
Abstract
Background
Despite the combination of surgical resection, radio- and chemotherapy, median survival of glioblastoma multiforme (GBM) patients only slightly increased in the last years. Disease recurrence is definite with no effective therapy existing after tumor removal. Dendritic cell (DC) vaccination is a promising active immunotherapeutic approach. There is clear evidence that it is feasible, results in immunological anti-tumoral responses, and appears to be beneficial for survival and quality of life of GBM patients. Moreover, combining it with the standard therapy of GBM may allow exploiting synergies between the treatment modalities. In this randomized controlled trial, we seek to confirm these promising initial results.
Methods
One hundred and thirty-six newly diagnosed, isocitrate dehydrogenase wildtype GBM patients will be randomly allocated (1:1 ratio, stratified by O6-methylguanine-DNA-methyltransferase promotor methylation status) after near-complete resection in a multicenter, prospective phase II trial into two groups: (1) patients receiving the current therapeutic “gold standard” of radio/temozolomide chemotherapy and (2) patients receiving DC vaccination as an add-on to the standard therapy. A recruitment period of 30 months is anticipated; follow-up will be 2 years. The primary objective of the study is to compare overall survival (OS) between the two groups. Secondary objectives are comparing progression-free survival (PFS) and 6-, 12- and 24-month OS and PFS rates, the safety profile, overall and neurological performance and quality of life.
Discussion
Until now, close to 500 GBM patients have been treated with DC vaccination in clinical trials or on a compassionate-use basis. Results have been encouraging, but cannot provide robust evidence of clinical efficacy because studies have been non-controlled or patient numbers have been low. Therefore, a prospective, randomized phase II trial with a sufficiently large number of patients is now mandatory for clear evidence regarding the impact of DC vaccination on PFS and OS in GBM.
Trial registration
Protocol code: GlioVax, date of registration: 17. February 2017.
Trial identifier: EudraCT-Number 2017–000304-14.
German Registry for Clinical Studies, ID: DRKS00013248 (approved primary register in the WHO network) and at ClinicalTrials.gov, ID: NCT03395587. Registered on 11 March 2017.
Similar content being viewed by others


Background
Glioblastoma multiforme (GBM) is the most frequent and aggressive malignant primary brain tumor. The yearly incidence is 3–4 per 100,000 adults [1]. The established therapeutic standard in the first-line therapy for GBM combines maximal safe resection, fractionated radiotherapy with concomitant alkylating temozolomide (TMZ) chemotherapy followed by adjuvant TMZ. This multimodal approach has improved the survival of patients significantly. Nevertheless, the prognosis of newly diagnosed GBM patients is still poor. Median overall survival (OS) is only 14.6 months, the 2-year survival rate is 27.2%, and disease recurrence is universal, with virtually all patients dying within 1.5 years after relapse [2, 3]. Thus, there is a clear need for novel therapeutic modalities.
Dendritic cell (DC)-based, active anti-tumoral cellular immunotherapy aims at generating autologous cytotoxic effector T cells, which kill tumor cells specifically. Because of their central role in initiating such T-cell responses, DC are used as cellular adjuvant for application of tumor antigens in tumor vaccines [4, 5]: Patients are vaccinated with tumor antigen-loaded DC, with the concept that they will migrate to local lymph nodes, present tumor antigen-derived peptides on human leukocyte antigens (HLA) and initiate an anti-tumoral T-cell response.
DC-based vaccination therapy was first applied in a clinical trial in 1996 for B-cell lymphoma [6], but only in 2006 has the clinical efficacy of this approach been proven in a phase III trial for hormone-refractory prostate cancer patients [7]. For GBM, since the first case report in 2000 [8], 38 studies and case reports have been published, initially applying DC vaccination in recurrent disease, but more recently as part of first-line therapy in newly diagnosed patients as well [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46]. Close to 500 GBM patients have been treated with DC vaccination in these studies, with the age of patients ranging from 1 to 80 years.
Previous studies have documented feasibility and safety. There have been no reports of severe side effects (≥ grade 3) attributable to vaccination, except for one patient with gross residual tumor after surgery, who suffered from peritumoral edema, which was controllable by glucocorticoids [16, 23]. Frequently observed mild and easily controllable toxicities (≤ grade 2), which may be attributable to DC vaccination, have been injection site reactions with itching, pain, erythema, induration and lymph node swelling as well as flu-like symptoms, fever, fatigue, myalgia, headache, edema and meningeal irritation. Thus, overall toxicity of DC vaccination therapy is limited, and vaccination has also no severe impact on the quality of life of patients. Indeed, the quality of life of patients has been reported to remain stable during vaccination therapy [27, 28, 42] or even to improve [35, 37, 44].
Induction of antigenic, target-directed immune responses have been observed in the course of vaccination, with detection of interferon (IFN)-γ responses being most informative [25], but anti-tumoral cytotoxic responses [10, 17,18,19] and an increase in tetramer positive cytotoxic T cells [18, 31, 36, 44, 45] have been reported as well. Several studies identified immunological responders based on delayed-type hypersensitivity-like reactions [20], IFN-γ responses [20, 25, 30, 39, 43, 45, 46] or cytotoxic responses [19] and reported longer survival times of responders [19, 20, 25, 30, 39]. Moreover, long-term survivors with durable IFN-γ responses have been identified [43]. Such associations of anti-tumoral immunity with survival are in line with the proposed mechanism of action of vaccination therapy and strong arguments for the efficacy of this type of active immunotherapy in GBM.
Prolonged survival of vaccinated patients has been reported compared to matched or historic controls [10, 18,19,20, 26, 29, 32, 40, 43]. In a recent meta-analysis of 9 studies, Cao et al. [47] concluded that vaccination significantly improves OS (1–5 years) as well as PFS rates (1–4 years), e.g., OS rates at 4 years were 20% and 1%, at 5 years 14% and 0% for vaccinated and non-vaccinated patients, respectively.
For newly diagnosed GBM, in a recent randomized phase II trial with 34 patients [35], the OS rate at 24 months (44.4%) as well as the median OS (31.9 months) of the vaccination group compared favorably to the control group (18.8%; 15.0 months). In another controlled study including 25 patients, median OS (17.0 vs. 10.0 months) as well as OS rates at 2 years (7.7% vs. 0%) compared favorably for the vaccinated patients [37]. In 16 non-randomized studies, the median OS of newly diagnosed GBM patients ranged from 11.0 to 38.4 months [10, 17, 19, 24, 26, 28,29,30, 32, 34, 38,39,40, 43, 45, 46], with one [10] of the three studies [10, 24, 29] reporting the lowest median OS (11.0–15.0 months), nevertheless describing an improved OS compared to matched controls (15.0 vs. 8.4 months).
Thus, results of previous DC vaccination trials have been encouraging, although they cannot yet provide robust evidence of clinical efficacy. Moreover, DC vaccination can be easily integrated into the current first-line therapy, and there is a rational for such an integration besides retaining a therapy with proven clinical efficacy for the patients; it may allow exploiting synergies between the treatment modalities: (1) after resection/radio-/chemotherapy, patients are in a state of minimal residual disease, which is probably beneficial for immunotherapy because of the lower tumor load and the depletion of immunosuppressive cells [19, 23], (2) TMZ appears to improve immunological responsiveness [48,49,50], probably by reducing regulatory T cells (Treg) [50, 51] and interfering with their recruitment to the tumor [52], (3) the recovering lymphocyte compartment after chemotherapy appears to be beneficial for induction of anti-tumoral responses [53], (4) dying tumor cells after radiochemotherapy result in local pro-inflammatory conditions and a release of tumor antigens, which could improve homing of tumor-specific effector T cells to the brain tumor [54] and (5) there is an increased responsiveness to TMZ chemotherapy after DC vaccination [17]. Therefore, it now appears mandatory to determine efficacy of DC vaccination in a randomized, controlled trial on a large cohort of newly diagnosed GBM patients with standard therapy as comparator. If the hypothesis is proven correct that DC vaccination as add-on to the standard therapy in GBM significantly prolongs survival with acceptable toxicity and without significantly reducing the quality of life of patients, this trial will contribute to its implementation as therapy in newly diagnosed GBM. Otherwise, clarification of efficacy is also urgently needed for counseling GBM patients, who are aware of the apparently promising results of this therapeutic approach.
