
Abstract
Approximately 20% of patients with acute brain injury (ABI) also experience acute kidney injury (AKI), which worsens their outcomes. The metabolic and inflammatory changes associated with AKI likely contribute to prolonged brain injury and edema. As a result, recognizing its presence is important for effectively managing ABI and its sequelae. This review discusses the occurrence and effects of AKI in critically ill adults with neurological conditions, outlines potential mechanisms connecting AKI and ABI progression, and highlights AKI management principles. Tailored approaches include optimizing blood pressure, managing intracranial pressure, adjusting medication dosages, and assessing the type of administered fluids. Preventive measures include avoiding nephrotoxic drugs, improving hemodynamic and fluid balance, and addressing coexisting AKI syndromes. ABI patients undergoing renal replacement therapy (RRT) are more susceptible to neurological complications. RRT can negatively impact cerebral blood flow, intracranial pressure, and brain tissue oxygenation, with effects tied to specific RRT methods. Continuous RRT is favored for better hemodynamic stability and lower risk of dialysis disequilibrium syndrome. Potential RRT modifications for ABI patients include adjusted dialysate and blood flow rates, osmotherapy, and alternate anticoagulation methods. Future research should explore whether these strategies enhance outcomes and if using novel AKI biomarkers can mitigate AKI-related complications in ABI patients.
Graphical abstract

Similar content being viewed by others

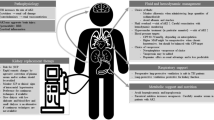
Background
Acute kidney injury (AKI) in intensive care units (ICUs) is an independent risk factor for death. Reported mortality rates from AKI with renal replacement therapy (RRT; 40–55%) are higher than the mortality rates due to ICU-related myocardial infarction (20%), sepsis without AKI (15–25%), or acute respiratory distress syndrome requiring mechanical ventilation (30–40%) [1]. This high mortality rate is attributed to the systemic impact of AKI on the brain, heart, lungs, liver, and gastrointestinal tract, linking AKI to various syndromes (Additional file 1) [2]. Beyond the acute phase, AKI increases the likelihood of chronic kidney disease (CKD), cardiovascular complications, recurrent AKI, and functional impairment [3].
In the context of neurocritical care, AKI diagnosis holds significance for acute brain injury (ABI) management. This article summarizes the epidemiology and outcomes of AKI in neurocritically ill adults admitted to the ICU, focusing on prevalent ABI conditions like traumatic brain injury (TBI), aneurysmal subarachnoid hemorrhage (SAH), intracerebral hemorrhage (ICH), and acute stroke. The review explores potential AKI mechanisms and their involvement in reciprocal ABI progression, while also highlighting essential principles in AKI management. Additionally, considerations for managing RRT in the context of ABI are discussed. Our literature search strategy is provided in Additional file 2.
AKI epidemiology and outcomes in neurocritical care
Studies on AKI prevalence in neurocritical care are few, and their results vary with AKI definition [4,5,6] (Additional file 3). Current AKI diagnosis and classification are based on the 2012 KDIGO (Kidney Disease: Improving Global Outcomes) consensus criteria [6] (Additional file 4; left panel). This three-stage severity classification system is based on changes in serum creatinine (SCr) concentration and urine output (UOP). However, most neurocritical care studies only used SCr-based definitions because SCr data are readily available in electronic health records, missing a substantial proportion of patients with AKI [7].
In a multinational study spanning 97 ICUs and encompassing more than 1800 patients, with 25.9% primarily diagnosed with neurological symptoms, 57% of the entire cohort developed AKI by KDIGO criteria within the initial week of admission [8]. Among these, 39% experienced stages 2–3 AKI, and 13.5% required RRT, accounting for 23.5% of AKI cases [8]. While insights into AKI incidence among neurocritical patients mainly stem from retrospective studies in single centers and exclude cases diagnosed with AKI upon admission, a recent study in a mixed neurological/neurosurgical ICU reported an overall AKI occurrence of 23.5% utilizing the SCr/UOP KDIGO criteria [9]. Interestingly, similar AKI rates were noted among specific brain pathologies, including TBI, 11.9–26.7% (KDIGO, SCr only) [10,11,12,13,14] and 17.7% (SCr/UOP KDIGO) [15]; acute stroke, 20.9–43.3% (KDIGO, SCr only) [16, 17]; non-traumatic aneurysmal SAH, 16.7–23.7% (KDIGO, SCr only) [18, 19], and ICH, 20.0–29.9% [18, 20,21,22]. The few studies assessing RRT in patients with ABI reported overall rates of 0.3–5.6% among neurocritical patients [17, 23]. Irrespective of the criteria employed and the specific brain condition under consideration, AKI severity is associated with increased mortality, obstacles to rehabilitation care, increased likelihood of disability at discharge, and potential downstream effects on healthcare services [8, 10, 24,25,26,27].
