
Abstract
Background
Malignant melanoma is a highly heterogeneous skin cancer with the highest mortality rate among dermatological cancers. Catenins form functional networks in the nucleus to regulate gene expression and determine cell fate. Dysregulation of catenin expression correlates with the malignant characteristics of the tumor. We aimed to investigate the regulatory mechanisms of catenins in melanoma and to further define the function of catenin-related molecular signaling in the tumor microenvironment.
Methods
In this study, a bioinformatics approach combined with experimental validation was used to explore the potential tumor biology mechanisms of catenin-related signaling.
Results
Melanoma patients can be divided into two catenin clusters. Patients defined by high Junction Plakoglobin (JUP), Plakophilin 1 (PKP1), Plakophilin 3 (PKP3) levels (C2) had shorter survival time than other patients (C1). We demonstrated that JUP regulates Anterior Gradient 2 (AGR2)/LY6/PLAUR Domain Containing 3 (LYPD3) to maintain melanoma stemness and promotes glycolysis. We also found that LYPD3 was co-expressed with S100A9 and associated with immunosuppressive tumor microenvironment (TME).
Conclusion
The JUP/AGR2/LYPD3 signaling axis plays an important role in the malignant features of melanoma. Targeting the JUP/AGR2/LYPD3 signaling axis can help develop promising drugs.
Similar content being viewed by others

Introduction
As one of the most aggressive cutaneous malignancies, melanoma poses a major public health challenge and its incidence has been increasing over the past decade [1, 2]. Although, due to prominent immunogens, significant breakthroughs have been made in melanoma immunotherapy, particularly with the use of immune checkpoint Immune checkpoint blockades (ICBs) [3,4,5]. Unfortunately, there is still a significant percentage of patients who struggle to benefit from ICBs and the frequent band of anti-Programmed Cell Death-1 (PD-1) drugs brings intolerable adverse effects [6]. Considering that the mechanism of tumor response to ICBs is regulated by multiple factors, it is necessary to further elucidate it at the molecular level [7,8,9]. Many studies have confirmed that the tumor microenvironment (TME), on which tumor cell survival depends, is highly heterogeneous and that multiple pathways exist to suppress anti-tumor immunity [10,11,12]. For example, tumor-infiltrating lymphocytes (TILs) with low abundance or dysfunctional tumors do not respond significantly to ICBs [13, 14].
Most vertebrates have 12 different types of gene encoding catenins that belong to four subfamilies, including alpha catenins, beta catenins, delta catenins, and p120 catenins [15]. Among them, β-catenins have been widely demonstrated to be targets of Wnt signaling [16], specifically, the degradation of cytoplasmic β-catenins upon binding of Wnt to its receptor, thereby facilitating its nuclear translocation [17]. Interestingly, nuclear β-catenins often co-exist with PD-1, and it has been shown that β-catenins promote immune escape by impairing T cell activity through defective recruitment of dendritic cells [18]. In addition to the classical β-catenins signaling pattern, other linker proteins have equally promising emerging functions [19,20,21]. Since all catenins possess a central armadillo domain that is well suited for nucleo-cytoplasmic shuttling, it is easy to understand that they play important and complex roles in the organism [22,23,24]. Indeed, the expression of many genes is regulated by multiple catenins that act synergistically or antagonistically [25]. Importantly, there is a frequent shared relationship between catenins and actin, with α-catenin binding to filamentous actin (F-actin) and thereby mediating the attachment of the cadherin-β catenins complex to the cytoskeleton [26, 27]. This reflects the importance of catenins in cell development and differentiation. However, the commonalities and differences in the functions of catenins and the extent to which they network or act independently remain largely unknown.
Cancer stem cells (CSCs) are subpopulations of cells in tumors that are in a stem cell state and have stem cell characteristics, which partly explain the heterogeneity of tumors [28]. It is well documented that CSCs are an important cause of treatment resistance, metastatic recurrence, and immunosuppression in tumor patients [29]. As an intrinsic tumorigenic mechanism, catenin signaling is associated with cancer Epithelial mesenchymal transition (EMT) and stem cell-like biological phenotypes [30, 31]. A previous study demonstrated that β-catenin-positive tumors were unresponsive to immune checkpoint therapy compared to murine tumors lacking β-catenin [32]. These studies suggest a potential link among catenins, stem cell-like phenotypes, and immunosuppression.
In this study, we analyzed the gene expression and transcriptional heterogeneity of 12 catenins in 32 cancer types and assessed their prognostic value. The results suggest that genomic changes, including DNA methylation and Copy number variation (CNV), affect the transcriptional levels of catenins. We focused on the regulatory mechanisms of catenins in melanoma. Linking catenins to stem cell-like features of melanoma cells and immunosuppressive TME was done by The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) bulk data. Our results suggest the presence of Plakoglobin (JUP)/Anterior Gradient 2 (AGR2)/LY6/PLAUR Domain Containing 3 (LYPD3) pro-oncogenic signaling in melanoma. We also found that LYPD3 was closely associated with S100 Calcium Binding Protein A9 (S100A9)-labelled myeloid cells, impairing immunotherapy response. We confirmed that the JUP/AGR2/LYPD3 signaling axis promotes F-actin expression, remodels the cytoskeleton and confers a robust invasive phenotype to melanoma cells.
Materials and methods
All specific bioinformatics methods and experimental approaches are described in the Supplementary Methods.
Antibodies, reagents, and cell lines
The details for the wet-lab experiments and all antibodies, reagents, and cell lines are summarized in Additional file 1: Table S1–S5.
Data source and process
RNA-seq transcriptome information and associated clinical data for 471 patients with skin cutaneous melanoma (SKCM) were downloaded from the TCGA portal, while a portion of the TCGA data was processed using the Gene Set Cancer Analysis (GSCA) online tool. And, additional independent validation cohorts (GSE22153, GSE59455, and GSE65904) were obtained from the Gene Expression Omnibus (GEO) database [33, 34]. In addition, two immunotherapy datasets (Nathanson_2017 and GSE120575) were processed through the Tumor Immunotherapy Gene Expression Resource (TIGER) online tool [35]. All gene expression data were exported as a standardised data matrix with the help of R software.
Statistical analysis
We used Spearman and Pearson correlation coefficients to determine the correlation between variables. The Mann–Whitney U-test (also known as the Wilcoxon rank sum test) was used to analyze non-normally distributed variables. One-way analysis of variance (ANOVA) test was used to compare two or more groups. The log-rank (Mantel-Cox) test was used for survival analysis. The univariate Cox regression was utilized to identify factors with independent prognostic value, and the corresponding hazard ratios (HR) were provided. Statistical analyses were performed using the programming language software R and Excel (Microsoft) and a P value (two-tailed) < 0.05 was considered statistically significant.
