
Abstract
Background
Overexpression of fibroblast growth factor receptor 1 (FGFR1), found in ≤8% of hormone receptor–positive (HR+), human epidermal growth factor receptor 2–negative (HER2−) breast cancer cases, is correlated with decreased overall survival and resistance to endocrine therapy (ET). Dovitinib, a potent FGFR inhibitor, has demonstrated antitumor activity in heavily pretreated patients with FGFR pathway–amplified breast cancer.
Methods
In this randomized, placebo-controlled phase II trial, we evaluated whether the addition of dovitinib to fulvestrant would improve outcomes in postmenopausal patients with HR+, HER2− advanced breast cancer that had progressed during or after prior ET. Patients were stratified by FGF pathway amplification and presence of visceral disease, and they were randomized 1:1 to receive fulvestrant plus dovitinib or placebo. The primary endpoint was progression-free survival (PFS).
Results
From 15 May 2012 to 26 November 2014, 97 patients from 36 centers were enrolled. The frequency of FGF pathway amplification was lower than anticipated, and the study was terminated early owing to slow accrual of patients with FGF pathway amplification. The median PFS (95% CI) was 5.5 (3.8–14.0) months vs 5.5 (3.5–10.7) months in the dovitinib vs placebo arms, respectively (HR, 0.68; did not meet predefined efficacy criteria). For the FGF pathway–amplified subgroup (n = 31), the median PFS (95% CI) was 10.9 (3.5–16.5) months vs 5.5 (3.5–16.4) months in the dovitinib vs placebo arms, respectively (HR, 0.64; met the predefined superiority criteria). Frequently reported adverse events in the dovitinib (diarrhea, nausea, vomiting, asthenia, and headache) and placebo (diarrhea, fatigue, nausea, and asthenia) arms were mostly low grade.
Conclusions
The safety profile of dovitinib plus fulvestrant was consistent with the known safety profile of single-agent dovitinib. Dovitinib in combination with fulvestrant showed promising clinical activity in the FGF pathway–amplified subgroup. However, the data reported herein should be interpreted with caution, given that fewer PFS events occurred in the FGF pathway–amplified patients than was expected and that an effect of dovitinib regardless of FGR pathway amplification status cannot be excluded, because the population was smaller than expected.
Trial registration
ClinicalTrials.gov identifier: NCT01528345. Registered 31 January 2012.
Similar content being viewed by others


Background
Breast cancer is the most common type of cancer and the leading cause of cancer deaths in women worldwide [1]. In most countries, 3% to 12% of breast cancers are advanced or metastatic at diagnosis [2]. Most breast cancers are hormone receptor–positive (HR+), with 75% to 83% of breast cancers expressing estrogen receptor (ER)-α and/or progesterone receptor [3–5]. Likewise, approximately 86% to 87% of breast cancers are negative for overexpression of human epidermal growth factor receptor 2 (HER2−) [6, 7].
Currently, endocrine therapy is recommended as initial therapy for patients with HR+, HER2− advanced breast cancer [8, 9]. Aromatase inhibitors are the standard of care for postmenopausal patients [9]. However, only 20% to 40% of patients respond to first-line therapy, and approximately one-half of responders relapse within 8–14 months [10]. Most patients eventually relapse because currently available treatments are not curative [11]. Second-line endocrine therapy (e.g., fulvestrant, aromatase inhibitors, tamoxifen) is recommended following relapse, but the response is generally short-lived. For example, the duration of response (DOR) to second-line fulvestrant or exemestane is approximately 3–5 months [12].
Several mechanisms of endocrine therapy resistance have been described, including activation of receptor tyrosine kinases (e.g., fibroblast growth factor receptor [FGFR]) and their downstream signaling pathways (e.g., phosphoinositide 3-kinase [PI3K]/Akt/mechanistic target of rapamycin [mTOR]), as well as activation of the cyclin-dependent kinases 4 and 6 that regulate cell cycle progression [13]. Efforts to improve outcomes and reduce endocrine therapy resistance have led to the development of combination therapies that included targeted agents against these resistance pathways. Positive results from the phase III Breast Cancer Trials of Oral Everolimus 2 (BOLERO-2) trial led to the approval of everolimus, an mTOR inhibitor, in combination with second-line exemestane in postmenopausal women with HR+, HER2− advanced breast cancer that progressed during prior nonsteroidal aromatase inhibitor therapy [14, 15]. Later, positive results from the Palbociclib: Ongoing Trials in the Management of Breast Cancer (PALOMA) studies led to the approval of palbociclib (a cyclin-dependent kinase 4 and 6 inhibitor) as first-line therapy in combination with letrozole and as second-line therapy in combination with fulvestrant [16, 17].
Aberrant regulation of fibroblast growth factor (FGF) and FGFR signaling is associated with tumorigenic activity [18], an increased risk of developing breast cancer [19–21], and resistance to endocrine therapy [13]. Amplifications in FGFR1 and FGFR4 are found in 9% to 10% and 10% of primary breast cancers overall, respectively [22–25]. FGFR1 amplification is more frequently associated with luminal B cancer, whereas FGFR4 amplification is more common in HR+ tumors and a subset of HER2+ tumors [22, 26, 27]. FGFR2 amplification is present in 4% of triple-negative breast cancers [28]. Overexpression of FGFR family members is associated with poor prognosis, including reduced overall survival (OS), disease-free survival, and relapse-free survival [24, 29–31]. FGFR overexpression is also associated with resistance to hormone therapy [26, 32] and chemotherapy [33, 34]. Importantly, FGFR1-induced tamoxifen resistance can be reversed by inhibiting FGFR1 expression [32]. Aberrant PI3K/Akt/mTOR signaling is also seen in cells with FGFR1 overexpression and amplification [26], and response to the PI3K inhibitor alpelisib is reduced in ER+/PIK3CA-mutant breast cancer cells that overexpress FGFR1 [35]. Taken together, these results provide rationale for the investigation of FGFR inhibitors in breast cancer therapy.