Data on renal recovery after AKI post-ABI are scarce [18], and long-term renal outcomes after AKI in ABI patients remain unknown.
AKI causes and pathophysiological mechanisms
Clinicians must consider various AKI causes specific to both neurocritical and non-neurological ICU patients (Additional file 1). The Graphical Abstract summarizes key brain-kidney interaction pathways in neurocritical care that could lead to reciprocal ABI and AKI progression.
CKD plays a significant role as a risk factor for AKI in neurocritical patients, with those having CKD displaying a fivefold higher prevalence of cerebrovascular disease compared to those without CKD [10, 26, 28]. These data are unsurprising, given that the kidneys and brain share common vascular risk factors, including hypertension, diabetes, and hyperlipidemia. Notably, proteinuria of ≥ 30 mg/dL and CKD stages 3–4 are established risk factors for cerebrovascular disease, which, in turn, increases the likelihood of AKI [29, 30]. A study on patients with spontaneous ICH found that individuals with CKD experience worse functional outcomes (OR 1.91; 95% CI 1.04–3.52) and higher mortality (OR 3.33; 95% CI 1.76–6.27) at 12 months post-ICH compared to those without CKD [31]. While TBI patients tend to be younger and possess normal pre-existing renal function, patients with acute stroke, aneurysmal SAH, and ICH are generally older and more likely to have underlying CKD.
ABI can be complicated by neurogenic stunned myocardium [32], which is a transient and diffuse form of left ventricular cardiomyopathy, likely triggered by autonomic nervous system activation with excess catecholamine release, impaired myocardial glucose metabolism secondary to neurocardiogenic injury, and coronary microvascular dysfunction [33, 34]. Patients with this condition exhibit a wide range of cardiac abnormalities including arrhythmias and ventricular dysfunction [33, 34], increasing the risk of AKI due to reduced cardiac output, pulmonary edema, and prolonged vasopressor support [35]. However, the prevalence of AKI in such patients has not been described. Autonomic nervous system imbalance also contributes to sympatho-adrenergic drive and renin–angiotensin–aldosterone system activation and vasopressin release, resulting in renal vasoconstriction, enhanced sodium and water reabsorption, and decreased renal blood flow and glomerular filtration rate (GFR) [36]. Neurogenic stunned myocardium prevalence in patients with SAH is high and estimated at ~ 30% [32].
The 2020 Neurocritical Care Society (NCS) guidelines suggest, with low evidence, using hyperosmolar therapy for initial management of cerebral edema in neurocritical patients with SAH, TBI, acute ischemic stroke, and ICH [37]. While mannitol is the most frequently administered hyperosmolar solution, its use, particularly at high doses, has been associated with an increased risk of AKI in such patients [38,39,40]. Risk factors for mannitol-associated AKI include higher illness severity, heart failure, diabetes, use of diuretics, and lower baseline GFR [38, 41]. The precise mechanism for mannitol-associated AKI is not well defined but may be related to increased serum osmolality [42] and appears to be dose-related [11]. The 2020 NCS guidelines recommend hypertonic sodium solution over mannitol as it might be more effective in reducing ICP or cerebral edema ABI [37]; however, its usage can lead to hypernatremia and hyperchloremia, both of which have been associated with an increased AKI risk [19, 43, 44]. However, the effects of high chloride content on the kidneys have been debated [45, 46]. While experimental studies suggest it causes renal vasoconstriction and decreased GFR [47,48,49], a post-hoc analyses of the EPO-TBI and COBI trials demonstrated no association between hypertonic sodium solution and AKI [11, 14]. Notably, randomized controlled studies specifically focusing on AKI and acid–base balance outcomes related to hyperosmolar therapy in neurocritical care are lacking.