Results
Genetic characterization, transcriptional variation and expression patterns of 12 catenin molecules
Information about the 12 catenins was downloaded from the Human Genome organization gene nomenclature committee (HGNC) portal. The 12 molecules are Catenin Alpha 1 (CTNNA1), Catenin Alpha 2 (CTNNA2), Catenin Alpha 3 (CTNNA3), Catenin Beta 1 (CTNNB1), JUP, Plakophilin 1 (PKP1), Plakophilin 2 (PKP2), Plakophilin 2 (PKP3), ARVCF Delta Catenin Family Member (ARVCF), Catenin Delta 1 (CTNND1), Catenin Delta 1 (CTNND2), and Plakophilin 4 (PKP4). To examine genetic variation in catenin molecules in cancer, we selected 1575 samples with at least one mutation from 12 catenin molecules in the TCGA pan-cancer database (Fig. 1A). Considering that only 14 cancer types had more than 10 paired tumor and normal samples, we provided differential mRNA expression of catenins in these 14 cancer types. The expression of different catenin molecules in different cancer tissues was inconsistent, and several molecules, including PKP3, PKP2, PKP1, and JUP, were overexpressed in several cancer tissues (Fig. 1B). We also explored the characteristics of catenin-related signaling pathways. At the pan-cancer level, most signaling pathways, particularly PKP4, PKP2, JUP, and CTNND2, showed high levels of activation in the Hormone AR signaling pathway, but consistent inhibition of apoptosis, cell cycle, and DNA damage responses (Fig. 1C). Importantly, we demonstrated that catenin expression may correlate with genetic variation. expression levels of CNV and mRNA were positively correlated in most cancer types, particularly for CNTTA1, CNTTB1, and CNTTD1 (Fig. 1D). In contrast, in most cancers, gene methylation levels were negatively correlated with mRNA expression levels (Fig. 1E). This part of the analysis showed that catenin expression patterns were highly heterogeneous across cancers and that its aberrant expression was associated with genomic variation.

Expression variation of catenin molecules. A The waterfall plot shows somatic mutations in the 10 catenins with the highest mutation frequency in the pan-cancer analysis. 95.62% is the percentage of 1575 cancer samples with 1506 mutations in at least 10 genes. The percentage plot (right side of the figure) is the number of samples with the corresponding gene mutation divided by 1575 samples with at least one mutation among the 10 catenins coding genes. B The color of these dots represents the degree of variance. Red dots represent high expression in cancerous tissues, blue dots represent the opposite. Fold change equals mean (tumor)/mean (normal), a t-test was used, and p values were False-discovery rate (FDR) corrected. The size of the bubble indicates the FDR. The larger the bubble, the lower the FDR. Genes with fold change (Fold change > 2) and significance (FDR > 0.05) were retained to generate plots. If there are no significant genes in a cancer type, omit that cancer type from the final graph. C The heatmap shows the correlation between the expression levels of 10 catenins and important cancer-related signaling pathways. The percentage of cancers in which each catenin-encoding gene has an effect on this pathway is shown for each of the 32 cancer types: (number of activated or repressed cancer types/total number*100%). Catenin-encoding genes that are functional (inhibited or activated) in at least 5 cancer types are shown. “Pathway activation” (red) indicates the percentage of cancers in which a pathway may be activated by a given gene, and inhibition is shown in a similar way as “pathway inhibition” (blue). D The bubble plots show the correlation between Copy number variation (CNV) and mRNA expression levels. Red color indicates positive correlation and blue color indicates negative correlation. Darker colors indicate larger correlation coefficients. The size of the bubbles represents the FDR. E The bubble plot shows the correlation between methylation of 10 catenin molecules and mRNA expression. Red color indicates positive correlation and blue color indicates negative correlation. Darker colors indicate larger correlation coefficients. The size of the bubbles represents FDR. F Hierarchical clustering dendrogram showing the Euclidean distance between genes calculated using the “dist” function. The 2 major clusters identified are indicated by green and pink boxes, respectively. G The forest plot shows the results of Cox regression analysis of overall survival (OS) for 10 catenin molecules in the The Cancer Genome Atlas (TCGA)- Skin cutaneous melanoma (SKCM) cohort. H mRNA level of catenin coding genes between cancer tissues in the TCGA-SKCM cohort (n = 469) and control tissues (n = 812). Normal tissues in both the TCGA and GTEx databases were included as negative controls. ***P < 0.001 I Mutation frequency of 10 catenin molecules in 104 cutaneous melanoma patients in the TCGA-SKCM cohort. J Mutational characterization of 10 catenin molecules in 104 patients with cutaneous melanoma in the TCGA-SKCM cohort; green indicates co-mutations and asterisks indicate P values (P < 0.05, *P < 0.01). K Cells were clustered into 6 types via Uniform Manifold Approximation and Projection (UMAP) plot dimensionality reduction algorithm. Different cell types are represented by different colors. L–M UMAP (L) and Violin (M) plots visualization of the catenin feature (obtained based on the “AddModuleScore” function in “Seurat” package)
The focus of this study is on malignant melanoma. We first performed a hierarchical clustering of 12 catenins based on the euclidean distance matrix in TCGA-SKCM and found that PKP1, JUP, PKP3 (pink) had a significantly different expression pattern from other catenins (green) (Fig. 1F). Further COX regression analysis indicated that PKP1, JUP, PKP3 were significant detrimental factors for Overall survival (OS) in SKCM patients (Fig. 1G). As there was only one normal skin sample in the TCGA-SKCM data, we included normal tissue from the Genotype Tissue Expression (GTEx) database as a negative control to measure the difference in catenins expression between diseased and normal tissue. As shown in Fig. 1H, aside from CTNNA1, CTNNA2, and CTNNB1, there was low expression of most catenins in the tumor samples (P < 0.001) compared with the controls. On the other hand, catenin molecular mutations occurred in 41 of the 104 samples, with a mutation frequency of 39.13%. Among the 104 cases, CTNNA2 had the highest mutation frequency, all of which were missense mutations (Fig. 1I). In TCGA-SKCM, there were significant co-mutations between CTNNA2 and CTNND2, CTNNA2 and PKP2, PKP2 and PKP3, CTNND2 and PKP4, and CTNNA3 and PKP4 (P < 0.05, Fig. 1J). Finally, to better understand the expression patterns of catenins in TME, we utilized a melanoma single-cell dataset (Additional file 1: Fig. S1). Annotation of cells by unique gene markers for each cell type revealed that catenins were expressed at low levels in immune cells, including T cells, B cells, and monocytes (Fig. 1K–M). Results strongly suggested that the expression of catenins was highly heterogeneous and associated with tumorigenesis.
Two different catenin expression patterns identified by unsupervised learning
To fully understand the intergratd mechanism of catenins in melanoma, we performed unsupervised clustering of 456 samples from TCGA-SKCM using the R package “ConsensusClusterPlus”. Two unique catenin-modified phenotypes were identified, named cluster 1 (C1, 410 cases), and cluster 2 (C2, 46 cases) (Fig. 2A, B, Additional file 1: Fig. S2). Principal component analysis (PCA) showed that the two clusters had significantly different catenins expression patterns, confirming the validity of the clustering (Fig. 2C). The heat map showed that PKP1, JUP, and PKP2 were significantly overexpressed in C2 than in C1 (Fig. 2D). To ensure stability of clustering, the same process was performed in merged GEO (mGEO) data (Additional file 1: Fig. S3). We then compared our phenotype with commonly used clinical staging indicators. In contrast to C1, there are mostly advanced subtypes in C2, particularly a high proportion of Breslow thickness ≥ 3 mm and Clark’s Level IV, and V tumor (Fig. 2E). Survival analysis showed that C1 provided a particularly significant survival advantage, whereas C2 had a poorer prognosis (log-rank, P = 0.000087, Fig. 2F). We further examined the 10 genes with the highest mutation frequencies in each subgroup (Additional file 1: Fig. S4). Interestingly, BRAF had a high mutation frequency in C2, and BRAF mutations have been shown to be a driver of poor prognosis in melanoma patients. The mutation profiles of genes frequently mutated in C1 and C2 are shown in oncoplots.