Methods
Study design
This is a national, prospective, multicenter, open-label, randomized phase II study with two parallel groups, with the participating hospitals being located in the state of North Rhine-Westphalia in the proximity of Düsseldorf (a list of participating centers can be obtained from the sponsor or the registry). In the active recruitment period of 30 months, 136 patients with newly diagnosed, isocitrate dehydrogenase (IDH) wildtype, primary GBM [55] will be randomized at a 1:1 ratio after near-complete resection and successful production of tumor lysate. For a patient to be enrolled, all eligibility criteria (see below) must be met and free vaccine production capacity has to be available (enrollment may have to be paused). Enrollment of a patient is initiated by an investigator submitting by fax the Randomization Request Form to the Coordination Center for Clinical Trials, which will perform the randomization and inform the investigator of the outcome and allocation of the patient to the control or the vaccination arm of the study. The randomization list has been generated by the Coordination Center for Clinical Trials, using randomly selected block sizes of 4, 6, 8 and 12 patients. It will be centrally applied with concealment by the Coordination Center for Clinical Trials. The assignment to the two groups will be stratified according to O6-methylguanine-DNA-methyltransferase promotor methylation status, because it is an independent prognostic factor in TMZ chemotherapy [2, 56].
All patients will receive the current standard of care with proven clinical efficacy [2, 3, 57] for newly diagnosed GBM consisting of (1) FGS, (2) fractionated RT (60 Gy, 2 Gy/day, 5/7 days, 6 weeks) with concurrent TMZ chemotherapy (75 mg/m2/day, 6 weeks) and (3) subsequent adjuvant TMZ chemotherapy (150–200 mg/m2/day, days 1–5 of 28-day cycle, six cycles). TMZ dose reduction due to toxicity follows standard rules: Concomitant TMZ chemotherapy will be interrupted when the absolute neutrophil count is 500–1500/μl or the platelet count is 10,000–100,000/μl or non-hematological toxicity grade 2 occurs. Concomitant TMZ chemotherapy will be discontinued when the absolute neutrophil count is < 500/μl or the platelet count is < 10,000/μl or non-hematological toxicity grade 3 or 4 occurs. Adjuvant TMZ chemotherapy dose will be reduced when the absolute neutrophil count is < 1000/μl or the platelet count is < 50,000/μl or non-hematological toxicity grade 3 occurs. Adjuvant TMZ chemotherapy will be discontinued when hematological and non-hematological toxicities persist despite the lowest dose of TMZ (75 mg/m2/day).
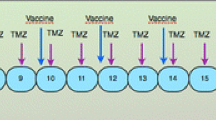
In the control group, patients receive this standard treatment with proven clinical efficacy (comparator). Patients in the vaccination arm of the study will receive as add-on up to seven tumor lysate-loaded, mature DC vaccines (2–10 × 106 cells each applied by intradermal injection): four priming vaccines in weekly intervals during the period between concomitant radiochemotherapy and adjuvant chemotherapy and three monthly boosting vaccines in the third week of the first three 28-day cycles of adjuvant TMZ (Fig. 1). Availability of less than three vaccines is regarded as treatment failure. Vaccines will be applied irrespective of whether concomitant TMZ has to be terminated or adjuvant TMZ has to be reduced or terminated due to toxicity. Corticosteroids should be avoided or kept to a minimum 1 week before until 1 week after vaccination. In case of temporary need for high-dose corticosteroids 1 week before vaccination (> 8 mg/day dexamethasone or equivalent), vaccination has to be postponed. Any other anti-cancer treatment, including agents with only suspected anti-cancer effects, any other investigational or experimental treatment, any other active immunotherapy or any treatment that interferes with immune function, including persistent immunosuppressive therapy (e.g., for rheumatism) should be avoided or kept to a minimum during the treatment period. If a patient misses two or more of the priming vaccinations, this is regarded as treatment failure. If one priming vaccination or one, two or all three boost vaccinations are missed, it will be attempted to add the missed vaccinations (maximum: 3) as additional boost vaccinations, always in the third week of subsequent adjuvant TMZ cycles (cycles 4–6).

GlioVax study design. After resection, patients are randomized 1:1 into the vaccination and standard arms of the study. Patients in the standard arm are treated with fractionated radiotherapy (RT) and concomitant temozolomide (TMZ) chemotherapy, followed by 6 cycles of adjuvant TMZ. Patients in the vaccination arm receive 4 priming vaccinations and 3 boosting vaccinations with tumor-lysate-loaded, mature DC as add-on therapy to the standard therapy, between radiochemotherapy and adjuvant TMZ and in the first 3 cycles of adjuvant TMZ, respectively. The overall intervention period in both arms is 38 weeks, follow-up is 2 years. After screening (visits − 3 to − 1), 18 study visits (visit 0 (baseline) to visit 17) and 8 3-monthly follow-up visits are planned, to assess safety, efficacy and quality of life. Visits indicated in blue are in the vaccination arm only. Other visits are for patients in both arms of the study
Patients are informed verbally and on the basis of the informed consent form in-depth about the importance of vaccination being performed in line with the protocol. They receive the contact information of the coordinating investigator and are invited to contact him in case of any questions. Standard therapy will be performed in all participating hospitals. Production and application of vaccines will be in Düsseldorf only. The intervention period is 38 weeks, follow-up is 24 months. Overall, a duration of 63 months (first patient in to last patient out 54 months) is anticipated for the trial (Fig. 2).

Visits for enrollment, treatment and assessments. After screening for eligibility and allocation (visits − 3 to − 1), 18 study visits (visits 0–17) are planned for treatment and assessments during the treatment period, followed by 8 additional visits (visits 18–25) during the 2-year follow-up. 1Resection is performed in week 0 (visit − 2); 2weeks 5–10: 6 cycles of concomitant radiochemotherapy; 3weeks 15, 19, 23, 27, 31, 35): 6 cycles (days 1–5 of 28-day cycle) of adjuvant temozolomide (TMZ) chemotherapy; 4weeks 11–14: 4 priming vaccinations; 5weeks 17, 21, 25: 3 boost vaccinations; 6weeks 16, 28, 37: magnetic resonance imaging (MRI) scans; 7weeks 5, 10: clinical, clinical chemistry, hematological, hemostaseological examinations and adverse events (AE), concomitant medication and KPS assessments; 8weeks 11–14: clinical, clinical chemistry, hematological, hemostaseological examinations and AE and concomitant medication assessments; 9weeks 15, 17, 21, 25, 29, 33, 38: clinical, clinical chemistry, hematological, hemostaseological examinations and AE and concomitant medication assessments; 10week 10: neurological performance and quality of life assessments; 11weeks 15, 29, 38: KPS, neurological performance and quality of life assessments; 12tumor tissue and blood for translational research studies are sampled during resection, leukapheresis and at the 7 time points of vaccination; 13pregnancy tests are performed at monthly intervals until 6 months after the treatment period; *vaccination arm only
A formal interim efficacy analysis will be performed when approximately 50% of the expected 87 events have occurred in the treatment and control groups combined. At the interim, a group-sequential methodology will be applied to the analysis. The O’Brien-Fleming approach [58] will be used and overwhelming efficacy will be concluded if the two-sided p value of the log-rank test for the difference in median OS between treatments is < 0.003. The decision to close the trial based on overwhelming or inferior efficacy of vaccination will be taken by the sponsor together with the coordinating investigator after critically assessing ethical and safety aspects.
