of the Lineweaver–Burk plot were utilized to calculate Km and Vmax values of RU3a3 at different concentrations (Table 4).

Michaelis–Menten curve for RU3a3 at different concentrations
A concentration-dependent decrease of Vmax was predicted in contrast to Km value which was found to increasing when concentration of RU3a3 was increased. The intersection of linear straight lines drawn against each concentration was located at same point, suggesting that RU3a3 reacts in competitive manner during the inhibition of xanthine oxidase.
In-vitro evaluation of antioxidant activity by DPPH and H2O2 method
The antioxidant potential of newly synthesized compounds was evaluated by DPPH and Hydrogen peroxide radical assay. The comparative analysis of IC50 values for both the assays was done and the results were found to be impressive (Table 5). The results evinced a noteworthy inhibition of DPPH almost all the compounds when compared with the positive control ascorbic acid. In case of DPPH assay compound RU4b1 was demonstrated as most potent compound against oxidative stress caused because of free radicals having an IC50 value of 02.647 ± 0.09 µM. Along with this compound RU3a1 also showed very good antioxidant potential with an IC50 value of 05.021 ± 0.10 µM. When the detailed structure activity relationship was developed between these compounds, it was concluded that both the compounds having hydrazine linkage derived from phenyl hydrazine and phenyl thiosemicarbazide. Similarly, during the analysis of hydrogen peroxide assay all the compounds with hydrazines substitution showed very good antioxidant potential having IC50 in range of 04.146 ± 0.01 to 09.134 ± 0.35 (Fig. 7). Compound RU3a2 having phenyl thiosemicarbazide substitution showed potential antioxidant activity among all the derivatives. Along with this phenyl hydrazine substituted rutin derivative (RU3a3) also showed very good scavenging activity with an IC50 value of 06.561 ± 0.10. When the detailed structure activity relationship was developed between these compounds, it was concluded that both the compounds having hydrazine linkage derived from phenyl hydrazine and phenyl thiosemicarbazide.
Conclusion
Starting from the structures of rutin as anti-XO hit previously identified, different series of novel analogues were designed and synthesized to explore the structure–activity relationships associated with these xanthine oxidase inhibitors along with their antioxidant potential. Different structural elements were identified as essential for antioxidant and anti-XO properties, such as the presence of rutinoside (RU3a1, RU3a2 and RU3a3) comes out as important skeleton for the inhibitory potential, presence of hydrazone linker along with phenyl group, while the associated xanthine oxidase inhibitory effect was found to follow a different trend for the two series hydrazine (RU3a1–3) and ester derivatives (RU7c1–3). The newly synthesized derivatives with anti-oxidant and ani-XO IC50 values in the low micromolar range and good selectivity indexes were identified. Contemporary synthetic efforts are focused towards the insertion of the hydrazones and ester linkage by replacing the side linkage rutinoside of rutin with more stable groups while maintaining the overall length of new derivatives. Molecular docking provide an improved trail to design the new molecules with an avantgarde stability and potency.
Availability of data and materials
Not applicable.
References
Berry CE, Hare JM (2004) Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol 555(3):589–606
Moriwaki Y, Yamamoto T, Higashino K (1997) Distribution and pathophysiologic role of molybdenum-containing enzymes. Histol Histopathol 12(2):513–524
Klinenberg JR, Goldfinger SE, Seegmiller JE (1965) The effectiveness of the xanthine oxidase inhibitor allopurinol in the treatment of gout. Ann Intern Med 62(4):639–647
Yu KH (2007) Febuxostat: a novel non-purine selective inhibitor of xanthine oxidase for the treatment of hyperuricemia in gout. Recent Pat Inflamm Allergy Drug Discov 1(1):69–75