Unsupervised clustering to identify two catenin classifications. A Unsupervised Machine Learning algorithms was used to identify 2 molecular subtypes in TCGA-SKCM. A Left: The cumulative distribution function (CDF) curves in consensus cluster analysis. CDF curves of consensus scores by different subtype numbers (k = 2, 3, 4, 5, and 6) were displayed. Right: Relative change in area under the CDF curve for k = 2–6. B The consensus score matrix of melanoma cases in TCGA-SKCM when k = 2. The higher the consensus score was, the more likely they were assigned to the same group. C Three-dimensional principal component analysis (PCA) plot showing the distribution of melanoma examples based on catenin levels when k = 2. Each point represents a sample and different sample clusters are marked using different colors. D Heatmap showing differences in expression of 10 catenin molecules between two melanoma clusters. E The Sankey diagram demonstrated the association between clinicopathological parameters (Breslow_depth, Clark_level, and T/N/M stage) and subtypes attributes. F OS analysis to the catenin subtype was performed. Log rank test was conducted. G Volcano map of differentially expressed genes (DEGs) between C1 and C2 in TCGA-SKCM dataset. Data on the abscissa are differences in gene expression (log2 fold change); data on the ordinate represent the significance of these differences (− log10 padj). Red indicates upregulation in C1, and blue indicates downregulation in C1. H Gene Ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis was performed using the “clusterProfiler” R package. The size of the bubbles represents the number of genes enriched and the color of the bubbles represents the significance level [− log10 (P.adjust)] of the enrichment. I Representative pictures of pathologic Hematoxylin and eosin (HE) staining of the two catenin phenotypes
We next analyzed the differentially expressed genes (DEGs) between different catenin subtypes and explored the characterization of the associated molecular signals. Based on the “limma” R package, we obtained 511 genes highly expressed in C2 and 40 genes highly expressed in C1 (Fig. 2G). We then performed Gene Ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) enrichment analyses based on DEGs (Fig. 2H). In KEGG terms, we observed that the B-cell receptor signaling pathway and hematopoietic cell lineage were down-regulated in C2, while the IL-17 signaling pathway, gastric cancer, and basal cell carcinoma were up-regulated in C2. In GO terms, immune system-related signals were downregulated in C2, whereas skin development and keratinocyte differentiation signals were upregulated in C2. Notably, histopathological sections confirmed that tumor cells in C2 showed more severe cytological heterogeneity compared to C1, suggesting a more significant degree of malignancy (Fig. 2I).
Identification of LYPD3 as one of the underlying regulators of catenin phenotype
To identify potential regulators in the catenin phenotype, the DEGs identified above were applied to a Multiscale Embedded Gene co-expression Network Analysis (MEGENA) (Fig. 3A). We identified 18 modules and 551 module genes. The largest module, C1_4, consisted of 142 genes, followed by C1_3 with 122 genes. Figure 3B, C demonstrated the network structure of C1_3 and C1_4. In addition, Metascape enrichment analysis showed that C1_3 and C1_4 exhibited consistent correlations with regulating epidermal development and cornified envelope (Fig. 3D).

Bulk data combined with single-cell data to identify LYPD3 as a study subject. A Co-expression network constructed based on DEGs between C1 and C2. B, C The Multiscale Embedded Gene co-expression Network Analysis (MEGENA) network shows the two largest gene modules. Different colors represent genes in different modules, and triangles represent hub genes in the modules. D GO and KEGG functional enrichment analysis demonstrated shared and unique biological signals of C1_3 and C1_4. E Violin plots show the expression of hub genes in C1_3 and C1_4 across 6 cell types in GSE189889. F UMAP plot showing the expression levels of LYPD3, defined for six cell types. G–I The forest map shows the results of Cox regression analysis on the OS (G), Disease Specific survival (DSS, H), and Progrssion Fre Interval (PFI, I) of 10 hub genes in the TCGA-SKCM cohort
To further understand the significance of C1_3 and C1_4 in TME, we extracted the hub genes in the module and demonstrated their expression levels by violin plots (Fig. 3E). Comparison of hub gene expression levels in different cell types was established and showed that LYPD3 was expressed in the highest abundance and upregulated in both melanoma cells and immune cells (Fig. 3F, green circle and box). Finally, associations between hub genes and OS, progression-free interval (PFI), and disease-specific survival (DSS) were modelled using Cox proportional risk regression (Fig. 3G–I). The results showed that, Nectin Cell Adhesion Molecule 4 (NECTIN4), Prominin 2 (PROM2), Transmembrane Protein 45B (TMEM45B), Short Chain Dehydrogenase/Reductase Family 16C Member 5 (SDR16C5), Grainyhead Like Transcription Factor 1 (GRHL1), Sulfotransferase Family 2B Member 1 (SULT2B1), LYPD3, Suprabasin (SBSN), and Family With Sequence Similarity 83 Member C (FAM83C) were significant unfavorable factors for OS, and NECTIN4, PROM2, SULT2B1, LYPD3, SBSN, and FAM83C were significant unfavorable factors for DSS (P < 0.05). Thus, we focus on LYPD3 in the next section.
Identification of the JUP/LYPD3/AGR2 signaling axis on melanoma cells
Herein, we investigated LYPD3-associated cancer biology functions. LYPD3 has been shown to be highly expressed in several human malignancies, suggesting a potential oncogenic role [36,37,38]. Open Targets software showed that LYPD3 plays an important regulatory role in cancer or benign tumor (Fig. 4A). Figure 4B showed correlation between LYPD3 expression and clinicopathological characteristics of melanoma patients, indicating that LYPD3 was positively correlated with several melanoma classifications, namely, T classification for TNM staging (p < 0.01), and Breslow depth (p < 0.001). The latter is considered the strongest predictor of melanoma mortality, with greater Breslow tumor thickness representing more severe local invasion [39]. Further, the level of LYPD3 was determined to be negatively correlated with survival (Fig. 4C, D, logrank P < 0.05). These results provide solid evidence for LYPD3 as a key regulator and stimulator of melanoma progression.