Eligibility criteria
All inclusion and exclusion criteria (Table 1) will be evaluated by the local investigators (neurosurgeons and/or neuro-oncologists) and in case of residual tumor volume after surgery and diagnosis/tumor cell content of tumor sample confirmed by central neuroradiologist and neuropathologist, respectively.
Study endpoints
The primary objective of the study is to determine whether survival of newly diagnosed GBM patients treated with lysate-loaded, mature DC vaccines as add-on to the standard of care is superior to the treatment with the standard of care alone. The primary efficacy endpoint is OS measured from the day of surgery until death. Secondary objectives are comparing (1) progression-free survival (PFS), (2) 6-, 12- and 24-month OS and PFS rates, (3) the safety profile, (4) overall and neurological performance and (5) the quality of life between the two treatment groups.
Following the baseline visit, 17 visits during the treatment period of 38 weeks and 8 visits during the 2-year follow-up period have been planned to assess primary and secondary endpoints. Study visits have been scheduled as close as possible to those of the standard treatment to limit any additional burden for the patients. Tumor progression will be assessed by magnetic resonance imaging (MRI) according to modified RANO (response assessment in neuro-oncology) criteria [59, 60]. Following standard protocol, MRI examinations after the 72-h post-surgical MRI scan will start in week 16 (6 weeks after completion of radiochemotherapy) and will be performed thereafter in 3-monthly intervals during the treatment as well as in the follow-up periods.
At each visit (for visit plan see Fig. 1), clinical examinations as well as laboratory assessments will be performed to monitor safety, with toxicity coded according to the Medical Dictionary for Regulatory Activities (MedDRA) and graded according to the National Cancer Institute (NCI) common terminology criteria for adverse events 4.03 (CTCAE 4.03). All adverse events (AE) occurring in the course of the trial will be documented. Each AE is rated by the investigator whether a correlation with the trial drug can be assumed (adverse drug reaction; ADR) or not. All severe adverse events (SAE), irrespective of whether associated with the trial drug (severe adverse drug reaction; SADR) or not and whether rated by the coordinating investigator as unexpected or a suspected unexpected severe drug reaction (SUSAR) will be reported to the sponsor within 24 h, and every SUSAR will be reported by the sponsor to the National Competent Authority and the Ethics Committee. Pregnancies are documented on a separate Pregnancy Report Form and reported to the sponsor immediately after learning of the pregnancy.
The Karnofsky performance status, neurological performance status (Minimal Mental State Examination-2; [61]), quality of life (EORTC QLQ-C30 and QLQ-BN20 questionnaires; [62, 63]) and psycho-oncological distress (Distress Thermometer [64] and Hospital Anxiety and Depression Scale [65] will be assessed.
Patients withdrawn from the trial prematurely by the investigator or at their own request are asked to participate in the Termination Visit (clinical, clinical chemistry, hematological, hemostaseological examinations, MRI, AE assessment, overall and neurological performance, quality of life) and to the 3-monthly follow-up according to study protocol until the end of the follow-up period. At a minimum, all AE should be followed until they are fully and permanently resolved or have stabilized. If the patient withdraws or declines consent for disclosure of future information, no further evaluations will be performed, and no additional data will be collected, but the use of any data collected before of the withdrawal will be retained, to be used further as may be necessary, e.g., to assess efficacy and safety.
Safety and efficacy data will also be assessed by an independent Data Safety and Monitoring Committee (DSMC) composed of a neurosurgeon, a neuro-oncologist, a neuropathologist and a biostatistician, who had no involvement in the design of the study, are not involved in its conduct other than through their role on the DSMC, and who have no financial interest in the outcome of the study. Assessments will be based on semiannually safety reports and the interim report. The DSMC may recommend to the sponsor at any time during the trial discontinuation of the trial or modification of the protocol for safety reasons. It may also recommend to stop the trial in case of insufficient enrollment or overwhelming/inferior efficacy. The DSMC Charter provides a detailed description of the responsibilities and activities of the DSMC.
Data management, monitoring and auditing
All data will be collected in electronic case report forms (eCRF). The eCRF will be implemented in a Good Clinical Practice (GCP)-compliant clinical data management system with electronic data capture functionality, which provides the capability to perform the major data management activities within a consistent, auditable and integrated electronic environment (query management, data entry, data validation). Range, validity and consistency checks will be implanted in the eCRF for application during data entry. All data entry, modification or deletion will be recorded automatically in an audit trail indicating the original value, the new value, the reason for change, who made the change and when the change was made. A digital signature is implemented and included in the audit trail. Periodically, central monitoring of the data is performed and actions are taken to ensure data quality (queries, re-training). Implementation of the eCRF has been performed by data managers in cooperation with biostatisticians from the Coordination Center for Clinical Trials. All procedures are documented in the protocol and in the Data Management Manual.
Source data remain in the respective hospital information system. Source documents, in particular documents with plain-text names of the participating patients, are kept under strict control of the investigator at the trial site. They will not be transferred to the sponsor or other persons that are not authorized by the patient in the data protection declaration part of the informed consent form. Data recorded in the eCRF are pseudonymized; the Patient Identification List is kept under strict control at the individual trial sites.
Periodic monitoring of the trial will be performed on site in the trial centers by clinical research associates to ensure the safety of the trial participants, the trial itself as well as the correctness of the collected data. They will check the availability of the patient’s informed consent, adherence to eligibility criteria, adherence to treatment according to the trial protocol, safety parameters and completeness of the trial documents in the trial center. The monitor confirms completed source data verification by electronic signature in the eCRF. Monitoring is described in detail in the Monitoring Manual. The on-site trial monitoring is combined with central monitoring with regular data checks.
To ensure sponsor oversight, audits will be performed by an independent auditor at the sponsor’s request. Especially, the vaccine production and its documentation as well as the correct delivery of the tumor and blood samples will be audited. In case of non-compliance of the study centers an audit can also be performed.
Advanced therapy medicinal product: DC vaccine
The DC vaccines for the GlioVax trial are produced specifically for each patient according to established standard operating procedures with production permission by the local authorities. The vaccines consist of two autologous cellular components: (1) tumor lysates as a whole-cell source of tumor antigens and (2) monocyte-derived, mature DC.
Lysates are produced from autologous tumor material of the patients obtained from fluorescein-guided surgery (FGS). FGS is an established standard surgical procedure for GBM [57]. Because the “solid” part of the tumor can be identified intraoperatively, high-purity tumor material can be obtained, and only samples with a tumor cell content of at least 60% are used for vaccine production. After resection, tumor material is stored frozen at − 80 °C until processing, which involves mechanical dissection and repeated freeze/thaw cycles to generate the lysate.
DC are generated from monocytes enriched immunomagnetically from non-mobilized leukapheresis products. A two-step serum-free culture system is used, to successively generate immature and mature DC [66, 67]. Monocytes are cultured for 6 days in Teflon bags in the presence of granulocyte-macrophage-colony-stimulating factor and interleukin-4 to generate immature DC, which are loaded with tumor antigens by adding the autologous tumor lysate to the cultures. Maturation of DC is induced during an additional 3-day culture period, using the classical pro-inflammatory maturation cocktail described by Jonuleit et al. [68], containing tumor necrosis factor α, interleukin-1β, interleukin-6 and prostaglandin E2, which allows generation of CD83+, fully mature DC with high purity and potent T-cell stimulatory and TH1-polarizing activity. After ex-vivo generation of the tumor-lysate-loaded, mature DC, they are stored frozen until the day of application, on which they are thawed, washed, resuspended in 0.9% sodium chloride solution and transferred into a 1-ml syringe for immediate application.