Intravenous contrast might be a concern in patients with ABI due to increased AKI risk, particularly in patients with SAH who might receive substantial amounts of contrast due to repeated computed tomography angiography scans for the diagnosis and/or management of cerebral vasospasm/delayed cerebral ischemia. However, existing evidence suggests that the risks associated with low- or iso-osmolar intravenous contrast agents are minimal, and AKI is infrequent in patients with eGFR of ≥ 30 mL/min/1.73 m2 [50, 51]. Intraarterial contrast administration poses a greater risk for AKI because such procedures often require larger doses that reach the renal arteries at high concentrations and potentially lead to atheroembolic complications [52].
Sepsis and nosocomial infections, especially ventilator-associated pneumonia, are common in neurocritical patients. A retrospective single-center study reported sepsis (75%) and respiratory infections (68%) as the main non-neurological complications in patients with TBI [53]. Although data on patients with ABI are limited, infectious complications are considered the main AKI disease modifiers during ICU stay [16, 26], in line with general ICU populations [8].
Myoglobin-associated rhabdomyolysis and renal toxicity should be considered in patients with TBI. A recent multicenter registry study found rhabdomyolysis present in 3.8% of patients with TBI and independently associated with AKI occurrence [54]. Moreover, while rhabdomyolysis and elevated serum/urinary myoglobin correlate with AKI occurrence, they also correlate with trauma severity and other renal insults. Hence, the direct contribution of rhabdomyolysis to kidney injury is often uncertain. The causative role of myoglobin toxicity in AKI is more certain in cases with severe rhabdomyolysis (creatinine kinase > 15,000 IU/L), which rarely occurs in patients with TBI.
Brain injury progression following AKI
Experimental evidence suggests that while AKI is an indicator of illness severity, it can cause further organ dysfunction and damage. Despite limitations due to interspecies differences, animal kidney injury models have been used extensively to elucidate the mechanisms leading to remote organ dysfunction after AKI (Table 1).
AKI causes systemic inflammation by generating proinflammatory cytokines and reducing cytokine clearance [63, 64]. These inflammatory processes contribute to AKI initiation and likely perpetuate and extend brain injury. Consequently, the brain and kidneys might interact during AKI by amplifying cytokine-induced damage and oxidative stress, extravasating leukocytes, and dysregulating cerebral aquaporin channels [65]. The various proinflammatory and metabolic changes observed in AKI (e.g., waste solute retention, disturbances of inorganic solute metabolism, and reduced drug clearance) can disrupt the blood–brain barrier. This disruption may lead to an influx of water and the accumulation of inflammatory cells, cytokines, and neurotoxic substances in the central nervous system. This accumulation can result in cerebral edema, inflammation, hemorrhage, and even death [66]. Animal models have supported this mechanism, demonstrating blood–brain barrier disruption [59], microvascular protein leakage, microglial cell activation [59], hippocampal injury, inflammation characterized by neuronal pyknosis [62], and increased soluble inflammatory proteins in the cerebral cortex and hippocampus [59] following renal ischemia–reperfusion injury-induced AKI. Importantly, neuronal pyknosis did not increase in corresponding animal models of acute liver injury, suggesting that some observed effects are relatively specific to AKI and not associated with generalized inflammation following acute organ injury [59]. Functionally, mice with AKI showed impaired locomotor function that correlated with renal ischemia duration [59]. Alterations in neurotransmitter secretion and uptake during AKI might worsen brain injury and dysfunction [66]. Furthermore, sepsis and liver failure, which are frequently observed in critical illness, can exacerbate brain injury and/or AKI and contribute to multiple organ dysfunction [67].
AKI management
The underlying cause of AKI should be identified promptly, paying special attention to reversible causes. AKI-related syndromes (Additional file 1) pose a major challenge to AKI management, so regular re-evaluation is required for adaptive management. Since current evidence does not suggest that AKI in neurocritical patients should be managed differently from AKI in other critically ill populations, KDIGO-bundle recommendations (i.e., reduce nephrotoxic agents, monitor SCr/UOP, discontinue renin-angiotensin-system blockers, optimize fluid status) are considered appropriate [68]. No externally-validated scoring system is available to evaluate AKI risk in neurocritical patients.
Hemodynamic management
Blood pressure (BP) targets should consider premorbid BP. The potential benefits of increased renal perfusion must be weighed against potentially deleterious effects on cerebral perfusion. Cerebral autoregulation is impaired in approximately one-third of patients with TBI [69], in whom a rise in mean arterial pressure (MAP) might increase the ICP due to hyperemia, while a drop in MAP might lead to cerebral hypoperfusion. Although targeting high MAP (≥ 80 mmHg) in patients with sepsis and chronic hypertension could benefit renal outcomes [70], careful evaluation of its effects on ICP and, consequently, on cerebral perfusion pressure (CPP) is required. As disease-specific data are lacking, CPP targets for ABI are usually derived from TBI guidelines, which recommend maintaining CPP at 60–70 mmHg and assessing cerebral autoregulation to individualized CPP targets [71, 72].