Bioinformatics combined with in vitro experiments to determine the presence of JUP/AGR2/LYPD3 signaling in melanoma cells. A Pathway and disease analysis of LYPD3. B Boxplot showing the expression level of LYPD3 among different pathologic stages (stage T, N, M and Breslow depth). C, D OS (C) and DSS (D) analysis according to the expression level of the LYPD3 gene were performed using melanoma cases in the TCGA-SKCM cohort. E The gene–gene interaction network for LYPD3 and neighboring genes was analyzed using the GeneMANIA database. Each node represents a gene. The line color represents possible relationship between the respective genes. F Correlation between LYPD3 level and four physically interacting genes as analyzed in TCGA-SKCM. Spearman method was applied. G Heatmap showing the correlation between LYPD3 and ten catenins, and the Spearman correlation coefficients are provided on the right side of the graph. H, I The positive correlation among LYPD3, JUP, and AGR2 was stronger in C2 than in C1. J UMAP plot demonstrating the expression of AGR2 and JUP. Higher expression is indicated by a greener color. (K-L) Verification of AGR2 and LYPD3 expression by Western blotting assay in melanoma cells transfected with oe and sh-JUP. One-way ANOVA was applied. ns not significant, *P < 0.05, **P < 0.01, ***P < 0.001
Subsequently, we utilized GENEMINIA to build a network of LYPD3 and its interacting genes. This tool identified four proteins that physically interact with LYPD3, including Anterior Gradient 3 (AGR3), Tripartite Motif-Containing Protein 69 (TRIM69), Anterior Gradient 2 (AGR2), and Polypeptide N-Acetylgalactosaminyltransferase 10 (GALNT10) (Fig. 4E). We computed transcriptome correlations between LYDP3 and the four interacting genes in TCGA-SKCM and found a significant positive correlation between LYPD3 and AGR2 levels (Fig. 4F, R = 0.361, P < 0.001). Interestingly, LYPD3 showed a significant positive correlation with JUP (R = 0.351, P < 0.001), PKP1 (R = 0.464, P < 0.001), and PKP3 (R = 0.440, P < 0.001) (Fig. 4G). These three genes were previously found to have similar expression patterns and were negatively associated with prognosis (Fig. 1F, G). It has been demonstrated that LYPD3 is a functional cell surface receptor for AGR2 and that AGR2 is highly regulated by β-catenin signaling [40, 41]. Considering that JUP is a homologue of β-catenin and is closely related to β-catenin in many cases to concertedly regulate cell fate, we speculate that there may be a mechanism of JUP/AGR2/LYPD3 signaling regulation in melanoma. We confirmed that JUP and AGR2 were associated with unfavourable prognosis of melanoma by KM curves (Additional file 1: Fig. S5A, B), and that AGR2 was associated with higher T stage and deeper Breslow depth (Additional file 1: Fig. S5C, D). Notably, compared to C1, we observed that the correlations among JUP, AGR2, LYPD3 was significantly strengthened in C2 (Fig. 4H, I) and that JUP and AGR2 were synchronously expressed on melanocytes (Fig. 4J). To verify our speculation, a stably transfected cell line for JUP was established and western blot was performed. As shown, the expression of JUP knockdown mediated by the shRNA group significantly reduced AGR2/LYPD3 signaling activity, whereas the opposite was true for oeRNA-mediating JUP gene overexpression, in agreement with our prediction (Fig. 4K, L, P < 0.05).
Upregulation of JUP/AGR2/LYPD3 signaling contributes to the maintenance of melanoma cell stemness and enhanced glycolysis levels
Cell stemness scores based on bulk data showed that C2 exhibited greater stemness (P < 0.001, Fig. 5A). Since JUP/AGR2/LYPD3 signaling is more prominent in C2, we speculate that the enhanced stemness phenotype of C2 may be related to this signaling. We performed cluster analysis on 19,547 melanoma cells and identified eight prominent cell subpopulations (Cluster 0-Cluster 7). CytoTRACE analysis indicateed that cluster 1 exhibited significantly less differentiated state than other melanoma cell populations (Fig. 5B–E). Figure 5F demonstrated the highly expressed genes in each cluster, where Brevican (BCAN), Long Intergenic Non-Protein Coding RNA 2303 (LINC02303), Flotillin 1 (FLOT1) were upregulated in cluster 1. We noted that JUP, AGR2, and LYPD3 were all expressed in cluster 1 (Fig. 5G), suggesting a potential correlation between them and poorly differentiated status. Clone formation experiments showed that significantly more colonies were formed in the oe-JUP group compared to the control group, and this effect was attenuated by sh-LYPD3 (Fig. 5H). In spheroidal cultures, JUP silencing leaded to decreased sphere-forming ability, whereas JUP overexpression produced the opposite effect (P < 0.01, Fig. 5I).

Bioinformatics combined with in vitro experiments to determine that JUP/AGR2/LYPD3 signaling in melanoma cells can maintain stemness. A Boxplot showing the difference in RNA stemness score between C1 and C2. B UMAP plot of melanoma cells. C, D tSNE plot demonstrating the degree of differentiation of each melanoma cell cluster assessed by CytoTRACE. E Boxplot showing the differentiation score of melanoma cell cluster. F Volcano graph demonstrating the feature genes of each melanoma cell cluster. G UMAP plot showing the co-localization of JUP, AGR2, and LYPD3. H Images of colony formation assay in control, NC, oe-JUP, sh-JUP, and oe-JUP + sh-LYPD3 melanoma cell group. I Images and quantitative analysis of sphere-forming assay in control, NC, oe-JUP, sh-JUP, and oe-JUP + sh-LYPD3 melanoma cell group. J–K Bubble and UMAP plots demonstrating upregulation of glycolysis levels in cluster 1 melanoma cells. L Lollipop plot demonstrating the Spearman correlation between JUP/AGR2/LYPD3 signaling and five key enzymes of glycolysis in TCGA-SKCM. Only SLC2A1 (GLUT1) is consistently and significantly correlated with JUP, AGR2, and LYPD3. (M–N) Verification of GLUT1 expression by Western blotting assay in control, NC, oe-JUP, sh-JUP, and oe-JUP + sh-LYPD3 melanoma cell group. One-way ANOVA was applied. **P < 0.01, ***P < 0.001, ****P < 0.0001
The Warburg effect is a common metabolic phenotype in tumors, where CSCs are heavily dependent on glycolysis [42]. We performed the scMetabolism tool to assess the metabolic profile of each melanoma cell subpopulation, and the results showed that the metabolic level of cluster 1 was significantly higher than that of the other subpopulations, especially glycolysis (Fig. 5J, K). To determine by what pathway JUP/AGR2/LYPD3 signaling affects metabolism, we calculated the correlation of signaling molecules with six key enzymes of glycolysis based on TCGA-SKCM (Fig. 5L). The results showed that Solute Carrier Family 2 Member 1 (SLC2A1, also named GLUT1) was significantly positively correlated with all three signaling molecules (P < 0.05). Western blot confirmed that GLUT1 is a downstream regulatory target of JUP/AGR2/LYPD3 signaling (Fig. 5M, N). Together, the results suggest that JUP/AGR2/LYPD3 signaling plays a role in maintaining tumor cell stemness and enhances the glycolytic phenotype.