Sample size considerations and statistical analysis
The sample size calculation is based on the primary outcome OS. A median OS of 14.6 m has been reported for the control intervention [2, 3]. In eight recent smaller phase I/II trials on newly diagnosed GBM treated with lysate-loaded DC as add-on to radiochemotherapy, median OS of 35.9 months [32], 34.4 months [17], 31.9 months (control group 15.0 months) [35], 30.5 months [45], 28.0 months [30], 27.4 months [38], 24.0 months [28], 18.4 months [34] and 17.0 months (control group 10.5 months) [37] have been reported. Based on a critical appraisal of these data, taken into consideration potential treatment dilution effects for the results from the smaller phase I/II trials, we have assumed a prolongation of OS of 12 months for the vaccination group (OS 26.6 months). When the sample size in each group is 61, with a total number of events required of 87, a 0.050-level two-sided log-rank test for equality of survival curves will have 80% power to detect the difference between a median OS in the control group of 14.6 months vs. 26.6 months in the test group (hazard ratio for death, 0.55).
In order to compensate for dropouts (i.e. patients dropping out prior to 24 months without having an event) the total sample size is planned at 136 patients (i.e. an approximately 10% dropout rate). Mainly due to a tumor content of samples for vaccine production below 60% or unsterile tumor material (7/37 (19%); determined at the Institute for Transplantation Diagnostics and Cell Therapeutics, Düsseldorf) or residual tumor volume above 5 ml upon radiological evaluation (17%; [57]), it is anticipated that approximately 35% of GBM patients will fail to meet inclusion/exclusion criteria after resection. Therefore, 210 patients have to be screened to allow for enrollment of 136 patients in the trial.
Each year, about 450–500 patients with newly diagnosed GBM are treated in the participating centers, of which about 280 (60%) would fulfill the inclusion/exclusion criteria of the trial. It is anticipated that about 260 of these patients would agree to participate and that it would be possible to recruit 210 patients in the 30-month recruitment period. Thus, recruitment of 136 patients seems feasible. The recruitment rate would be about one patient/center/month and centers would have to assess one to two/patients/month for eligibility.
Evaluation of results will be based on the Statistical Analysis Plan. Efficacy is measured by OS computed from the day of surgery until event or censored at last follow-up according to the Kaplan-Meier method with two-sided log-rank statistics for comparison. Primary efficacy analysis will be based on the intention-to-treat population; patients lost to follow-up are censored at the time of last contact. If required, a model-based Bayesian analysis for imputation of missing data will be used, and missing values imputed using the posterior predictive distribution of the model. In addition, a sensitivity analysis based on the per-protocol population will be performed.
All patients will be evaluated for safety; assessment of safety will be based on descriptive and explorative methods. For explorative analyses of secondary endpoints, classical statistical tests will be applied (e.g., log-rank test for survival data (PFS), chi-square/Fischer’s exact test for categorical data and t test/Mann-Whitney U test for continuous data). In addition, 95% confidence intervals will be calculated when applicable.
A Cox regression model will be used for explorative analysis of overall survival and progression-free survival with the covariates (1) treatment group, (2) recursive partitioning analysis classes I–IV as defined by Mirimanoff et al. [69] and de Vleeschouwer et al. [70], which already have been documented to influence outcome of standard as well as of vaccination therapy, the prognostic markers, (3) MGMT promotor methylation status, (4) extent of resection (complete vs. near-complete), (5) age, (6) center, (7) dexamethasone medication and (8) the therapeutic intervention in case of tumor progression. Subgroups of vaccinated patients will also be defined based on their immunological responsiveness as well as immunosuppressive scores (see below).
Discussion
Trial design
The trial design attempts to minimize parameters, which have been identified to affect clinical and immunological outcome, in order to generate informative results on efficacy of DC vaccination in GBM. Only adults with confirmed newly diagnosed primary GBM after near-complete resection will be included, with all patients undergoing standard radio/TMZ chemotherapy. Moreover, DC vaccination with high-purity, tumor-lysate-loaded, mature DC will be fully integrated into standard therapy.
Randomization will be the main method against bias. DC vaccines are produced specifically for the individual patient, using the patient’s own tumor and leukocytes as starting material. We have considered it to be unethical to subject the patients of the control group to the potential risks and the stress of leukapheresis merely for blinding purposes. Thus, only patients of the vaccination group are subjected to leukapheresis and, therefore, there will apparently be different procedures applied to patients in the control and vaccination groups, which also requires different logistics. Therefore, blinding the study has not been possible.
Tumor-induced immunosuppression
After activation by the DC, effector T cells can cross the blood-brain barrier and exert their effector function in the brain [71], with a contribution of luminal antigen presentation by endothelial cells to identify the specific site for effector T cells to enter the brain parenchyma [54] and a final tuning of T-cell effector functions by the brain microenvironment [71, 72]. Thus, the basic pre-requisites for DC vaccination therapy of GBM are in place, and numerous animal studies could confirm that it is a a promising therapeutic approach in brain tumors [73,74,75,76,77]. However, there is also tumor-induced immunosuppression, and several contributing mechanisms have been identified [78, 79], including an increase in regulatory T cells in the blood as well as in the tumor microenvironment, which is associated with impaired T-cell responsiveness [80]. Such immunosuppression may hamper DC vaccination therapy and, therefore, has to be overcome, to improve or even allow efficacy. In GBM, it correlates with the size of the tumor, and surgical cytoreduction can restore T-cell responsiveness, at least partially [81,82,83,84]. Certain chemotherapeutics, including TMZ, which is used in the standard treatment of GBM, have also been reported to reduce Treg in the periphery as well as in the tumor [50,51,52], which might be important to increase the vaccine efficacy [85, 86]. Thus, there is a rationale to maximally reduce the tumor burden, prior to DC vaccination therapy and combine it with TMZ chemotherapy.
Lysate as a source of tumor antigens
For the GlioVax trial, autologous tumor lysates are used as a whole-tumor cell source of tumor antigens. Even when high-purity tumor tissue is collected by FSG, there is an abundance of normal self-proteins in tumor lysates. Nevertheless, induction of autoimmunity or other severe side effects attributable to the use of lysates for vaccine production have not been reported. Therefore, lysates can be considered a safe source of tumor antigens.
Lysates most likely will contain multiple tumor antigens, which are present in the individual tumor of a patient (including the various cellular subsets within the tumor), i.e. the patient’s full antigenic repertoire, ensuring antigenic diversity, thereby reducing the risk of escape of tumor-antigen-loss variants [87]. Since the respective proteins are endogenously processed in the DC, presentation on HLA-class I and II molecules is possible and independent of the HLA type of the patient, thereby allowing induction of cytotoxic as well as TH-responses at the same time, which is a pre-requisite for the development of an efficient cytotoxic T-cell response. Furthermore, lysate may provide in addition to the tumor antigens the necessary signals for guiding effector T cells to the brain [88]. Whether lysates are superior to molecularly defined tumor antigens in inducing anti-tumoral immune responses or more beneficial clinically in GBM is currently unknown. However, Neller et al. concluded from the analysis of 173 published immunotherapy trials on various tumor entities, including melanoma, renal cell and hepatocellular carcinomas, lung, prostate, breast, colorectal, cervical, pancreatic and ovarian cancers, a higher objective response rate (8.1% vs. 3.6%) when whole-tumor-cell antigens compared to molecularly defined tumor antigens were used [89]. Therefore, lysates represent an immunologically potent source of tumor antigens.