Norepinephrine is the first-line vasopressor used in sepsis with organ dysfunction and ABI [73, 74]. Data suggest that vasopressin could benefit some sepsis-associated AKI subtypes, but its role in patients with ABI and AKI is not fully known [75, 76]. Notably, vasopressin should be used cautiously as it might increase the risk of hyponatremia (and subsequent cerebral edema).
The magnitude of acute systolic blood pressure (SBP) reduction in patients with ICH, aimed at limiting hematoma growth, requires careful monitoring. According to the 2022 guidelines from the American Heart Association/American Stroke Association, reducing acute SBP to 140 mmHg and maintaining it within the range of 130–150 mmHg is regarded as safe and potentially beneficial for enhancing functional outcomes in patients with mild-to-moderate ICH who initially present with an SBP between 150–220 mmHg [77]. Because chronic hypertension shifts the plateau of the renal autoregulatory curve to higher levels [78], a sudden decrease in blood pressure (BP) could lead to significantly compromised tissue perfusion. A targeted stepwise BP reduction rather than absolute targeted value could optimize renal perfusion and mitigate AKI. In a post-hoc analysis of ATACH-II, which included patients with initial systolic blood pressure (SBP) of ≥ 220 mmHg (22.8% of the group), a significant reduction in SBP (110–139 mmHg) resulted in a higher rate of neurological dysfunction at 24 h and more renal complications by the seventh day of discharge [79]. This was compared to standard SBP lowering (140–179 mmHg), without any benefits in reducing hematoma growth at 24 h or rates of death or severe disability at 90 days [79]. This suggests that cautious lowering of blood pressure might be necessary for this specific subgroup. Subsequent analysis of ATACH-II indicated that a baseline SCr ≥ 1.25 mg/dL and higher intravenous nicardipine doses were associated with increased risk for AKI [80]. Accordingly, another study on patients with ICH indicated that an intensive SBP reduction to > 90 mmHg in the first 12 h increases the risk of AKI regardless of preexisting CKD [21].
Fluid management
Much of the evidence on fluid management in ABI is derived from TBI guidelines. A negative fluid balance has been associated with adverse outcomes in patients with TBI [81], whereas fluid overload can cause systemic complications or cerebral edema and increased ICP [81, 82]. A multicenter study evaluating variable fluid management in patients with TBI found that incrementally positive fluid balance was associated with increased ICU mortality (OR, 1.10 per 0.1 L increase; 95% CI 1.07–1.12) and poor functional outcomes (OR, 1.04 per 0.1 L increase; 95% CI 1.02–1.05) [83]. Notably, these data likely represent confounding by indication, as sicker patients are more likely to receive additional volume. While the study did not assess renal outcomes, patients receiving a mean daily fluid balance of ≥ 0.37 L were more likely to undergo RRT than those with < 0.37 L (4% vs. 2%; P = 0.021) [83]. Reports on the association between fluid balance and renal outcomes in ABI are scarce. However, in non-neurological critically ill patients, the relationship between fluid overload and AKI is well-established due to factors like venous congestion, increased renal interstitial pressure, and decreased renal blood flow and GFR as observed in patients with cardiorenal syndrome [84].
The 2018 ESICM (European Society of Intensive Care Medicine) consensus statement recommends using MAP, fluid balance, and multimodal monitoring (e.g., ICP, brain tissue oxygen tension, autoregulatory status) to optimize fluid therapy in neurocritically ill patients [85]. Point-of-care focused ultrasonography is increasingly used to determine fluid status rapidly, facilitating personalized fluid management when appropriate [86]; however, their role in the neurocritical care setting has not been defined.