High-dimension weighted correlation network analysis (hdWGCNA) analysis highlights the biological properties of cluster 1
Herein, hdWGCNA pipeline was used to explore the potential features of cluster 1. As shown in Fig. 6A, B, nine gene modules (M1-M9) were obtained, with the top hub gene distributed along the hdWGCNA pipeline. Figure 6C illustrates the correlation between modules. We focused on modules significantly associated with cluster 1 and found that M1 is highly expressed on cluster 1 (red box, Fig. 6D, E, Additional file 1: Fig. S6). Enrichment analysis showed that M1 is involved in a large number of cancer-associated signaling pathways, including glycolysis, cadherin binding, and hypoxia-inducible factor (HIF)-1 signaling (Fig. 6F–G). Further analysis showed that all hub genes in M1 were overexpressed in tumor tissues and were detrimental to OS (Fig. 6H, I, HR > 1). Parallelly, the cell–cell communication analysis was also performed using cellchat tool (Fig. 6J, K). Interestingly, compared to the subpopulation of highly differentiated cells (cluster 0), the subpopulation of poorly differentiated cells (cluster 1) had a relatively weak overall ability to communicate with T cells (Fig. 6L–O). This implies that cluster 1 may contribute to the immunosuppressive state of TME.

High-dimension Weighted Correlation Network Analysis (hdWGCNA) of melanoma cell reveals the crucial roles of C1. A Dendrogram demonstrating nine gene modules in the scale-free network. B Nine gene modules and the associated top 10 hub gene were displayed according to the hdWGCNA pipeline. C Correlation between nine gene modules. Green represents negative correlation and purple represents positive correlation. D Nine module levels in different melanoma cell clusters. E Violin plot demonstrating that Module 1 (M1) activity is significantly higher in Cluster 1 than in the other melanoma cell clusters. F Protein–protein interaction (PPI) network for the top 25 genes in M1. G GO and KEGG enrichmnt analysis of M1. H Expression level of M1 hub genes between melanoma tissues in the TCGA-SKCM cohort (n = 469) and control tissues in in both the TCGA and GTEx databases (n = 813). Wilcoxon test, ***P < 0.05. I Forest plot showing the results of Cox regression analysis of the mean survival (overall survival) of the 10 M1 hub genes in the TCGA-SKCM cohort. J–K All cell types were analyzed by Cellchat and melanoma cells were classified according to stemness level as cluster 1 (high), cluster 0 (low), and other melanoma cells (medium). The results showed both interaction numbers and interaction strengths. L–O Cellchat analysis showed weaker communication strength between melanoma cell cluster 1 and T cells compared to melanoma cell cluster 0
JUP/AGR2/LYPD3 signaling promotes tumor growth and remodels the cytoskeleton
We first demonstrated in vivo that upregulation of JUP levels significantly increased tumor growth rate and weight compared to controls, and that this effect was partially reversed by sh-LYPD3 (Fig. 7A–C, P < 0.05). In addition, JUP/AGR2/LYPD3 signaling was shown to have a regulatory role for GLUT1 in vivo (Fig. 7D, P < 0.0001).

JUP/AGR2/LYPD3 signaling on melanoma cells promotes tumor growth and controls cytoskeletal remodeling. A–C In vivo experiments showed that overexpression of JUP significantly promoted tumor growth and that this effect could be attenuated by sh-LYPD3. D JUP/AGR2/LYPD3 signaling significantly promoted the expression of the key glycolytic enzyme GLUT1 in vivo. E–H JUP/AGR2/LYPD3 signaling significantly promoted the expression of the cytoskeletal protein F-actin in vivo (E, F) and had similar effects in vitro (G, H). One way ANOVA was conducted. I Phalloidin staining showed that JUP/AGR2/LYPD3 signaling remodelled the cytoskeleton and generated more pseudopod structures (yellow arrows). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Alterations in gene transcription and biological function of cells are often accompanied by morphological changes. It has been established that CSCs drive tumor progression by promoting an invasive phenotype [43]. Furthermore, the highly dynamic and polarized actin cytoskeleton is required for tumor cell migration and invasion into healthy tissue [44]. We demonstrated in vivo that upregulation of JUP/AGR2/LYPD3 signaling contributes to the regulation of the actin cytoskeleton (Fig. 7E, F, P < 0.05) and obtained similar results in in vitro experiments (Fig. 7G, H, P < 0.05). More importantly, cytoskeletal remodeling induced by polymerization of newly generated actin means that cells are able to form specialized cytoarchitectures such as denser filopodia, lamellipodia, and invadopodia. We stained and visualized the cytoskeleton using phalloidin and found that JUP/AGR2/LYPD3 can help melanoma cells form pseudopodia (Fig. 7I).
Identification of LYPD3 as an immunosuppressive factor
As a well-known immunogenic cancer, melanoma is impressive for its abundant antigen and antigen-specific lymphocytes [45]. Therefore, in order to survive and metastasize, melanoma cells must acquire robust immune evasion properties, usually through various pathways mediating T cell dysfunction thereby shaping a broadly immunosuppressive TME [46, 47]. In this study, the “ssGSEA” method was used to estimate immune cell infiltration. We observed a weak negative correlation between LYPD3 level and CD8 + T cell abundance (Fig. 8A, R = − 0.118, P < 0.05). Interestingly, the negative correlation between LYPD3 and CD8 + T cells was more obvious in C2 compared to C1, and the abundance of immune cell infiltration was lower in C2 (Fig. 8B–D). These data implied that LYPD3 appeared to correlate with the immunosuppressive profile of C2. Considering that we have demonstrated the regulatory function of LYPD3 on the maintenance of tumor cell stemness, and that CSCs are usually accompanied by increased expression of immunosuppressive pathways, we speculated that LYPD3 may be an immunosuppressive factor. To test this conjecture, we included an ICB dataset to examine the relationship between LYPD3 expression and immunotherapy response. The results showed that LYPD3 expression was lower in responsive patients than in non-responsive patients and that LYPD3 levels decreased in patients after ICB treatment (Fig. 8E, F). In addition, patients with high LYPD3 expression had a poor prognosis (Fig. 8G). Although statistical significance was not reached due to the relatively small sample size, we observed a positive correlation between LYPD3 levels and T-cell dysfunction score (Fig. 8H, R = 0.29, P = 2.5e−10).