Maturation state of DC
In the GlioVax trial, mature DC rather than immature or semi-mature DC are used as vaccine cells, because (1) they have potent T-cell stimulatory activity [67], (2) they polarize responses towards T-helper cell (TH1) [67], which is required for efficient induction of effector cytotoxic T cells [90], (3) they express CC-motive chemokine receptor type 7 [67]), which is required for lymph node homing [91, 92], (4) they are phenotypically stable upon withdrawal of cytokines [67], (5) they are resistant against immunosuppressive cytokines like interleukin(IL)-10 or transforming growth factor(TGF)-β2, which may be produced by the tumors [93, 94] and (6) only immature and semi-mature DC, but not fully mature DC have been implicated in tolerance induction [95, 96], which would be detrimental to the intended induction of anti-tumoral immune responses. Indeed, Yamanaka et al., who used mature as well as immature DC in their study, reported a trend towards a better outcome in GBM patients vaccinated with the mature DC [20] and de Vries and colleagues reported that tumor-antigen-loaded, mature DC are superior to immature DC in the induction of immunological responses in melanoma patients [97]. Moreover, Dhodapkar et al. showed a decline of the influenza-matrix-peptide-specific T-cell response in healthy individuals after vaccination with matrix-peptide-loaded, immature DC and urge caution with the use of immature DC to enhance tumor immunity [98].
Route of vaccine administration
Depending on the route of application, DC can be detected in different organs. Intravenous application results in a rapid enrichment in liver, lung and kidney, but is highest in spleen, whereas after subcutaneous application, there is a marked accumulation of DC in the draining lymph nodes, with a preferential paracortical localization in the T-cell areas [99]. When intradermal application is used instead, even more DC reach the T-cell areas of lymph nodes in mice [100] as well as in humans [101], with only mature, but not immature DC, efficiently migrating to the lymph nodes in melanoma patients [102], probably due to their expression of the CC-motive chemokine receptor 7, which is missing on immature DC [91, 92]. DC can be detected already after 30 min in the lymph nodes; they are at a maximum after 48 h, and they appear to persist for up to 14 days [103,104,105]. Although only 4% of injected DC may reach the lymph nodes, this appears to be sufficient for effective induction of anti-tumoral immune responses [106, 107]. Due to these advantages, intradermal application of DC vaccines is used in the GlioVax trial.
MRI and pseudoprogression
Standard radiochemotherapy of GBM may result in a transient increase in contrast enhancement in a large fraction of patients, which will subside eventually without any change in therapy [59]. Therefore, early MRI diagnostics at week 10 after completion of radiochemotherapy is not feasible. Similarly, immunotherapy may result in inflammatory reactions, which mimic tumor progression and may even include the appearance of “new lesions” on MRI scans [60]. This pseudoprogression, irrespective of whether due to standard radiochemotherapy or immunotherapy, is difficult to differentiate from true tumor progression. Moreover, immunotherapy may result in delayed responses, i.e. patients may still benefit from therapy although showing signs of progressive disease initially [60]. To minimize premature declaration of tumor progression and discontinuation of therapy, in the situation of suspected progression, thus when patients meet the radiological RANO criteria for progression initially, patients may continue study medication until progression has been confirmed on a subsequent MRI scan performed within 4–6 weeks [59]. However, there still may be uncertainty regarding progression. Due to the sometimes long-lasting nature of particularly immune-related effects, distinguishing progression from pseudoprogression unequivocally can require a prolonged (≥ 3 months) observation period as summarized by Okada et al. [60]. Therefore, subsequent confirmatory MRI scans in 4–6-week intervals will be performed and patients can continue study medication as long as the clinical and neurological status of the patient has not been worsening substantially and the study medication is well tolerated, does not interfere with therapeutic interventions for imminent complications, and the investigator assesses an overall possible clinical benefit [60]. Re-operation will be considered, to allow for final histological confirmation of tumor progression [59, 60], if the suspected lesion if surgically accessible and it is the investigator’s medical judgement that the patient may benefit from re-operation as reported previously [108, 109]. The use of additional imaging techniques (e.g., fluoroethyl-tyrosine positron-emission tomography (FET-PET)) to confirm MRI findings is also possible. The exact time of progression is defined retrospectively, when pseudoprogression and pseudoresponses can be excluded. Further treatment in case of progression is at the discretion of the investigator.
Translational research program
GBM-mediated immunosuppression may interfere with the development of potent anti-tumor immune responses and, thus, could be inversely correlated with clinical and immunological outcome of DC vaccination [79, 110, 111]. Moreover, detection of induction of anti-tumoral immune responses after vaccination and superior clinical outcome in immunological responders compared to non-responders would be in line with the proposed mechanism of action of DC vaccination therapy and could be a strong argument to mechanistically support the efficacy of this type of active immunotherapy in GBM. Therefore, our translation research program aims to identify immunological responders and non-responders within the group of vaccinated patients and to evaluate the association between individual anti-tumor immune responses, the degree of peripheral and local immunosuppression before, during and after the vaccination procedure, and clinical outcome.
At defined time points during priming and boost vaccination, phenotypical and functional analysis of peripheral blood mononuclear cells (PBMC) will be performed. Multicolor flow cytometric analysis will focus on the assessment of the frequencies of CD4+ and CD8+ T-cell subsets including regulatory T cells, natural-killer-cell subsets and myeloid-derived suppressor cells (MDSC) and the expression of PD-1, PD-L1, CTLA-4 and CD107a [112,113,114]. Furthermore, interferon (INF)-γ ELISPOT and qPCR responses of peripheral blood mononuclear cells (PBMC) and multicytokine responses (IFN-γ, IL-2, IL-10, TGF-β2, IL-17) in patient serum and conditioned media of specifically restimulated cells in the course of vaccination will be assessed. In addition, we will try to establish short-term cultured tumor cell lines from individual patients that will be used as targets for GBM-specific cytotoxic responses in CD107a degranulation assays [115].
For selected HLA-A*0201- and HLA-A*2402-positive immunological responders, we will further attempt to identify the target antigen of the anti-GBM immune response. Tetramer analysis and INF-γ responses towards molecularly defined antigens (e.g., EphA2, MAGE1, HER2, TRP2, IL-13R2, EGFRvIII, survivin, CMVp65 peptides and the IMA950 targets [43, 116, 117] will be performed after stimulation of PBMC with peptide-loaded and unloaded autologous DC. In addition, we will make use of selected Peptivators® (Miltenyi Biotec, Bergisch Gadbach, Germany) in non-HLA-A*0201 or non-HLA-A*2404 patients.
Moreover, tumor tissue will be assessed by immunohistochemistry and tissue microarrays to determine local immunosuppressive factors in vaccinated patients. Analysis will include, but is not necessarily limited to, the frequency of regulatory T cells and MDSC and the expression levels of TGF-β2, IL-10, indolamin-2,3-dioxigenase, PD-L1 and arginin-1. If surgical resection is performed at the time of progression, tumor material will be reanalyzed as mentioned above to evaluate whether the composition and phenotype of the tumor microenvironment at baseline and after treatment correlate with clinical efficacy (Additional file 1).
Trial status
Protocol version: GlioVax_Prüfplan_V02_F, 01.08.2017.
Approval by the national regulatory authority (Paul-Ehrlich Institute): #3011/01.
Patient recruitment is planned for January 2018.