Battelli MG, Bolognesi A, Polito L (2014) Pathophysiology of circulating xanthine oxidoreductase: new emerging roles for a multi-tasking enzyme. Biochim Biophys Acta Mol Basis Dis 1842(9):1502–1517
Brass CA, Narciso J, Gollan JL (1991) Enhanced activity of the free radical producing enzyme xanthine oxidase in hypoxic rat liver. Regulation and pathophysiologic significance. J Clin Invest 87(2):424–431
Chambers DE, Parks DA, Patterson G, Roy R, McCord JM, Yoshida S, Parmley LF, Downey JM (1985) Xanthine oxidase as a source of free radical damage in myocardial ischemia. J Mol Cell Cardiol 17(2):145–152
Desco MC, Asensi M, Márquez R, Martínez-Valls J, Vento M, Pallardó FV, Sastre J, Viña J (2002) Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes 51(4):1118–1124
Kuppusamy P, Zweier JL (1989) Characterization of free radical generation by xanthine oxidase. Evidence for hydroxyl radical generation. J Biol Chem 264(17):9880–9884
Dawson J, Walters M (2006) Uric acid and xanthine oxidase: future therapeutic targets in the prevention of cardiovascular disease? Br J Clin Pharmacol 62(6):633–644
Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, Krotova K, Block ER, Prabhakar S, Johnson RJ (2005) Hyperuricemia induces endothelial dysfunction. Kidney Int 67(5):1739–1742
Kaynar H, Meral M, Turhan H, Keles M, Celik G, Akcay F (2005) Glutathione peroxidase, glutathione-S-transferase, catalase, xanthine oxidase, Cu–Zn superoxide dismutase activities, total glutathione, nitric oxide, and malondialdehyde levels in erythrocytes of patients with small cell and non-small cell lung cancer. Cancer Lett 227(2):133–139
Griguer CE, Oliva CR, Kelley EE, Giles GI, Lancaster JR, Gillespie GY (2006) Xanthine oxidase-dependent regulation of hypoxia-inducible factor in cancer cells. Cancer Res 66(4):2257–2263
Kanellis J, Kang DH (2005) Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Seminars in nephrology, vol 25. New York, WB Saunders, pp 39–42
Miesel R, Zuber M (1993) Elevated levels of xanthine oxidase in serum of patients with inflammatory and autoimmune rheumatic diseases. Inflammation 17(5):551–561
Wijermars LG, Bakker JA, de Vries DK, van Noorden CJ, Bierau J, Kostidis S, Mayboroda OA, Tsikas D, Schaapherder AF, Lindeman JH (2016) The hypoxanthine–xanthine oxidase axis is not involved in the initial phase of clinical transplantation-related ischemia–reperfusion injury. Am J Physiol Renal Physiol 312(3):F457–F464
Poles MZ, Bódi N, Bagyánszki M, Fekete É, Mészáros AT, Varga G, Szűcs S, Nászai A, Kiss L, Kozlov AV, Boros M (2018) Reduction of nitrosative stress by methane: neuroprotection through xanthine oxidoreductase inhibition in a rat model of mesenteric ischemia–reperfusion. Free Radic Biol Med 120:160–169
Osada Y, Tsuchimoto M, Fukushima H, Takahashi K, Kondo S, Hasegawa M, Komoriya K (1993) Hypouricemic effect of the novel xanthine oxidase inhibitor, TEI-6720, in rodents. Eur J of Pharmacol 241(2–3):183–188
Krakoff IH, Meyer RL (1965) Prevention of hyperuricemia in leukemia and lymphoma: use of allopurinol, a xanthine oxidase inhibitor. JAMA 193(1):1–6
Pacher PA, Nivorozhkin A, Szabó C (2006) Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev 58(1):87–114
Inkster ME, Cotter MA, Cameron NE (2007) Treatment with the xanthine oxidase inhibitor, allopurinol, improves nerve and vascular function in diabetic rats. Eur J Pharmacol 561(1–3):63–71
Sagor M, Taher A, Tabassum N, Potol M, Alam M (2015) Xanthine oxidase inhibitor, allopurinol, prevented oxidative stress, fibrosis, and myocardial damage in isoproterenol induced aged rats. Oxid Med Cell Longev. https://doi.org/10.1155/2015/478039
Min HK, Lee B, Kwok SK, Ju JH, Kim WU, Park YM, Park SH (2015) Allopurinol hypersensitivity syndrome in patients with hematological malignancies: characteristics and clinical outcomes. Korean J Intern Med 30(4):521
Quach C, Galen BT (2018) HLA-B* 5801 testing to prevent allopurinol hypersensitivity syndrome: a teachable moment. JAMA Int Med 178(9):1260–1261
Takano Y, Hase-Aoki K, Horiuchi H, Zhao L, Kasahara Y, Kondo S, Becker MA (2005) Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci 76(16):1835–1847