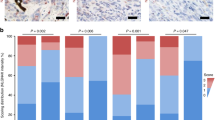
LYPD3 positively correlates with myeloid immunosuppressive marker S100A9. A In TCGA-SKCM, LYPD3 levels were weakly negatively correlated with CD8 + T cell infiltration abundance. B, C In C2 (C), the level of LYPD3 was significantly negatively correlated with the number of CD8 + T cells compared to in C1 (B). Green represents negative correlation and red represents positive correlation. D Boxplot indicating the infiltration level of immune cells in different catenin phenotypes in the TCGA-SKCM database. Wilcoxon test was conducted. E Boxplot showing different LYPD3 expression between pre-therapy and post-therapy in 24 melanoma patients studied by Tavi Nathanson et al. F Boxplot showing different LYPD3 expression between responder and non-responder after anti-CTLA4 treatment in 24 melanoma patients studied by Tavi Nathanson et al. Wilcoxon test was conducted. G Kaplan–Meier plot showing different overall survival outcomes between high and low LYPD3 patient groups in all patients by using data from Tavi Nathanson et al. Logrank test was applied. H Correlation between LYPD3 level and T cell dysfunction score in data of Tavi Nathanson et al. Spearman method was used. I UMAP plot showing cell types identified by using single-cell RNA-seq dataset GSE120575. J–K Distribution of LYPD3 expression in different myeloid cell clusters and LYPD3 was co-expressed with S100A9. (L-M) UMAP plot (L) and histogram (M) showing that myeloid cells were highly enriched in non-responder patients. N Histogram showing the proportional expression values of LYPD3 in myeloid cells of responder (0%) and non-responder (6.18%) patients. O–P Trajectory analysis of myeloid cells and cellular LYPD3 expression profiles in a two-dimensional space. Each point represents an individual cell, coloured by cell type. The solid black line indicates the main diameter path of the minimum spanning tree (MST). Q Spearman Correlation between levels of LYPD3 and S100A9 in TCGA-SKCM (left) and merged GEO (mGEO, right) cohort. R Left: Immunohistochemical (IHC) images of high/low protein expression of LYPD3 and S100A9. Scale bars: 100 µM. Right: Quantifications of LYPD3 and S10A9 IHC staining (H-score) in melanoma cohort (n = 30) showing the positive association between LYPD3 and S100A9 (R = 0.723, P < 0.001). S The IF staining for LYPD3 and S100A9 in sections derived from melanoma biopsies. Scale bar, 50 µM. T The co-localization correlation between LYPD3 and S100A9 was estimated by calculating co-localization coefficients using IamgeJ software. Greater correlation coefficients represent a greater degree of co-localization (Person R = 0.7423, P < 0.0001). *P < 0.05, **P < 0.01, ***P < 0.001
To further resolve the immunosuppressive mechanism of LYPD3, we examined the relationship between LYPD3 expression pattern and ICB response at single-cell resolution. As shown in Fig. 8I–K, LYPD3 was expressed on myeloid cells and T cells. In particular, LYPD3 was highly expressed on S100A9 + myeloid cells (red box, Mye_C6_S100A9). S100A9 is now thought to be a calcium-binding protein that promotes the accumulation of myeloid-derived suppressor cells (MDSCs) and mediates the formation of malignant loops between MDSCs and CD8 + T cells [48,49,50]. Further analysis showed that myeloid cells were more abundant in non-responsive patients (87.25%) compared to responder patients (12.75%) and that LYPD3 was almost exclusively expressed in non-responsive myeloid cells (Fig. 8L–N), reinforcing the evidence that LYPD3 is a detrimental factor for immunotherapy. Pseudotime analysis showed that LYPD3 is involved in the differentiation trajectory of myeloid cells (Fig. 8O-P). Finally, we analyzed the correlation between S100A9 and LYPD3. Bulk data showed a significant positive correlation (Fig. 8Q; left, TCGA-SKCM, R = 0.493, P < 0.001; right, mGEO, R = 0.120, P = 0.015). In addition to the findings at the transcriptional level, Immunohistochemical (IHC) staining confirmed that the protein level of S100A9 increased with increasing LYPD3 protein expression (Fig. 8R, R = 0.723, P < 0.001). The co-expression pattern of LYPD3 and S100A9 was confirmed by multiple immunofluorescence (mIF) staining of the same melanoma resection specimen (Fig. 8S-T). Thus, LYPD3 was identified to exert immunosuppressive functions through S100A9-related signaling.
Discussion
Β-catenins are now considered to be classical downstream targets of Wnt signaling and play a key role in cell–cell junctions [51]. Disturbances in Wnt/β-catenin signaling often lead to tumorigenesis [52, 53]. In most cases, there are several additional catenins that are structurally as well as functionally related to β-catenin, as evidenced in particular by the fact that other catenins are responsive to classical Wnt signaling [54]. There is growing evidence that multiple catenin proteins localized in the nucleus form interactive networks involved in the regulation of gene expression and protein interactions, particularly in the context of actin-based cell morphology and cell motility [55,56,57,58,59].
In the present study, we described the genomic and transcriptomic heterogeneity of 12 catenins at the pan-cancer level and found that the aberrant expression of catenins may be associated with genomic variants, which is consistent with current reports. Interestingly, there are 2 RNA expression patterns of catenins in TCGA-SKCM, where PKP1, JUP, PKP3 belong to the same group and are deleterious for survival in melanoma patients. Among them, JUP belongs to the β-catenin superfamily and is structurally similar to β-catenin, while PKP1 and PKP3 belong to the plakophilin branch, which belongs to the largest subfamily, δ-catenins [60]. This seems to suggest a potential heterogeneity of catenins. In TCGA-SKCM, we found that the collective up-regulation of JUP, PKP1, and PKP3 could define a distinct catenin subgroup in melanoma (C2), which possesses poorer survival and more aggressive clinical features compared to another subgroup (C1). Previous studies confirmed that PKP1 and PKP3 levels are elevated in several cancer tissues compared to normal tissues and regulate the proliferation and invasive capacity of cancer cells. Our study suggests that imbalanced expression of catenins correlates with the malignant features of melanoma.
Transcriptional differences between different catenin subtypes have been shown to involve a wide range of biological signals [61, 62]. To mine key regulatory mechanisms, MEGENA was performed to characterize the correlations between genes and genes in the module. Based on the correlation network, we obtained the 10 most significant hub genes, among which LYPD3 had the highest expression abundance and was therefore identified for study. LYPD3, also known as C4.4A, is a membrane protein that is partially anchored to the cell surface by glycosylphosphatidylinositol (GPI) [63]. LYPD3 has been shown to be highly expressed in several human malignancies and is associated with poor prognosis [37, 38, 64]. Interestingly, LYPD3 expression is markedly attenuated in the non-invasive stage of cutaneous malignant lesions (carcinoma in situ), but re-activated upon transformation to malignant invasive squamous cell carcinoma, especially in the invasive front region. This wonderful phenomenon links LYPD3 to the process of tumor infiltration. In the present study, we found that LYPD3 levels were accompanied by a progressive increase in the depth of melanoma infiltration and confirmed the statistical correlation between high LYPD3 level and poor OS and DSS. A study showed that LYPD3 stimulates proliferation, migration, invasion and drug resistance of pancreatic ductal adenocarcinoma (PDAC) cells by interacting with AGR2 [41]. In the present study, we demonstrated that JUP can regulate the level of AGR2/LYPD3 and constructed the JUP/AGR2/LYPD3 signaling axis. It was shown that hyperactivation of the JUP/AGR2/LYPD3 signaling axis contributes to the regulation of melanoma cell stemness and promotes glycolysis through a Glucose Transporter Type 1 (GLUT1)-dependent pathway. In colorectal cancer models, LYPD3 was found to be preferentially expressed in CSCs and to play a role in maintaining CSCs [65]. Notably, the correlation between JUP, AGR2, and LYPD3 was significantly stronger in C2 than in C1, which may be able to partially explain the higher stemness score in C2 than in C1.