Abbreviations
- ADR:
-
Adverse drug reaction(s)
- AE:
-
Adverse event(s)
- CTCAE:
-
Common terminology criteria for adverse events
- DC:
-
Dendritic cell(s)
- DSMC:
-
Data Safety and Monitoring Committee
- eCRF:
-
Electronic case report form(s)
- FET-PET:
-
Fluoroethyl-tyrosine positron emission tomography
- FGS:
-
Fluorescence-guided surgery
- GBM:
-
Glioblastoma multiforme
- GCP:
-
Good Clinical Practice
- HLA:
-
Human leukocyte antigen(s)
- IDH:
-
Isocitrate dehydrogenase
- IFNγ:
-
Interferon- γ
- IL-2, -10, -17:
-
Interleukin-2, -10, -17
- MDSC:
-
Myeloid-derived suppressor cells
- MedDRA:
-
Medical Dictionary for Regulatory Activities
- MRI:
-
Magnetic resonance imaging
- NCI:
-
National Cancer Institute
- OS:
-
Overall survival
- PBMC:
-
Peripheral blood mononuclear cells
- PFS:
-
Progression-free survival
- RANO:
-
Response assessment in neuro-oncology
- RT:
-
Radiotherapy
- SADR:
-
Severe adverse drug reaction(s)
- SAE:
-
Severe adverse event(s)
- SUSAR:
-
Suspected unexpected severe drug reaction(s)
- TGF-β2:
-
Transforming growth factor-β2
- TH1:
-
T-helper cell 1
- TMZ:
-
Temozolomide
- Treg :
-
Regulatory T cell(s)
References
Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro-Oncology. 2014;16:896–913.
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96.
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–77.
Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39:38–48.
Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–8.
Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–94.
Liau LM, Black KL, Martin NA, Sykes SN, Bronstein JM, et al. Treatment of a patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I-matched tumor peptides. Case report. Neurosurg Focus. 2000;e8:9.
Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, et al. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol Immunother. 2001;50:337–44.
Yu JS, Wheeler CJ, Zeltzer PM, Ying H, Finger DN, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842–7.
Wheeler CJ, Black KL, Liu G, Ying H, Yu JS, et al. Thymic CD8+ T cell production strongly influences tumor antigen recognition and age-dependent glioma mortality. J Immunol. 2003;171:4927–33.
Yamanaka R, Abe T, Yajima N, Tsuchiya N, Homma J, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer. 2003;89:1172–9.
Caruso DA, Orme LM, Neale AM, Radcliff FJ, Amor GM, et al. Results of a phase 1 study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro-Oncology. 2004;6:236–46.
De Vleeschouwer S, Van Calenbergh F, Demaerel P, Flamen P, Rutkowski S, et al. Transient local response and persistent tumor control in a child with recurrent malignant glioma: treatment with combination therapy including dendritic cell therapy. Case report. J Neurosurg. 2004;100:492–7.
Kikuchi T, Akasaki Y, Abe T, Fukuda T, Saotome H, et al. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J Immunother. 2004;27:452–9.
Rutkowski S, De Vleeschouwer S, Kaempgen E, Wolff JE, Kuhl J, et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br J Cancer. 2004;91:1656–62.
Wheeler CJ, Das A, Liu G, Yu JS, Black KL. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin Cancer Res. 2004;10:5316–26.
Yu JS, Liu G, Ying H, Yong WH, Black KL, et al. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64:4973–9.
Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11:5515–25.
Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005;11:4160–7.
Khan JA, Yaqin S. Dendritic cell therapy with improved outcome in glioma multiforme—a case report. J Zhejiang Univ Sci B. 2006;7:114–7.
Okada H, Lieberman FS, Walter KA, Lunsford LD, Kondziolka DS, et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J Transl Med. 2007;5:67.
de Vleeschouwer S, Fieuws S, Rutkowski S, Van Calenbergh F, Van Loon J, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res. 2008;14:3098–104.
Walker DG, Laherty R, Tomlinson FH, Chuah T, Schmidt C. Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: potential interaction with adjuvant chemotherapy. J Clin Neurosci. 2008;15:114–21.
Wheeler CJ, Black KL, Liu G, Mazer M, Zhang XX, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008;68:5955–64.
Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE, 2nd, , et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther 2009;8:2773–2779.
Ardon H, De Vleeschouwer S, Van Calenbergh F, Claes L, Kramm CM, et al. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr Blood Cancer. 2010;54:519–25.
Ardon H, Van Gool S, Lopes IS, Maes W, Sciot R, et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: a pilot study. J Neuro-Oncol. 2010;99:261–72.
Chang CN, Huang YC, Yang DM, Kikuta K, Wei KJ, et al. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci. 2011;18:1048–54.
Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother. 2011;34:382–9.
Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29:330–6.
Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17:1603–15.
Akiyama Y, Oshita C, Kume A, Iizuka A, Miyata H, et al. alpha-type-1 polarized dendritic cell-based vaccination in recurrent high-grade glioma: a phase I clinical trial. BMC Cancer. 2012;12:623.
Ardon H, Van Gool SW, Verschuere T, Maes W, Fieuws S, et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: results of the HGG-2006 phase I/II trial. Cancer Immunol Immunother. 2012;61:2033–44.
Cho DY, Yang WK, Lee HC, Hsu DM, Lin HL, et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: a phase II clinical trial. World Neurosurg. 2012;77:736–44.
Iwami K, Shimato S, Ohno M, Okada H, Nakahara N, et al. Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor alpha2 chain in recurrent malignant glioma patients with HLA-A*24/A*02 allele. Cytotherapy. 2012;14:733–42.
Jie X, Hua L, Jiang W, Feng F, Feng G, et al. Clinical application of a dendritic cell vaccine raised against heat-shocked glioblastoma. Cell Biochem Biophys. 2012;62:91–9.
Valle RD, de Cerio AL, Inoges S, Tejada S, Pastor F, et al. Dendritic cell vaccination in glioblastoma after fluorescence-guided resection. World J Clin Oncol. 2012;3:142–9.
Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62:125–35.
Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tonnesen P, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. 2013;62:1499–509.
Olin MR, Low W, McKenna DH, Haines SJ, Dahlheimer T, et al. Vaccination with dendritic cells loaded with allogeneic brain tumor cells for recurrent malignant brain tumors induces a CD4(+)IL17(+) response. J Immunother Cancer. 2014;2:4.
Hunn MK, Bauer E, Wood CE, Gasser O, Dzhelali M, et al. Dendritic cell vaccination combined with temozolomide retreatment: results of a phase I trial in patients with recurrent glioblastoma multiforme. J Neuro-Oncol. 2015;121:319–29.
Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366–9.
Sakai K, Shimodaira S, Maejima S, Udagawa N, Sano K, et al. Dendritic cell-based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent malignant glioma. J Neurosurg. 2015;123(4):989–97.
Akasaki Y, Kikuchi T, Homma S, Koido S, Ohkusa T, et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol Immunother. 2016;65:1499–509.
Inoges S, Tejada S, de Cerio AL, Gallego Perez-Larraya J, Espinos J, et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J Transl Med. 2017;15:104.
Cao JX, Zhang XY, Liu JL, Li D, Li JL, et al. Clinical efficacy of tumor antigen-pulsed DC treatment for high-grade glioma patients: evidence from a meta-analysis. PLoS One. 2014;9:e107173.
Kim CH, Woo SJ, Park JS, Kim HS, Park MY, et al. Enhanced antitumour immunity by combined use of temozolomide and TAT-survivin pulsed dendritic cells in a murine glioma. Immunology. 2007;122:615–22.
Park SD, Kim CH, Kim CK, Park JA, Sohn HJ, et al. Cross-priming by temozolomide enhances antitumor immunity of dendritic cell vaccination in murine brain tumor model. Vaccine. 2007;25:3485–91.