Dovitinib (TKI258), a small-molecule inhibitor of FGFR1, FGFR2, and FGFR3 and other receptor tyrosine kinases [36], has shown preclinical activity in FGFR-expressing breast cancer models in vivo and in vitro [37]. Dovitinib inhibited cell proliferation in FGFR-amplified cell lines and showed antitumor activity in FGFR-amplified xenograft models [38]. In a phase II trial of single-agent dovitinib, encouraging clinical activity was observed in patients with HR+, HER2− FGF pathway–amplified breast cancer [38]. FGF pathway amplification status was determined using in situ hybridization as part of the eligibility criteria (FGFR1 only) and using quantitative polymerase chain reaction (qPCR) as an exploratory analysis (FGFR1, FGFR2, and FGF3). Correlative studies between FGF pathway amplification markers and antitumor activity indicated that dovitinib activity was higher in patients who had FGF pathway amplification measured by qPCR, particularly in those who had higher levels of FGFR1 amplification (i.e., at least six copies of FGFR1) [38]. The combination of fulvestrant and dovitinib could potentially overcome resistance to endocrine therapy, thereby reducing the need for cytotoxic chemotherapy in relapsed patients. Together, these data and hypotheses prompted the initiation of this phase II, placebo-controlled trial of dovitinib plus fulvestrant in postmenopausal patients with HR+, HER2− locally advanced or metastatic breast cancer.
The primary objective of this study was to determine the effect of treatment with dovitinib in combination with fulvestrant vs placebo plus fulvestrant on progression-free survival (PFS) in postmenopausal patients with HR+, HER2− breast cancer that had progressed during or after prior endocrine therapy in all evaluable patients, regardless of FGF pathway amplification status, and in patients with FGF pathway amplification (as measured by qPCR using a cutoff of at least six copies of FGFR1, FGFR2, or FGF3). The key secondary objective was overall response rate (ORR). Additional secondary objectives included DOR, OS, safety, and pharmacokinetics of dovitinib.
Methods
Study design
We conducted a phase II, multicenter, international, randomized, double-blind, placebo-controlled trial (ClinicalTrials.gov identifier: NCT01528345) designed to evaluate the efficacy and safety of dovitinib in combination with fulvestrant in postmenopausal women with HR+, HER2− locally advanced or metastatic breast cancer who had evidence of disease progression. Enrolled patients were randomized in a 1:1 ratio to receive dovitinib plus fulvestrant or placebo plus fulvestrant, stratified by FGF pathway amplification status (amplified vs nonamplified) and presence of visceral disease (yes vs no). All patients received fulvestrant 500 mg (intramuscular injection once every 4 weeks, with an additional dose 2 weeks after the initial dose) and dovitinib (500 mg) or placebo orally following a weekly 5 days on and 2 days off schedule until death, loss to follow-up, disease progression, or consent withdrawal.
Patients and medications were randomized using automated systems. At the time of initial screening, enrolled patients received a patient number, which was used as the primary identifier for the patient throughout the study. The interactive response technology provider generated a randomized patient list, using a validated automated system, by randomly assigning patient numbers to randomization numbers. Each randomization number was linked to a treatment arm and a medication number. Patients were randomized 1:1 to each of the study arms, with 45 FGF pathway–amplified and 30 FGF pathway–nonamplified patients planned in each arm. Medications were separately randomized by the study sponsor using a validated automated system that randomly assigned medication numbers to medication packs containing each of the study treatments. In this double-blind study, patients, investigators, study team members, and anyone involved in the conduct of the study remained blinded to the identity of the treatment from the time of randomization until database lock. The study medication and placebo had identical packaging, labeling, appearance, and administration schedules to conceal the identity of the treatments.
Patients
Eligible patients were postmenopausal women with HR+, HER2− locally advanced or metastatic breast cancer who had evidence of disease progression. Progression was defined as at least one measurable lesion per Response Evaluation Criteria In Solid Tumors (RECIST) version 1.1 or at least one nonmeasurable lytic or mixed bone lesion in the absence of measurable disease. Progression could have occurred during or after prior endocrine therapy, within 12 months of the end of adjuvant endocrine therapy, or within 1 month of the end of any endocrine therapy for localized advanced or metastatic breast cancer. Eligible patients had confirmed postmenopausal status (i.e., aged ≥55 years with ≥1 year of amenorrhea, aged <55 years with ≥1 year of amenorrhea in the absence of ovarian suppression with an estradiol assay result of <20 pg/ml, or surgical menopause with bilateral oophorectomy), Eastern Cooperative Oncology Group (ECOG) performance status ≤2, and available archival or fresh tumor tissue for FGF pathway status determination in the primary tumor by the central laboratory. FGF pathway amplification was determined by a designated Clinical Laboratory Improvement Amendments–certified laboratory using a TaqMan PCR assay (Life Technologies, Carlsbad, CA, USA), as previously described [38]. Positive amplification for each FGF pathway marker tested (i.e., FGFR1, FGFR2, or FGF3) was defined as a copy number ≥6; copy number was quantified by comparison with a reference gene (human ribonuclease P RNA component H1) and calculated using CopyCaller software (version 1.1; Applied Biosystems, Foster City, CA, USA). Samples were considered to be FGF pathway–amplified if they had positive amplification of FGFR1, FGFR2, and/or FGF3. Given that the previously defined amplification cutoff of at least six copies for FGFR1 was associated with higher sensitivity to dovitinib monotherapy [38], the same cutoff was used in this combination therapy study. Exclusion criteria included HER2 overexpression (assessed by immunohistochemistry), prior therapy with fulvestrant (as a single agent or in combination with other therapies) or FGFR inhibitors, or chemotherapy or more than one line of any prior hormone therapy for locally advanced or metastatic breast cancer. An estimated 1000 patients were expected to be screened for FGF pathway amplification in order to identify and randomize a total of 150 patients stratified by FGF pathway amplification and presence of visceral disease.
Assessments
Radiographic assessments (computed tomography, magnetic resonance imaging, or radiography) were performed at screening, on day 5 of weeks 8 and 16, before fulvestrant administration every 8 weeks for the remainder of study treatment, and at the end of treatment (if not assessed within 8 weeks before visit). Safety assessments were performed continually until 30 days after the last study treatment. No additional tumor assessments were required to confirm response (complete response [CR] or partial response [PR]) outside the protocol-specified 8-week tumor assessment.