A central feature of catenins is their association with calreticulin, and the two form protein complexes that directly or indirectly regulate cytoskeletal shaping, which allows them to be extensively involved in cell adhesion, motility, and material transfer [59, 66,67,68]. In addition, CSCs are morphologically characterized as more invasive. In the present study, we demonstrated that upregulation of JUP/AGR2/LYPD3 signaling promotes melanoma cytoskeleton remodeling and stimulates the formation of new pseudopods.
The expression of immunosuppressive cytokines and inhibitory co-stimulatory molecules in CSCs gives CSCs an immunosuppressive potential [69]. Our data suggest that melanoma cells in an undifferentiated state have weak communication with immune cells. Notably, C2 presented an immunosuppressed state (low level of immune infiltration) and we observed a negative correlation between LYDP3 level and CD8 + T cells only in C2, but not in C1. These results imply a link between LYPD3 and immunosuppressive TME. Subsequent analysis showed that LYPD3 was co-expressed on myeloid cells with S100A9, which has been shown to mediate dysfunction of effector T cells thereby affecting the response to immunotherapy. Thus, LYPD3 is a potential immunosuppressive factor.
This study extends the biological significance of the JUP/AGR2/LYPD3 signaling axis to melanoma. The development of an anti-tumour drug (bay 1129980) targeting C4.4A is currently underway and has shown effective therapeutic effects in a human-derived tumour xenograft model [36]. We hope to provide some basis for expanding the clinical applications of this drug. We recognise that our study has some limitations. The clinical studies included in this study are based on public databases and may be biased. We are collecting patients in a multicentre clinical cohort to further analyse and validate the significance of LYPD3 in immunotherapy, and large-scale sequencing analyses are necessary. Our findings suggest that the JUP/AGR2/LYPD3 signaling axis plays an important role in melanoma, but its in-depth mechanism of action is still lacking. Our team is conducting further research on this topic.
Conclusion
The JUP/AGR2/LYPD3 signaling axis plays an important role in the malignant features of melanoma. Targeting the JUP/AGR2/LYPD3 signaling axis can help develop promising drugs.
Data Availability
The data and materials in the current study are available from the corresponding author: Song-yang Xi: 20215011@njucm.edu.cn.
References
Ahmed B, Qadir MI, Ghafoor S. Malignant melanoma: skin cancer-diagnosis, prevention, and treatment. Crit Rev Eukaryot Gene Expr. 2020;30(4):291–7.
Dzwierzynski WW. Melanoma risk factors and prevention. Clin Plast Surg. 2021;48(4):543–50.
Eddy K, Chen S. Overcoming immune evasion in melanoma. Int J Mol Sci. 2020;21(23):8984.
Marzagalli M, Ebelt ND, Manuel ER. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin Cancer Biol. 2019;59:236–50.
Mao L, et al. Immunotherapy in acral and mucosal melanoma: current status and future directions. Front Immunol. 2021;12: 680407.
Baxi S, et al. Immune-related adverse events for anti-PD-1 and anti-PD-L1 drugs: systematic review and meta-analysis. BMJ. 2018;360: k793.
Li B, Chan HL, Chen P. Immune checkpoint inhibitors: basics and challenges. Curr Med Chem. 2019;26(17):3009–25.
Morad G, et al. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021;184(21):5309–37.
Topalian SL, et al. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–87.
Zhang A, et al. Tumor heterogeneity reshapes the tumor microenvironment to influence drug resistance. Int J Biol Sci. 2022;18(7):3019–33.
Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. 2020;30(16):R921-r925.
Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell. 2020;78(6):1019–33.
Mihm MC, Mulé JJ. Reflections on the histopathology of tumor-infiltrating lymphocytes in melanoma and the host immune response. Cancer Immunol Res. 2015;3(8):827–35.
Paijens ST, et al. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol Immunol. 2021;18(4):842–59.
Gul IS, et al. Evolution and diversity of cadherins and catenins. Exp Cell Res. 2017;358(1):3–9.
Chai G, et al. Complete functional segregation of planarian beta-catenin-1 and -2 in mediating Wnt signaling and cell adhesion. J Biol Chem. 2010;285(31):24120–30.
Jackson BM, Eisenmann DM. β-catenin-dependent Wnt signaling in C. elegans: teaching an old dog a new trick. Cold Spring Harb Perspect Biol. 2012;4(8):a007948.
Muto S, et al. Wnt/β-catenin signaling and resistance to immune checkpoint inhibitors: from non-small-cell lung cancer to other cancers. Biomedicines. 2023;11(1):190.
McCrea PD, Gu D. The catenin family at a glance. J Cell Sci. 2010;123(Pt 5):637–42.
Hu G. p120-Catenin: a novel regulator of innate immunity and inflammation. Crit Rev Immunol. 2012;32(2):127–38.
Huang F, et al. δ-Catenin promotes tumorigenesis and metastasis of lung adenocarcinoma. Oncol Rep. 2018;39(2):809–17.
Lu Q, et al. Genetic alterations of δ-catenin/NPRAP/Neurojungin (CTNND2): functional implications in complex human diseases. Hum Genet. 2016;135(10):1107–16.
McCrea PD, Park JI. Developmental functions of the P120-catenin sub-family. Biochim Biophys Acta. 2007;1773(1):17–33.
Miller RK, et al. Beta-catenin versus the other armadillo catenins: assessing our current view of canonical Wnt signaling. Prog Mol Biol Transl Sci. 2013;116:387–407.
Zang X, et al. Circular RNA EIF4G3 suppresses gastric cancer progression through inhibition of β-catenin by promoting δ-catenin ubiquitin degradation and upregulating SIK1. Mol Cancer. 2022;21(1):141.
Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778(3):660–9.
Stepniak E, Radice GL, Vasioukhin V. Adhesive and signaling functions of cadherins and catenins in vertebrate development. Cold Spring Harb Perspect Biol. 2009;1(5): a002949.
Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol. 2016;11:47–76.
Clarke MF. Clinical and therapeutic implications of cancer stem cells. N Engl J Med. 2019;380(23):2237–45.
Babaei G, Aziz SG, Jaghi NZZ. EMT, cancer stem cells and autophagy: the three main axes of metastasis. Biomed Pharmacother. 2021;133: 110909.
Lambert AW, Weinberg RA. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer. 2021;21(5):325–38.
Ruiz de Galarreta M, et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9(8):1124–41.
Liu Y, et al. Construction and validation of a ferroptosis-related prognostic signature for melanoma based on single-cell RNA sequencing. Front Cell Dev Biol. 2022;10: 818457.
Yan K, et al. 9-gene signature correlated with CD8(+) T cell infiltration activated by IFN-γ: a biomarker of immune checkpoint therapy response in melanoma. Front Immunol. 2021;12: 622563.
Chen, Z., et al., TIGER: a web portal of tumor immunotherapy gene expression resource. Genomics Proteomics Bioinformatics, 2022.
Willuda J, et al. Preclinical antitumor efficacy of BAY 1129980-a novel Auristatin-based anti-C44A (LYPD3) antibody-drug conjugate for the treatment of non-small cell lung cancer. Mol Cancer Ther. 2017;16(5):893–904.
Hu T, et al. LYPD3, a new biomarker and therapeutic target for acute myelogenous Leukemia. Front Genet. 2022;13: 795820.