Kim TG, Kim CH, Park JS, Park SD, Kim CK, et al. Immunological factors relating to the antitumor effect of temozolomide chemoimmunotherapy in a murine glioma model. Clin Vaccine Immunol. 2010;17:143–53.
Banissi C, Ghiringhelli F, Chen L, Carpentier AF. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol Immunother. 2009;58:1627–34.
Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, et al. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother. 2008;57:123–31.
Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology. 2011;13:324–33.
Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, et al. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. 2007;204:2023–30.
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803–20.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003.
Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, et al. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006;7:392–401.
DeMets DL, Lan KK. Interim analysis: the alpha spending function approach. Stat Med. 1994;13:1341–52.
Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28:1963–72.
Okada H, Weller M, Huang R, Finocchiaro G, Gilbert MR, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol. 2015;16:e534–42.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98.
Aaronson NK, Ahmedzai S, Bergman B, Bullinger M, Cull A, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85:365–76.
Taphoorn MJ, Claassens L, Aaronson NK, Coens C, Mauer M, et al. An international validation study of the EORTC brain cancer module (EORTC QLQ-BN20) for assessing health-related quality of life and symptoms in brain cancer patients. Eur J Cancer. 2010;46:1033–40.
Mehnert A, Müller D, Lehmann C, Koch U. Die deutsche Version des NCCN Distress-Thermometers. Empirische Prüfung eines Screening-Instruments zur Erfassung psychosozialer Belastung bei Krebspatienten. Zeitschrift Psychiatr Psychol Psychother. 2006;54:213–23.
Herrmann C, Buss U, Snaith RP. HADS-D: Hospital Anxiety and Depression Scale—Deutsche Version; Ein Fragebogen zur Erfassung von Angst und Depressivitäit in der somatischen Medizin. Bern: Hans Huber Verlag; 1995.
Rapp M, Ozcan Z, Steiger HJ, Wernet P, Sabel MC, et al. Cellular immunity of patients with malignant glioma: prerequisites for dendritic cell vaccination immunotherapy. J Neurosurg. 2006;105:41–50.
Sorg RV, Ozcan Z, Brefort T, Fischer J, Ackermann R, et al. Clinical-scale generation of dendritic cells in a closed system. J Immunother. 2003;26:374–83.
Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27:3135–42.
Mirimanoff RO, Gorlia T, Mason W, Van den Bent MJ, Kortmann RD, et al. Radiotherapy and temozolomide for newly diagnosed glioblastoma: recursive partitioning analysis of the EORTC 26981/22981-NCIC CE3 phase III randomized trial. J Clin Oncol. 2006;24:2563–9.
De Vleeschouwer S, Ardon H, Van Calenbergh F, Sciot R, Wilms G, et al. Stratification according to HGG-IMMUNO RPA model predicts outcome in a large group of patients with relapsed malignant glioma treated by adjuvant postoperative dendritic cell vaccination. Cancer Immunol Immunother. 2012;61:2105–12.
Calzascia T, Di Berardino-Besson W, Wilmotte R, Masson F, de Tribolet N, et al. Cutting edge: cross-presentation as a mechanism for efficient recruitment of tumor-specific CTL to the brain. J Immunol. 2003;171:2187–91.
Masson F, Calzascia T, Di Berardino-Besson W, de Tribolet N, Dietrich PY, et al. Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J Immunol. 2007;179:845–53.
Maes W, Van Gool SW. Experimental immunotherapy for malignant glioma: lessons from two decades of research in the GL261 model. Cancer Immunol Immunother. 2011;60:153–60.
Pellegatta S, Poliani PL, Corno D, Grisoli M, Cusimano M, et al. Dendritic cells pulsed with glioma lysates induce immunity against syngeneic intracranial gliomas and increase survival of tumor-bearing mice. Neurol Res. 2006;28:527–31.
Ni HT, Spellman SR, Jean WC, Hall WA, Low WC. Immunization with dendritic cells pulsed with tumor extract increases survival of mice bearing intracranial gliomas. J Neuro-Oncol. 2001;51:1–9.
Liau LM, Black KL, Prins RM, Sykes SN, DiPatre PL, et al. Treatment of intracranial gliomas with bone marrow-derived dendritic cells pulsed with tumor antigens. J Neurosurg. 1999;90:1115–24.
Heimberger AB, Crotty LE, Archer GE, McLendon RE, Friedman A, et al. Bone marrow-derived dendritic cells pulsed with tumor homogenate induce immunity against syngeneic intracerebral glioma. J Neuroimmunol. 2000;103:16–25.
Magana-Maldonado R, Chavez-Cortez EG, Olascoaga-Arellano NK, Lopez-Mejia M, Maldonado-Leal FM, et al. Immunological evasion in glioblastoma. Biomed Res Int. 2016;2016:7487313.
Razavi SM, Lee KE, Jin BE, Aujla PS, Gholamin S, et al. Immune evasion strategies of glioblastoma. Front Surg. 2016;3:11.
Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294–302.
Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. 1999;100:216–32.
Morford LA, Elliott LH, Carlson SL, Brooks WH, Roszman TL. T cell receptor-mediated signaling is defective in T cells obtained from patients with primary intracranial tumors. J Immunol. 1997;159:4415–25.
Brooks WH, Latta RB, Mahaley MS, Roszman TL, Dudka L, et al. Immunobiology of primary intracranial tumors. Part 5: correlation of a lymphocyte index and clinical status. J Neurosurg. 1981;54:331–7.
Braun DP, Penn RD, Flannery AM, Harris JE. Immunoregulatory cell function in peripheral blood leukocytes of patients with intracranial gliomas. Neurosurgery. 1982;10:203–9.
Grauer OM, Nierkens S, Bennink E, Toonen LW, Boon L, et al. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int J Cancer. 2007;121:95–105.
Grauer OM, Sutmuller RP, van Maren W, Jacobs JF, Bennink E, et al. Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. Int J Cancer. 2008;122:1794–802.
Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722–9.
Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, et al. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity. 2005;22:175–84.
Neller MA, Lopez JA, Schmidt CW. Antigens for cancer immunotherapy. Semin Immunol. 2008;20:286–95.
Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med. 1999;190:617–27.
Sallusto F, Schaerli P, Loetscher P, Schaniel C, Lenig D, et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol. 1998;28:2760–9.
Caux C, Ait-Yahia S, Chemin K, de Bouteiller O, Dieu-Nosjean MC, et al. Dendritic cell biology and regulation of dendritic cell trafficking by chemokines. Springer Semin Immunopathol. 2000;22:345–69.
Steinbrink K, Wolfl M, Jonuleit H, Knop J, Enk AH. Induction of tolerance by IL-10-treated dendritic cells. J Immunol. 1997;159:4772–80.
Grauer O, Poschl P, Lohmeier A, Adema GJ, Bogdahn U. Toll-like receptor triggered dendritic cell maturation and IL-12 secretion are necessary to overcome T-cell inhibition by glioma-associated TGF-beta2. J Neuro-Oncol. 2007;82:151–61.
Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711.
Dudek AM, Martin S, Garg AD, Agostinis P. Immature, semi-mature, and fully mature dendritic cells: toward a DC-cancer cells interface that augments anticancer immunity. Front Immunol. 2013;4:438.
de Vries IJ, Lesterhuis WJ, Scharenborg NM, Engelen LP, Ruiter DJ, et al. Maturation of dendritic cells is a prerequisite for inducing immune responses in advanced melanoma patients. Clin Cancer Res. 2003;9:5091–100.
Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193:233–8.
Eggert AA, Schreurs MW, Boerman OC, Oyen WJ, de Boer AJ, et al. Biodistribution and vaccine efficiency of murine dendritic cells are dependent on the route of administration. Cancer Res. 1999;59:3340–5.