Patients who did not discontinue study treatment owing to disease progression or death, or who were not lost to follow-up or did not withdraw consent, were assessed every 8 weeks for disease status, ECOG performance status, and patient-reported outcomes until the start of new anticancer therapy, disease progression, death, loss to follow-up, or consent withdrawal. Survival follow-up was performed every 3 months until death, loss to follow-up, or consent withdrawal for patients who discontinued the treatment.
Study endpoints
The coprimary endpoints were PFS in the overall patient population regardless of FGF pathway amplification status and PFS in the subgroup of patients with FGF pathway amplification. PFS was defined as the time from date of randomization to the date of first radiologically documented, investigator-assessed disease progression per RECIST v1.1 or death due to any cause. The key secondary endpoint was ORR, defined as the percentage of patients with best overall response of CR or PR. Additional secondary endpoints included DOR, OS, safety, and pharmacokinetics of dovitinib. Safety analysis, by treatment arm, was based on the frequency of adverse events (AEs), summarized by system organ class, severity (based on the Common Terminology Criteria for Adverse Events version 4.03), type, and relationship to study treatment.
Analysis sets
The full analysis set, which consisted of all patients who were randomized and assigned study treatment, was the primary population for the efficacy endpoint analyses. The safety set consisted of all randomized patients who received at least one dose of any compound of the study treatment (dovitinib plus fulvestrant or placebo plus fulvestrant).
Data analysis
The primary endpoint, PFS, was evaluated in each of the treatment arms using three sets of comparisons, following a Bayesian design: (1) all patients regardless of FGF pathway amplification status, (2) FGF pathway–amplified, and (3) FGF pathway–nonamplified (Additional file 1). Patients who did not have a PFS event at the time of analysis or who had received further antineoplastic therapy were censored at the time of the last tumor assessment. Kaplan-Meier plots were generated by treatment arm for the full population, FGF pathway–amplified subpopulation, and FGF pathway–nonamplified subpopulation. The HR of PFS in the full population was estimated using a Cox proportional hazards model stratified by FGF pathway amplification status and presence of visceral disease (yes vs no).
Efficacy of dovitinib plus fulvestrant over placebo plus fulvestrant was established if the estimated HR was <0.68 for the full population (i.e., improvement of approximately 3.0 months in median PFS) or <0.65 for the FGF pathway–amplified subpopulation (i.e., improvement of approximately 3.5 months in median PFS). Futility criteria in the FGF pathway–nonamplified subpopulation was determined if the posterior probability (HR >0.81) was >50% (i.e., improvement of <1.5 months in median PFS). The number of PFS events needed for the final analysis was calculated by assuming a 10% prevalence of FGF pathway amplification and a median PFS of 6.5 months with fulvestrant and placebo. To achieve the required number of PFS events for the final analysis (≥90 in the full population and ≥50 in the FGF pathway–amplified subgroup, whichever occurred later), a total of 150 patients had to be randomized as follows: 75 patients per treatment arm (45 FGF pathway–amplified and 30 FGF pathway–nonamplified).
Separate interim analyses were planned for patients with and without FGF pathway amplifications, owing to the faster enrollment expected for the FGF pathway–nonamplified subgroup. The first interim analyses occurred when 36 PFS events had been documented in the FGF pathway–nonamplified subgroup, and the second interim analysis occurred when ≥10 (20%) of 50 PFS events had been documented in the FGF pathway–amplified subgroup. The intent of these interim analyses was to assess the efficacy or futility of the study treatment. If the futility criteria were met (HR >0.81 in the first interim analysis; HR >0.7 in the second interim analysis), the study could be terminated early by the data monitoring committee.
The key secondary endpoint, ORR, was summarized as a percentage rate with 95% CI. OS was estimated using Kaplan-Meier analysis for each treatment arm; patients still alive at the time of analysis were censored at the last contact date.
Results
Patient demographics
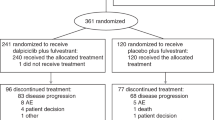
From 15 May 2012 to 26 November 2014, a total of 97 postmenopausal patients with HR+, HER2− locally advanced or metastatic breast cancer that had progressed during or after hormone therapy were enrolled in 36 centers. The last patient’s last visit was on 3 April 2015. All patients received fulvestrant; 47 were randomized to receive dovitinib and 50 were randomized to receive placebo (Fig. 1). Overall, 31 patients were classified as FGF pathway amplified (15 in the dovitinib arm vs 16 in the placebo arm); 725 patients were screened to enroll 31 FGF pathway–amplified patients. Although the data monitoring committee recommended continuing the study after 2 interim analyses, it subsequently recommended early termination of the study on 30 October 2014 due to lower than anticipated frequency of FGF pathway amplification and slow enrollment of patients with FGF pathway–amplified status. A total of 25 patients in each arm were classified as having visceral disease (53.2% vs 50.0% of patients in the dovitinib and placebo arms, respectively).

Consolidated Standards of Reporting Trials (CONSORT) diagram of patient disposition for patients randomized to receive dovitinib plus fulvestrant or placebo plus fulvestrant. FGF Fibroblast growth factor
Baseline characteristics, including age, ECOG performance status, disease characteristics, and type and number of prior therapies were comparable between the study arms (Table 1). The median age for all patients was 63 (range 38–82) years; the median weight for all patients was 66.0 (range 38.0–135.5) kg; and the majority (57.7%) of patients had an ECOG performance status of 0. For patients with metastatic disease, the most common metastatic site was bone (77.3%), followed by lymph nodes (48.5%) and liver (39.2%). Most patients (76.3%) had been initially diagnosed ≥24 months before study start. Overall, 48.5% of patients had de novo stage IV disease, which was balanced between the dovitinib and placebo arms (48.9% vs 48.8% overall; 19.1% vs 16.0% FGF pathway–amplified; 29.8% vs 32.0% FGF pathway–nonamplified). The majority of patients relapsed at or within 12 months of the end of adjuvant treatment with any endocrine therapy (50 [51.5%] of 97 patients), and 48.5% (47 of 97) of patients progressed at or within 1 month of end of any endocrine therapy treatment for first-line treatment of metastatic disease. All patients had received prior antineoplastic therapy, including surgery (100%), hormone therapy (100%), radiotherapy (77.3%), and chemotherapy (66.0%). Approximately one-half (49.5%) of patients had received prior tamoxifen, and most patients had received prior aromatase inhibitors, including letrozole (42.3%), anastrozole (35.1%), and exemestane (17.5%). The majority (62.9%) of patients had received hormone therapy as their last prior treatment.