Mayer MD, Khosravan R, Vernillet L, Wu JT, Joseph-Ridge N, Mulford DJ (2005) Pharmacokinetics and pharmacodynamics of febuxostat, a new non-purine selective inhibitor of xanthine oxidase in subjects with renal impairment. Am J Ther 12(1):22–34
Nepali K, Singh G, Turan A, Agarwal A, Sapra S, Kumar R, Banerjee UC, Verma PK, Satti NK, Gupta MK, Suri OP (2011) A rational approach for the design and synthesis of 1-acetyl-3, 5-diaryl-4, 5-dihydro (1H) pyrazoles as a new class of potential non-purine xanthine oxidase inhibitors. Bioorg Med Chem 19(6):1950–1958
Becker MA, Kisicki J, Khosravan R, Wu J, Mulford D, Hunt B, MacDonald P, Joseph-Ridge N (2004) Febuxostat (TMX-67), a novel, non-purine, selective inhibitor of xanthine oxidase, is safe and decreases serum urate in healthy volunteers. Nucleosides Nucleotides Nucleic Acids 23(8–9):1111–1116
Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L (2006) Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet 45(8):821–841
Malik N, Dhiman P, Khatkar A (2017) In-silico design and ADMET studies of natural compounds as inhibitors of xanthine oxidase (XO) enzyme. Curr Drug Metab 18(6):577–593
Muhammad A, Arthur DE, Babangida S, Erukainure OL, Malami I, Sani H, Abdulhamid AW, Ajiboye IO, Saka AA, Hamza NM, Asema S (2018) Modulatory role of rutin on 2, 5-hexanedione-induced chromosomal and DNA damage in rats: validation of computational predictions. Drug Chem Toxicol 10:1–4
Roleira FM, Varela CL, Costa SC, Tavares-da-Silva EJ (2018) Phenolic derivatives from medicinal herbs and plant extracts: anticancer effects and synthetic approaches to modulate biological activity. Nat Prod Chem 57:115–156
Baldisserotto A, Vertuani S, Bino A, De Lucia D, Lampronti I, Milani R, Gambari R, Manfredini S (2015) Design, synthesis and biological activity of a novel Rutin analogue with improved lipid soluble properties. Bioorg Med Chem 23(1):264–271
Gullon B, Lú-Chau TA, Moreira MT, Lema JM, Eibes G (2017) Rutin: a review on extraction, identification and purification methods, biological activities and approaches to enhance its bioavailability. Trends Food Sci Technol 67:220–235
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47(7):1739–1749
Dhiman P, Malik N, Verma PK, Khatkar A (2015) Synthesis and biological evaluation of thiazolo and imidazo N-(4-nitrophenyl)-7-methyl-5-aryl-pyrimidine-6 carboxamide derivatives. Res Chem Intermed 41(11):8699–8711
Patel A, Patel A, Patel A, Patel NM (2010) Determination of polyphenols and free radical scavenging activity of Tephrosia purpurea linn leaves (Leguminosae). Pharmacogn Res 2:152–154
Li P, Tian Y, Zhai H, Deng F, Xie M, Zhang X (2013) Study on the activity of non-purine xanthine oxidase inhibitor by 3D-QSAR modeling and molecular docking. J Mol Struct 5(1051):56–65
Shen L, Ji HF (2009) Insights into the inhibition of xanthine oxidase by curcumin. Bioorg Med Chem Lett 19(21):5990–5993
Wan Y, Zou B, Zeng H, Zhang L, Chen M, Fu G (2016) Inhibitory effect of verbascoside on xanthine oxidase activity. Int J Biol Macromol 1(93):609–614
Acknowledgements
The authors are highly thankful to the Head, Department of Pharmaceutical Sciences, M. D. University, Rohtak for providing essential facilities to accomplish this research study. The authors are also thankful to Dr. Vinod Devaraji Application Scientist Schrödinger LLC for his support to carry out the computational work.
Funding
No funding received for this research work from outside sources.
Author information
Authors and Affiliations
Contributions
Authors NM and AK have designed, synthesized and carried out the xanthine oxidase inhibitory and antioxidant activity and the author PD, have carried out the docking simulations with in silico ADMET studies. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1.
HNMR spectra of compound RU3a3
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Malik, N., Dhiman, P. & Khatkar, A. In silico design and synthesis of targeted rutin derivatives as xanthine oxidase inhibitors. BMC Chemistry 13, 71 (2019). https://doi.org/10.1186/s13065-019-0585-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-019-0585-8