Eggert AA, van der Voort R, Torensma R, Moulin V, Boerman OC, et al. Analysis of dendritic cell trafficking using EGFP-transgenic mice. Immunol Lett. 2003;89:17–24.
Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, et al. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999;59:56–8.
De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63:12–7.
Macatonia SE, Knight SC, Edwards AJ, Griffiths S, Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. Functional and morphological studies. J Exp Med. 1987;166:1654–67.
Ruedl C, Koebel P, Bachmann M, Hess M, Karjalainen K. Anatomical origin of dendritic cells determines their life span in peripheral lymph nodes. J Immunol. 2000;165:4910–6.
Garg S, Oran A, Wajchman J, Sasaki S, Maris CH, et al. Genetic tagging shows increased frequency and longevity of antigen-presenting, skin-derived dendritic cells in vivo. Nat Immunol. 2003;4:907–12.
Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res. 2009;15:2531–40.
Lesterhuis WJ, de Vries IJ, Schreibelt G, Lambeck AJ, Aarntzen EH, et al. Route of administration modulates the induction of dendritic cell vaccine-induced antigen-specific T cells in advanced melanoma patients. Clin Cancer Res. 2011;17:5725–35.
Ringel F, Pape H, Sabel M, Krex D, Bock HC, et al. Clinical benefit from resection of recurrent glioblastomas: results of a multicenter study including 503 patients with recurrent glioblastomas undergoing surgical resection. Neuro-Oncology. 2016;18:96–104.
Suchorska B, Weller M, Tabatabai G, Senft C, Hau P, et al. Complete resection of contrast-enhancing tumor volume is associated with improved survival in recurrent glioblastoma-results from the DIRECTOR trial. Neuro-Oncology. 2016;18:549–56.
Okada H, Kohanbash G, Zhu X, Kastenhuber ER, Hoji A, et al. Immunotherapeutic approaches for glioma. Crit Rev Immunol. 2009;29:1–42.
Grauer OM, Wesseling P, Adema GJ. Immunotherapy of diffuse gliomas: biological background, current status and future developments. Brain Pathol. 2009;19:674–93.
Dubinski D, Wolfer J, Hasselblatt M, Schneider-Hohendorf T, Bogdahn U, et al. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro-Oncology. 2015;18:807–18.
Fong B, Jin R, Wang X, Safaee M, Lisiero DN, et al. Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PLoS One. 2012;7:e32614.
Jacobs JF, Idema AJ, Bol KF, Nierkens S, Grauer OM, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro-Oncology. 2009;11:394–402.
Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78.
Dutoit V, Herold-Mende C, Hilf N, Schoor O, Beckhove P, et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain. 2012;135:1042–54.
Pollack IF, Jakacki RI, Butterfield LH, Hamilton RL, Panigrahy A, et al. Immune responses and outcome after vaccination with glioma-associated antigen peptides and poly-ICLC in a pilot study for pediatric recurrent low-grade gliomas. Neuro-Oncology. 2016;18:1157–68.
Funding
The study is funded by the Federal Ministry of Education and Research (BMBF; grant 01KG1242).
Availability of data and materials
There are no contractual agreements, which limit access by the investigators to the final trial data. Access to data is at the discretion of the coordinating investigator and the sponsor. Results of the trial, irrespective of positive or negative outcome, will be published in peer-reviewed journals.
Insurance
There is a clinical trials insurance as well as a travel accident insurance covering patients participating in the trial.
Responsibilities in the course of the trial
The coordinating investigator (MS)/deputy coordinating investigator (MR) are responsible for the conduct of the trial, including safety evaluation and the revision of trial documents (e.g., amendment of the study protocol, including approval by the Ethics Committees and the National Regulatory Authority), if required. The co-investigator for vaccine aspects (RVS) is responsible for vaccine production. The co-investigator for immunomonitoring (MOG) is responsible for immunomonitoring. For central histopathological reviewing, central radiological reviewing, sterility and environmental hygiene monitoring, viral infectious disease testing and bacterial infectious disease testing, the appropriate departments of the University Hospital Düsseldorf are responsible. The Coordinating Center for Clinical Trials at the University Hospital Düsseldorf is responsible for project management, quality management, randomization, data management, adverse event management, monitoring and data analysis.
In each participating center, an investigator (neurosurgeon, neurologist) is responsible for the composition and instruction of his team, for the identification, recruitment, treatment of patients and data collection adherent to the study protocol. For histopathological and radiological as well as laboratory assessments, the local departments of the participating centers are responsible.
Sponsor
Heinrich Heine University Düsseldorf.
Universitätsstr. 1.
40,225 Düsseldorf.
Germany.
represented by.
Prof. Dr. med. Michael C. Sabel.
Department of Neurosurgery.
Heinrich Heine University Hospital, Düsseldorf.
Moorenstr. 5, 40,225 Düsseldorf, Germany.
Phone: + 49 (0) 211 / 81–07334.
Fax: + 49 (0) 211 / 81–04658.
E-mail: Michael.Sabel@med.uni-duesseldorf.de
Author information
Authors and Affiliations
Contributions
MR is the deputy coordinating investigator. She has written the manuscript, has provided clinical expertise and contributed to the development of the protocol. OMG has contributed to the development and writing of the protocol and manuscript, particularly to the parts on immunomonitoring procedures and evaluation of tumor progression. MK has provided clinical expertise and contributed to the development of the protocol. NS is responsible for clinical project management. She has contributed to the development and writing of the protocol, particularly to the parts on study visits and the various examination procedures as well as to the patient’s information documents. NZ is the administrative trial project manager. She has contributed to the development and writing of the protocol, particularly to the parts on adverse event management, data management and GCP as well as biometric aspects. MS is the coordinating investigator and grant holder. He has been responsible for the design and development of the protocol and the implementation of the trial. RVS is responsible for all vaccine and regulatory aspects. He shared responsibility for the protocol design and development and contributed to writing the protocol and manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol has been approved by the Ethics Committee of the Medical Faculty of the Heinrich Heine University (leading Ethics Committee) and the respective Advisory Ethics Committees of the participating centers (MC-LKP-921, protocol ID: GlioVax). Written informed consent will be obtained from each patient, using the consent form approved by the Ethics Committees. It will be ensured that patients understand the procedures and risks of participating in the clinical trial, and only patients in a cognitive state to understand and sign the informed consent, indicating that they are aware of the investigational nature and the procedures of the study, will be eligible. Patients are informed that medical records may be examined by representatives of the sponsor (monitor, auditor, members of the DSMC), by the appropriate Ethics Committee members and by inspectors from the regulatory authorities. Moreover, must give their consent that tumor material and blood are transferred to the vaccine production site, processed and assessed for suitability for vaccine production, which includes infectious disease testing. Furthermore, patients agree that blood samples are used for immunomonitoring and that cells may be stored for later assessment of anti-tumoral immunity and tumor immunosuppressiveness.
Consent for publication
All authors have read and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Sponsor and funder are non-commercial. The sponsor keeps responsibility and oversight in the planning, initiation and conduct of the clinical trial regarding compliance to GCP and applicable laws. The funder had no role in the design of this study and will not have any role during its execution, analysis, interpretation of the data or decision to submit results.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 Checklist: recommended items to address in a clinical trial protocol and related documents. (DOCX 63 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Rapp, M., Grauer, O.M., Kamp, M. et al. A randomized controlled phase II trial of vaccination with lysate-loaded, mature dendritic cells integrated into standard radiochemotherapy of newly diagnosed glioblastoma (GlioVax): study protocol for a randomized controlled trial. Trials 19, 293 (2018). https://doi.org/10.1186/s13063-018-2659-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2659-7