Efficacy
Progression-free survival
The median (95% CI) PFS for the full population were 5.5 (3.8–14.0) months and 5.5 (3.5–10.7) months in the dovitinib and placebo arms, respectively, with an estimated HR of 0.68 (95% CI 0.41–1.14) (Fig. 2a), with 30 events in the dovitinib arm and 34 events in the placebo arm. The estimated HR did not meet the criterion for superior efficacy of dovitinib vs placebo (i.e., HR <0.68). The median (95% CI) PFS values were 10.9 (3.5–16.5) months and 5.5 (3.5–16.4) months in the FGF pathway–amplified subgroup and 5.5 (3.8–16.8) months and 5.5 (1.9–12.8) months in the FGF pathway–nonamplified subgroup for the dovitinib and placebo arms, respectively (Fig. 2b and c). The HRs (95% CIs) were 0.64 (0.22–1.86) for the FGF pathway–amplified subgroup and 0.69 (0.38–1.26) for the FGF pathway–nonamplified subgroup, which was sufficient to meet the efficacy criteria for the FGF pathway–amplified subgroup (i.e., HR <0.65) and pass the futility criteria for the FGF pathway–nonamplified subgroup (i.e., HR >0.81). Fewer PFS events than expected occurred in FGF pathway–amplified patients (18 vs planned 50 events); thus, results in that subgroup should be interpreted with caution.

Investigator-assessed PFS by treatment for all patients (a), FGF pathway–amplified patients (b), and FGF pathway–nonamplified patients (c). FGF Fibroblast growth factor, PFS Progression-free survival
Best overall response
The ORRs (95% CIs) per local investigator assessment in all patients were 27.7% (15.6% to 42.6%) in the dovitinib arm and 10.0% (3.3% to 21.8%) in the placebo arm (Table 2). According to FGF pathway amplification status, the ORRs (95% CIs) for each treatment arm (dovitinib vs placebo) were 20.0% (4.3% to 48.1%) vs 12.5% (1.6% to 38.3%) in the FGF pathway–amplified subgroup and 31.3% (16.1% to 50.0%) vs 8.8% (1.9% to 23.7%) in the FGF pathway–nonamplified subgroup. In an exploratory analysis, we found no correlation between the FGFR1 copy number and response to dovitinib in the FGF pathway–amplified subgroup, with the caveat that the analyzed patient population was small. When considering only those patients who had measurable disease at baseline (n = 41 [87.2%] in the dovitinib arm; n = 39 [78.0%] in the placebo arm), the ORRs (95% CIs) per local investigator assessment were 31.7% (18.1% to 48.1%) in the dovitinib arm and 12.8% (4.3% to 27.4%) in the placebo arm.
Time to response and duration of response
In patients who responded, the median (95% CI) time to first response in the dovitinib arm vs placebo arm was 2.0 (1.5–18.3) months vs 3.7 (1.6–9.1) months in the full population. Of the 13 patients who responded in the dovitinib arm, 11 patients (84.6%) responded within the first 4 months, 1 patient (7.7%) responded between 4 and <6 months, and 1 patient (7.7%) responded after 18 months of receiving the first dose of study treatment. Of the five patients who responded in the placebo arm, three patients (60.0%) responded within the first 4 months, and two patients (40.0%) responded between 6 and <12 months of initiating study treatment. The median (95% CI) values for DOR in the dovitinib arm vs placebo arm were 13.5 (5.5–16.6) months vs 14.7 (3.3–not estimable [NE]) months in the full population, 5.5 (3.2–16.3) months vs 14.7 (NE–NE) months in the FGF pathway–amplified subgroup, and 14.8 (5.5–NE) months vs 10.9 (3.3–NE) months in the FGF pathway–nonamplified subgroup. These data should be interpreted with caution owing to the small sample size.
Overall survival
The median (95% CI) OS was not reached (18.6 months–NE) in the dovitinib arm and was 25.9 (18.4–NE) months in the placebo arm (Fig. 3).

OS for all patients who received dovitinib plus fulvestrant or placebo plus fulvestrant. NE Not estimable, OS Overall survival
Safety
All patients received at least one dose of dovitinib or placebo and have discontinued study treatment (see Fig. 1). Of note, on the basis of the intention-to-treat principle, one patient with FGF pathway amplification who was misclassified as nonamplified at randomization remained in the nonamplified group in all analyses. The reasons for discontinuation in the dovitinib vs placebo arms were progressive disease (55.3% vs 80.0%), AEs (21.3% vs 2.0%), patient or guardian decision (10.6% vs 0%), termination by sponsor (8.5% vs 16.0%), death (2.1% vs 2.0%), and nonadherence to study treatment (2.1% vs 0%). The majority of patients who received dovitinib required at least one dose reduction or interruption (74.5% vs 26.0% for those who received placebo) and/or a dose change (57.4% vs 8.0% for those who received placebo); most patients required a dose interruption or delay or a dose change owing to experiencing an AE (70.2% and 53.2%, respectively) (Additional file 2: Table S1).