Hu P, et al. Elevated expression of LYPD3 is associated with lung adenocarcinoma carcinogenesis and poor prognosis. DNA Cell Biol. 2020;39(4):522–32.
Moreno-Ramírez D, et al. Association between tumor size and Breslow’s thickness in malignant melanoma: a cross-sectional, multicenter study. Melanoma Res. 2015;25(5):450–2.
Jiang Y, et al. Crystal structures of human C44A reveal the unique association of Ly6/uPAR/α-neurotoxin domain. Int J Biol Sci. 2020;16(6):981–93.
Arumugam T, et al. New blocking antibodies against novel AGR2-C44A pathway reduce growth and metastasis of pancreatic tumors and increase survival in mice. Mol Cancer Ther. 2015;14(4):941–51.
Schwartz L, Supuran CT, Alfarouk KO. The Warburg effect and the hallmarks of cancer. Anticancer Agents Med Chem. 2017;17(2):164–70.
Valent P, et al. Redistribution, homing and organ-invasion of neoplastic stem cells in myeloid neoplasms. Semin Cancer Biol. 2020;60:191–201.
Klein RM, Aplin AE. Rnd3 regulation of the actin cytoskeleton promotes melanoma migration and invasive outgrowth in three dimensions. Cancer Res. 2009;69(6):2224–33.
Kelderman S, et al. Antigen-specific TIL therapy for melanoma: a flexible platform for personalized cancer immunotherapy. Eur J Immunol. 2016;46(6):1351–60.
Umansky V, Sevko A. Melanoma-induced immunosuppression and its neutralization. Semin Cancer Biol. 2012;22(4):319–26.
Petrova V, et al. Modern aspects of immunotherapy with checkpoint inhibitors in melanoma. Int J Mol Sci. 2020;21(7):2367.
Huang M, et al. S100A9 regulates MDSCs-mediated immune suppression via the RAGE and TLR4 signaling pathways in colorectal carcinoma. Front Immunol. 2019;10:2243.
Li J, et al. S100A9-CXCL12 activation in BRCA1-mutant breast cancer promotes an immunosuppressive microenvironment associated with resistance to immunotherapy. Nat Commun. 2022;13(1):1481.
Feng PH, et al. CD14(+)S100A9(+) monocytic myeloid-derived suppressor cells and their clinical relevance in non-small cell lung cancer. Am J Respir Crit Care Med. 2012;186(10):1025–36.
Tsang JY, et al. Involvement of α- and β-catenins and E-cadherin in the development of mammary phyllodes tumours. Histopathology. 2012;61(4):667–74.
Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: update on effectors and inhibitors. Cancer Treat Rev. 2018;62:50–60.
Zhou Y, et al. Wnt signaling pathway in cancer immunotherapy. Cancer Lett. 2022;525:84–96.
Duñach M, Del Valle-Pérez B, García de Herreros A. p120-catenin in canonical Wnt signaling. Crit Rev Biochem Mol Biol. 2017;52(3):327–39.
Kosik KS, et al. Delta-catenin at the synaptic-adherens junction. Trends Cell Biol. 2005;15(3):172–8.
Aberle H, Schwartz H, Kemler R. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem. 1996;61(4):514–23.
Wang A, Dunn AR, Weis WI. Mechanism of the cadherin-catenin F-actin catch bond interaction. Elife. 2022;11:e80130.
Gielata M, et al. Emerging roles of the α-catenin family member α-catulin in development, homeostasis and cancer progression. Int J Mol Sci. 2022;23(19):11962.
Zhurinsky J, Shtutman M, Ben-Ze’ev A. Plakoglobin and beta-catenin: protein interactions, regulation and biological roles. J Cell Sci. 2000;113(Pt 18):3127–39.
Nieset JE, et al. Characterization of the interactions of alpha-catenin with alpha-actinin and beta-catenin/plakoglobin. J Cell Sci. 1997;110(Pt 8):1013–22.
Palacios J, et al. Beta- and gamma-catenin expression in endometrial carcinoma. Relationship with clinicopathological features and microsatellite instability. Virchows Arch. 2001;438(5):464–9.
Kolligs FT, et al. gamma-catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of beta-catenin. Genes Dev. 2000;14(11):1319–31.
Ohtsuka M, et al. Concurrent expression of C4.4A and Tenascin-C in tumor cells relates to poor prognosis of esophageal squamous cell carcinoma. Int J Oncol. 2013;43(2):439–46.
Paret C, et al. C4.4A as a candidate marker in the diagnosis of colorectal cancer. Br J Cancer. 2007;97(8):1146–56.
Wang L, et al. LY6/PLAUR domain containing 3 has a role in the maintenance of colorectal cancer stem-like cells. Biochem Biophys Res Commun. 2017;486(2):232–8.
Fadel MP, et al. Calreticulin affects beta-catenin-associated pathways. J Biol Chem. 2001;276(29):27083–9.
Opas M, et al. Calreticulin modulates cell adhesiveness via regulation of vinculin expression. J Cell Biol. 1996;135(6 Pt 2):1913–23.
Liu X, et al. HIF-1-regulated expression of calreticulin promotes breast tumorigenesis and progression through Wnt/β-catenin pathway activation. Proc Natl Acad Sci U S A. 2021;118(44):2109144.
Tsuchiya H, Shiota G. Immune evasion by cancer stem cells. Regen Ther. 2021;17:20–33.
Acknowledgements
We thank the TCGA and GEO databases for permitting the authors to use the data.
Funding
The present study was supported by Youth Science and Technology Project of Zhangjiagang (No. ZJGQNKJ202309), Social Development Guiding Science and Technology Plan Project of Zhenjiang (No. FZ2021065), Science and Technology Project of Jiangsu Provincial Administration of Traditional Chinese Medicine (No. YB201843), the Youth Science and Technology Project of Zhangjiagang (No. ZJGQNKJ202036), Suzhou Science and Education Promotion Health Project (No. KJXW2021065), Talent and Technology Construction Fund Project of Zhenjiang Hospital of Chinese Traditional and Western Medicine (No. ZXY202301), Natural Science Foundation of China (82205212).
Author information
Authors and Affiliations
Contributions
YH and KW wrote the manuscript and performed all experiments. YL, QZ analyzed the data. HS and JJ support and providing equipment/instrumentation. DF and SX developed the experimental plan and provided the research funding. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The Zhangjiagang TCM Hospital, an affiliate Hospital of the Nanjing University of Traditional Chinese Medicine, Ethics and Research Committee, accepted the research (Approval Number: 2022-03-014).
Informed consent
The research complies with the Helsinki Declaration. All patients have provided their written, voluntarily informed consent.
Competing interest
The authors declare no conflicts of interest. All authors contributed to data analysis, drafting, or revising of the article; agree on the journal to which the article is being submitted; provided final approval of the version to be published; agree to be accountable for all aspects of the work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hu, Yd., Wu, K., Liu, Yj. et al. LY6/PLAUR domain containing 3 (LYPD3) maintains melanoma cell stemness and mediates an immunosuppressive microenvironment. Biol Direct 18, 72 (2023). https://doi.org/10.1186/s13062-023-00424-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13062-023-00424-3