The most common any-grade AEs in the dovitinib arm, regardless of cause, were diarrhea (78.7%), nausea (72.3%), vomiting (57.4%), asthenia (38.8%), and headache (36.2%) (Table 3). In the placebo arm, diarrhea (32.0%), fatigue (26.0%), nausea and asthenia (22.0% each), and decreased appetite (16.0%) were the most common any-grade AEs. The most common grade 3 AEs (occurring in ≥10% of patients) in the dovitinib vs placebo arms were hypertension (21.3% vs 6.0%), diarrhea (14.9% vs 4.0%), alanine aminotransferase increase (14.9% vs 2.0%), fatigue (12.8% vs 2.0%), blood alkaline phosphatase increase (12.8% vs 0%), and γ-glutamyltransferase increase (10.6% vs 6.0%). The median (range) time to the onset of hypertension or blood pressure increase was 2.9 (0.1–28.1) weeks in the dovitinib arm and 2.7 (0.1–20.1) weeks in the placebo arm. In general, grade 4 AEs were infrequent and were comparable in the two arms, occurring in eight patients (17.0%) in the dovitinib arm and six patients (12.0%) in the placebo arm. Serious AEs suspected to be related to the study drug were reported in six patients (12.8%) in the dovitinib plus fulvestrant arm and included grade 3 pulmonary embolism, deep vein thrombosis, dehydration, esophageal varices hemorrhage, pneumonia, and varices esophageal (2.1% each), and grade 4 pulmonary embolism, ischemic cerebral infarction, and thrombocytopenia (2.1% each). In the placebo plus fulvestrant arm, two patients (4.0%) reported serious AEs suspected to be related to study drug (grade 4 hypotension [2.0%] and pancreatitis [2.0%]). The overall incidence of proteinuria and thyroid function abnormality was low in the dovitinib arm (two patients [4.3%] and one patient [2.1%], respectively; all grade 1/2 AEs) and was not reported in the placebo arm.
More patients discontinued study treatment owing to AEs in the dovitinib arm than in the placebo arm (38.3% vs 8.0%). The most frequently reported AEs (occurring in ≥3% of patients) leading to discontinuation were diarrhea (6.4% vs 0%), alanine aminotransferase increase (4.3% vs 2.0%), aspartate aminotransferase increase (4.3% vs 2.0%), and rash (4.3% vs 0%) (Additional file 2: Table S2). A total of four on-treatment deaths were reported (two deaths in each treatment arm); of the two on-treatment deaths that occurred in the dovitinib arm, one patient died as a result of breast cancer progression and one patient died because of a pulmonary embolism suspected to be related to study treatment. Overall, 14 patients (29.8%) in the dovitinib arm and 18 patients (36.0%) in the placebo arm died during the entire study period (i.e., including the time beyond the 30-day end of treatment follow-up period).
Discussion
In this randomized, double-blind trial, we evaluated the safety and efficacy of dovitinib plus fulvestrant compared with placebo plus fulvestrant in postmenopausal patients with HR+, HER2− advanced breast cancer that progressed during or after prior endocrine therapy. The final analysis was initially planned to occur when 90 PFS events were recorded in the full population, including ≥50 PFS events in the FGF-amplified subgroup. However, the study was terminated early because of slow enrollment in the FGF-amplified subgroup.
In this study, patients in the FGF pathway–amplified subgroup who received dovitinib plus fulvestrant had prolonged median PFS (10.9 vs 5.5 months), with an estimated 36% risk reduction compared with patients who received placebo plus fulvestrant. However, a similar trend in risk reduction with dovitinib plus fulvestrant treatment (vs placebo plus fulvestrant) was seen in all patients (32%) and in patients without FGF pathway amplification (31%). This suggests that dovitinib plus fulvestrant may have antineoplastic activity regardless of FGF pathway amplification status in the evaluated patient population, although the estimated risk reduction reached statistical significance (as defined in the study protocol) only in the FGF pathway–amplified cohort. Furthermore, patients in the dovitinib plus fulvestrant arm had a higher ORR than patients in the placebo plus fulvestrant arm (27.7% vs 10.0%), regardless of FGF pathway amplification status (ORR 20.0% vs 12.5% in FGF pathway–amplified subgroup; ORR 31.3% vs 8.8% in FGF pathway–nonamplified subgroup). Nevertheless, these data should be interpreted cautiously. First, the small sample size in the FGF pathway–amplified subgroup contributed to a lower-than-expected number of PFS events and very large CIs. Second, we cannot exclude that dovitinib had an effect regardless of FGF pathway amplification status, given that the number of events for the full study population was 64 (30 in the dovitinib arm and 34 in the placebo arm), which was less than the 90 planned events. One potential explanation for the activity of dovitinib plus fulvestrant regardless of FGF pathway amplification status is that, as a multitargeted tyrosine kinase inhibitor, dovitinib targeted other pathways [36], such as signaling through vascular endothelial growth factor receptor or c-Kit, which are overexpressed in 10% to 11% and 11% to 17% of breast cancers, respectively [39, 40].
Safety data were consistent with the known safety profile of dovitinib [38, 41–44], with no new safety concerns identified with the use of dovitinib in combination with fulvestrant in patients with HR+, HER2− advanced breast cancer. The use of FGFR inhibitors in breast cancer merits further investigation because other studies of single-agent FGFR inhibitors showed encouraging results in patients with breast cancer [38, 45–47]. Resistance to hormone therapy (i.e., tamoxifen) is potentially mediated by FGFR signaling through activation of the mitogen-activated protein kinase (MAPK) and PI3K pathways [26, 32]. For example, resistance to tamoxifen has been associated with constitutive activation of MAPK and the subsequent expression of cyclin D1 in FGFR1-amplified breast cancer cell lines [26]. Similarly, in ER+ cell lines, activation of FGFR3 reduced sensitivity to tamoxifen and fulvestrant through activation of MAPK and PI3K signaling pathways [32]. Furthermore, the combination of dovitinib and the dual PI3K/mTOR inhibitor dactolisib (BEZ235) showed strong inhibition of PI3K pathway activation in vitro and in vivo, as well as antitumor activity in FGFR-expressing breast cancer models [48]. Currently, researchers in a phase Ib trial (ClinicalTrials.gov identifier: NCT01928459) are investigating the pan-FGFR inhibitor BGJ398 in combination with the selective PI3K inhibitor alpelisib (BYL719) in patients with solid tumors with FGFR1, FGFR2, and FGFR3 alterations and phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit α mutations. Thus, further exploration of the use of FGFR inhibitors in combination with other agents is warranted.
The present study was terminated early because of slow accrual. The rates of FGF pathway amplification observed in this study were lower than previously reported. In the previous phase II monotherapy study, 10% of the patients screened who were enrolled in the study had FGFR1 amplification [38], whereas approximately 5% of patients screened and randomized in this study had FGF pathway amplifications. In the phase II monotherapy study, many of the patients were prescreened by the French cooperative group, which reduced the overall number of patients needed to be screened. In addition, the eligibility criteria allowed more heavily pretreated patients to be enrolled, which expanded the pool of potential patients. Conducting clinical trials in molecularly selected patient populations is challenging, particularly because the screening failure rate is high with current trial designs [49], accrual can be slow when the molecular aberration is very rare, and patient dropout rates [50] and costs [51] can be high. In this study, accrual of FGF-amplified patients was very slow and resulted in the early termination of the trial, thereby confounding interpretation of the results. Several strategies have been proposed to overcome these challenges. A novel idea to increase rapid recruitment of patients with rare molecular markers is to develop molecular screening programs that screen several genes at a time in a large patient pool, using next-generation sequencing assays, and then to guide patients to specific clinical trials on the basis of their specific biomarkers [52, 53]. One example is the National Cancer Institute’s Molecular Analysis for Therapy Choice (NCI MATCH) trial (ClinicalTrials.gov identifier: NCT02465060); patients are screened for approximately 200 genes, assigned to a study arm on the basis of a molecular abnormality, and followed for response and PFS [52]. The NCI MATCH study currently includes 24 arms, in one of which investigators are evaluating the FGFR inhibitor AZD4547 in patients with FGFR pathway aberrations (FGFR1–FGFR3 amplification, mutation, or translocation). Following progression, patients may be rescreened and enrolled in a second study arm; patients may also receive their screening results and decide, together with their doctor, to receive alternative therapy [52]. It remains to be seen whether new trial designs will have widespread support [50].
Conclusions
In this placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR+, HER2− advanced or metastatic breast cancer that progressed during or after prior endocrine therapy did not identify any new safety findings. Dovitinib in combination with fulvestrant showed promising clinical activity in the FGF pathway–amplified subgroup. However, the reported data should be interpreted with caution, given that fewer PFS events than expected occurred in the FGF pathway–amplified patients and that we cannot exclude an effect of dovitinib regardless of FGR pathway amplification status, owing to the smaller-than-expected sample size.
Abbreviations
- AE:
-
Adverse event
- CONSORT:
-
Consolidated Standards of Reporting Trials
- CR:
-
Complete response
- DOR:
-
Duration of response
- ECOG:
-
Eastern Cooperative Oncology Group
- ER:
-
Estrogen receptor
- ET:
-
Endocrine therapy
- FGF:
-
Fibroblast growth factor
- FGFR:
-
Fibroblast growth factor receptor
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hormone receptor
- MAPK:
-
Mitogen-activated protein kinase
- mTOR:
-
Mechanistic target of rapamycin
- NCI MATCH:
-
National Cancer Institute’s Molecular Analysis for Therapy Choice trial
- NE:
-
Not estimable
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PCR:
-
Polymerase chain reaction
- PD:
-
Progressive disease
- PFS:
-
Progression-free survival
- PI3K:
-
Phosphoinositide 3-kinase
- PR:
-
Partial response
- qPCR:
-
Quantitative polymerase chain reaction
- RECIST:
-
Response Evaluation Criteria In Solid Tumors
- SD:
-
Stable disease
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
American Cancer Society. Global cancer facts & figures. 3rd ed. Atlanta: American Cancer Society; 2015.
Anderson E. The role of oestrogen and progesterone receptors in human mammary development and tumorigenesis. Breast Cancer Res. 2002;4(5):197–201.
Nadji M, Gomez-Fernandez C, Ganjei-Azar P, Morales AR. Immunohistochemistry of estrogen and progesterone receptors reconsidered: experience with 5,993 breast cancers. Am J Clin Pathol. 2005;123(1):21–7.
Howlader N, Altekruse SF, Li CI, Chen VW, Clarke CA, Ries LA, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst. 2014;106(5):dju055.
Schwartz TL, Veerapong J, Hinyard L. Her2 positivity and race predict higher mastectomy rates: a SEER database analysis. Springerplus. 2015;4:715.
Beltjens F, Bertaut A, Pigeonnat S, Loustalot C, Desmoulins I, Charon-Barra C, et al. HER2-positivity rates in breast cancer: no variation over time when clinicopathological features and testing are stable. Eur J Cancer Care (Engl). doi:10.1111/ecc.12404.
Cardoso F, Costa A, Norton L, Senkus E, Aapro M, Andre F, et al. ESO-ESMO 2nd International Consensus Guidelines for Advanced Breast Cancer (ABC2). Ann Oncol. 2014;25(10):1871–88.
National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology: breast cancer. Version 2. Fort Washington, PA: NCCN; 2016.
Johnston SR. New strategies in estrogen receptor-positive breast cancer. Clin Cancer Res. 2010;16(7):1979–87.
Cardoso F, Costa A, Norton L, Cameron D, Cufer T, Fallowfield L, et al. 1st International Consensus Guidelines for Advanced Breast Cancer (ABC 1). Breast. 2012;21(3):242–52.
Gnant M. The role of mammalian target of rapamycin (mTOR) inhibition in the treatment of advanced breast cancer. Curr Oncol Rep. 2013;15(1):14–23.
Rugo HS, Vidula N, Ma C. Improving response to hormone therapy in breast cancer: new targets, new therapeutic options. Am Soc Clin Oncol Educ Book. 2016;35:e40–54.
Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–9.
Yardley DA, Noguchi S, Pritchard KI, Burris 3rd HA, Baselga J, Gnant M, et al. Everolimus plus exemestane in postmenopausal patients with HR+ breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30(10):870–84.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16(1):25–35.
Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209–19.
Heinzle C, Sutterluty H, Grusch M, Grasl-Kraupp B, Berger W, Marian B. Targeting fibroblast-growth-factor-receptor-dependent signaling for cancer therapy. Expert Opin Ther Targets. 2011;15(7):829–46.
Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447(7148):1087–93.
Ruiz-Narvaez EA, Haddad SA, Lunetta KL, Yao S, Bensen JT, Sucheston-Campbell LE, et al. Gene-based analysis of the fibroblast growth factor receptor signaling pathway in relation to breast cancer in African American women: the AMBER Consortium. Breast Cancer Res Treat. 2016;155(2):355–63.
André F, Cortes J. Rationale for targeting fibroblast growth factor receptor signaling in breast cancer. Breast Cancer Res Treat. 2015;150(1):1–8.
Jaakkola S, Salmikangas P, Nylund S, Partanen J, Armstrong E, Pyrhönen S, et al. Amplification of fgfr4 gene in human breast and gynecological cancers. Int J Cancer. 1993;54(3):378–82.
Jacquemier J, Adelaide J, Parc P, Penault-Llorca F, Planche J, DeLapeyriere O, et al. Expression of the FGFR1 gene in human breast-carcinoma cells. Int J Cancer. 1994;59(3):373–8.
Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res. 2007;9(2):R23.
André F, Job B, Dessen P, Tordai A, Michiels S, Liedtke C, et al. Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res. 2009;15(2):441–51.
Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70(5):2085–94.
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70.
Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29(14):2013–23.
Tomiguchi M, Yamamoto Y, Yamamoto-Ibusuki M, Goto-Yamaguchi L, Fujiki Y, Fujiwara S, et al. Fibroblast growth factor receptor-1 protein expression is associated with prognosis in estrogen receptor-positive/human epidermal growth factor receptor-2-negative primary breast cancer. Cancer Sci. 2016;107(4):491–8.
Templeton AJ, Diez-Gonzalez L, Ace O, Vera-Badillo F, Seruga B, Jordan J, et al. Prognostic relevance of receptor tyrosine kinase expression in breast cancer: a meta-analysis. Cancer Treat Rev. 2014;40(9):1048–55.
Sun S, Jiang Y, Zhang G, Song H, Zhang X, Zhang Y, et al. Increased expression of fibroblastic growth factor receptor 2 is correlated with poor prognosis in patients with breast cancer. J Surg Oncol. 2012;105(8):773–9.
Tomlinson DC, Knowles MA, Speirs V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int J Cancer. 2012;130(12):2857–66.
Roidl A, Foo P, Wong W, Mann C, Bechtold S, Berger HJ, et al. The FGFR4 Y367C mutant is a dominant oncogene in MDA-MB453 breast cancer cells. Oncogene. 2010;29(10):1543–52.
Thussbas C, Nahrig J, Streit S, Bange J, Kriner M, Kates R, et al. FGFR4 Arg388 allele is associated with resistance to adjuvant therapy in primary breast cancer. J Clin Oncol. 2006;24(23):3747–55.
Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, et al. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2− metastatic breast cancer. Clin Cancer Res. 2017;23(1):26–34.
de Menezes DE L, Peng J, Garrett EN, Louie SG, Lee SH, Wiesmann M, et al. CHIR-258: a potent inhibitor of FLT3 kinase in experimental tumor xenograft models of human acute myelogenous leukemia. Clin Cancer Res. 2005;11(14):5281–91.
Dey JH, Bianchi F, Voshol J, Bonenfant D, Oakeley EJ, Hynes NE. Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res. 2010;70(10):4151–62.
André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia J, Hurvitz SA, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res. 2013;19(13):3693–702.
Kashiwagi S, Yashiro M, Takashima T, Aomatsu N, Kawajiri H, Ogawa Y, et al. c-Kit expression as a prognostic molecular marker in patients with basal-like breast cancer. Br J Surg. 2013;100(4):490–6.
Jansson S, Bendahl PO, Grabau DA, Falck AK, Fernö M, Aaltonen K, et al. The three receptor tyrosine kinases c-KIT, VEGFR2 and PDGFRα, closely spaced at 4q12, show increased protein expression in triple-negative breast cancer. PLoS One. 2014;9(7):e102176.
Angevin E, Lopez JA, Pande A, Moldovan C, Shi M, Soria JC, et al. TKI258 (dovitinib lactate) in metastatic renal cell carcinoma (mRCC) patients refractory to approved targeted therapies: a phase I/II dose finding and biomarker study. J Clin Oncol. 2009;27:(Suppl 15). [abstract 3563].
Kang YK, Yoo C, Ryoo BY, Lee JJ, Tan E, Park I, et al. Phase II study of dovitinib in patients with metastatic and/or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib. Br J Cancer. 2013;109(9):2309–15.
Milowsky MI, Dittrich C, Duran I, Jagdev S, Millard FE, Sweeney CJ, et al. Phase 2 trial of dovitinib in patients with progressive FGFR3-mutated or FGFR3 wild-type advanced urothelial carcinoma. Eur J Cancer. 2014;50(18):3145–52.
Infante JR, Ramanathan RK, George D, Tan E, Quinlan M, Liu A, et al. A randomized, crossover phase 1 study to assess the effects of formulation (capsule vs tablet) and meal consumption on the bioavailability of dovitinib (TKI258). Cancer Chemother Pharmacol. 2015;75:729–37.
Dienstmann R, Andre F, Soria J, Tabernero J, De Braud FGM, Cereda R, et al. Significant antitumor activity of E-3810, a novel FGFR and VEGFR inhibitor, in patients with FGFR1 amplified breast cancer. Ann Oncol. 2012;23:(Suppl 9). [abstract 3190]. ᅟ
Dienstmann R, Bahleda R, Adamo B, Rodon J, Varga A, Gazzah A, et al. First in human study of JNJ-42756493, a potent pan fibroblast growth factor receptor (FGFR) inhibitor in patients with advanced solid tumors. Cancer Res. 2014;74:(Suppl 19). [abstract CT325]. ᅟ
Sequist LV, Cassier P, Varga A, Tabernero J, Schellens JHM, Delord J, et al. Phase I study of BGJ398, a selective pan-FGFR inhibitor in genetically preselected advanced solid tumors. Cancer Res. 2014;74:(Suppl 19). [abstract CT326].
Issa A, Gill JW, Heideman MR, Sahin O, Wiemann S, Dey JH, et al. Combinatorial targeting of FGF and ErbB receptors blocks growth and metastatic spread of breast cancer models. Breast Cancer Res. 2013;15:R8.
Andre F, Delaloge S, Soria JC. Biology-driven phase II trials: what is the optimal model for molecular selection? J Clin Oncol. 2011;29(10):1236–8.
Catenacci DVT. Next-generation clinical trials: novel strategies to address the challenge of tumor molecular heterogeneity. Mol Oncol. 2015;9(5):967–96.
Le Tourneau C, Kamal M, Alt M, Verlingue L, Servois V, Sablin M, et al. The spectrum of clinical trials aiming at personalizing medicine. Chin Clin Oncol. 2014;3(2):13.
Abrams J, Conley B, Mooney M, Zwiebel J, Chen A, Welch JJ, et al. National Cancer Institute’s precision medicine initiates for the new National Clinical Trials Network. Am Soc Clin Oncol Educ Book. 2014:71–6. doi:10.14694/EdBook_AM.2014.34.71.
Arnedos M, André F, Farace F, Lacroix L, Besse B, Robert C, et al. The challenge to bring personalized cancer medicine from clinical trials into routine clinical practice: the case of the Institut Gustave Roussy. Mol Oncol. 2012;6(2):204–10.
Acknowledgements
We thank Alejandro Yovine, Seven Gogov, Andrea Kay, Ying Wang, and Beth McGrain for their involvement in the study design, statistical analysis, and data analysis. Editorial assistance was provided by Pamela Tuttle, PhD, CMPP, and Katherine Mills-Luján, PhD, of ArticulateScience LLC and was funded by Novartis Pharmaceuticals. This study was previously reported in abstract and poster format at the American Society of Clinical Oncology 2012 and 2013 congresses in Chicago, IL, USA, and at the American Association for Cancer Research San Antonio Breast Cancer Symposium in San Antonio, TX, USA, in 2012.
Funding
This study was sponsored and funded by Novartis Pharmaceuticals (Basel, Switzerland).
Availability of data and materials
The datasets generated and/or analyzed during the present study are not publicly available in order to protect patient information in the clinical trial database, but they are available from the corresponding author on reasonable request.
Authors’ contributions
The study was designed by the sponsor (Novartis Pharmaceuticals) and by the study steering committee (chair: FA; members: AM, MC, PN, ND, CHB, JC, and KB). MS, SD, and MMS contributed to the design of the study. As members of the study steering committee, FA, AM, MC, PN, ND, CHB, JC, and KB oversaw the conduct of the study. HS, ZK, and HB contributed substantially to patient recruitment. AM, MC, PN, ND, CHB, JC, KB, HS, ZK, HB, and FA contributed to data collection. AM, MC, PN, ND, JC, KB, HS, MS, YZ, MMS, and FA analyzed and interpreted the data. YZ performed the statistical analyses. AM and FA wrote the manuscript with medical editorial support from ArticulateScience LLC, funded by the sponsor. All authors contributed to draft revisions, had full access to the data, attest to the accuracy and integrity of the data, and read and approved the final manuscript.
Competing interests
FA and CHB have received research funding from AstraZeneca (London, UK), Eli Lilly and Co. (Indianapolis, IN, USA), Novartis Pharmaceuticals, and Pfizer (New York, NY, USA). CHB has also received research funding from AbbVie (Chicago, IL, USA), Abraxis BioScience (Los Angeles, CA, USA), Amgen (Thousand Oaks, CA, USA), Asana BioSciences (Lawrenceville, NJ, USA), BioMarin (Novato, CA, USA), Boehringer Ingelheim (Ingelheim am Rhein, Germany), Bristol-Myers Squibb (New York, NY, USA), Daiichi Sankyo (Tokyo, Japan), GlaxoSmithKline (Brentford, UK), Merck (Kenilworth, NJ, USA), Merrimack Pharmaceuticals (Cambridge, MA, USA), Mylan (Canonsburg, PA, USA), Roche/Genentech (South San Francisco, CA, USA), Sanofi (Paris, France), and Taiho Pharmaceuticals (Tokyo, Japan). ND has received research funding from Genentech, GTx (Memphis, TN, USA), and Novartis. CHB and MC have been consultants for and received honoraria from Novartis Pharmaceuticals and Pfizer. CHB has also been a consultant for and/or received honoraria from AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Roche/Genentech, Eisai (Tokyo, Japan), and Samsung Bioepis (Incheon, Republic of Korea). JC has been a consultant for AstraZeneca, Biothera Pharmaceuticals (Eagan, MN, USA), Celgene (Summit, NJ, USA), Cellestia Biotech (Basel, Switzerland), and Roche/Genentech, and he has received honoraria from Eisai, Novartis Pharmaceuticals, and Roche/Genentech. KB has been a consultant for and has received funding from Novartis Pharmaceuticals. HS has been a consultant for AstraZeneca, Biotheranostics (San Diego, CA, USA), and Celgene. MS, YZ, SD, and MMS are current or former employees of Novartis Pharmaceuticals. AM, PN, ZK, and HB declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study was reviewed and approved by the relevant independent ethics committees/institutional review boards at each study center (Additional file 2: Table S3) and was conducted in accordance with the Declaration of Helsinki, good clinical practice guidelines, and applicable laws. All patients provided written informed consent.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Figure S1.
Primary endpoint data analysis. (PDF 163 kb)
Additional file 2: Table S1.
Dose changes and dose delays by treatment arm. Table S2. Adverse events leading to study drug discontinuation regardless of study drug relationship. Table S3. Study center. (DOCX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Musolino, A., Campone, M., Neven, P. et al. Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR+, HER2− breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res 19, 18 (2017). https://doi.org/10.1186/s13058-017-0807-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-017-0807-8