Several studies have focused on the administered fluid type. Current data do not conclusively support routine use of balanced crystalloid solutions over 0.9% saline to reduce the risk of AKI and RRT in critically ill patients. A recent meta-analysis showed that the risk ratios of developing AKI and of being treated with RRT with balanced crystalloids compared with 0.9% saline were 0.96 (95% CI 0.89–1.02) and 0.95 (95% CI 0.81–1.11), respectively [87]. However, the 2018 ESICM consensus statement recommends crystalloids as maintenance and resuscitation fluids in neurocritical care patients while not recommending albumin and hypotonic solutions [85]. Small single-center randomized trials in patients with SAH [88] and TBI [89] found that balanced crystalloids reduced the hyperchloremia rate compared to 0.9% saline. Synthetic colloids (e.g., starch, gelatin) should be avoided in patients with ABI as they increase the risk of AKI and death [85, 90].
Hyperosmolar therapy
Hypernatremia and hyperchloremia are common complications in patients with ABI and risk factors for AKI and excess mortality [18, 19]. Consequently, fluid choice should be informed by the need to correct the specific electrolyte and acid–base imbalances. SCr and UOP should be closely monitored in patients receiving hyperosmolar therapy due to its strong association with AKI occurrence [6, 37]. The 2020 NCS guidelines recommend an upper serum sodium range of 150–155 mmol/L and chloride range of 110–115 mmol/L to decrease the risk of AKI In patients receiving hypertonic sodium solutions [37]. Bolus administration could be considered over continuous hypertonic sodium solution infusion as it could lead to fewer chloride values aberrations [20, 91]. An osmolality threshold of ≥ 320 mOsm/L has been suggested to increase the risk of AKI in patients receiving mannitol infusion [42]; however, this threshold has recently been questioned [41].
Nephrotoxic exposure and drug dosing
Medications should be closely reviewed for potentially nephrotoxic agents, which should be discontinued or substituted with less nephrotoxic drugs. However, potentially nephrotoxic agents, e.g., intravenous contrast, should still be used in patients with ABI if the information gained could have important therapeutic implications. The 2020 American College of Radiology and National Kidney Foundation guidelines recommend prophylactic 0.9% saline before and after intravenous contrast exposure in patients with eGFR < 30 mL/min/1.73 m2 or recent AKI to reduce the risk of AKI [50]. Drug-induced acute interstitial nephritis (e.g., due to anticonvulsants or antibiotics) must also be considered [92, 93].
Augmented renal clearance of > 130 mL/min/1.73 m2 is common in patients with ABI. The 74% (95% CI 55–87) pooled prevalence of augmented renal clearance in neurocritical care patients reported in a recent meta-analysis is higher than in any other critical care population [94]. Postulated mechanisms that promote an augmented renal clearance in ABI patients include increased cardiac output, high serum atrial natriuretic peptides, and increased hypothalamus–pituitary–adrenal axis activity with elevated levels of cortisol and catecholamines [94, 95]. Augmented renal clearance has important implications for drug dosing in patients with ABI and may lead to underdosing of levetiracetam and vancomycin [96, 97]; therefore, clinicians should monitor renal-eliminated medications to achieve target trough concentrations.
Biomarkers for AKI risk assessment
Factors contributing to AKI development and progression could be modified by incorporating novel biomarkers of early tubular stress or damage when clinical interventions or exposures increase the risk of AKI progression. Whether early identification of these patients could help reduce ABI-associated morbidity remains to be determined. A recent Acute Dialysis Quality Initiative consensus statement suggested that the AKI definition should be augmented by integrating novel AKI biomarkers into its risk-classification (Additional file 4; right panel) [98]. Although many candidate biomarkers exist (Additional file 5), prospective validation and implementation are needed. Among the few biomarkers studied in neurocritical care, cystatin C, neutrophil gelatinase-associated lipocalin, and liver-type fatty acid-binding protein at admission have been associated with increased risk of AKI in patients with TBI or stroke [99,100,101,102].
Renal replacement therapy
Patients with ABI may require RRT to manage the consequences of impaired renal function, including electrolyte imbalances, metabolic acidosis, and fluid overload. However, the use of RRT can impact cerebral blood flow, CPP, ICP, and brain tissue oxygenation, potentially leading to neurological complications. The effect on these factors depends on the specific type of RRT and its outcomes, which calls for special consideration in ABI patients [103].
Effects of RRT on the brain
RRT might lead to exacerbation of cerebral edema through the “reverse urea effect” or dialysis disequilibrium syndrome (DDS). During the initial stages of RRT, effective osmolytes such as urea are rapidly removed from the blood, creating an osmotic gradient between the blood and brain tissue. As brain cells have a relatively slow transport rate through cell membranes, this gradient causes water to move into the brain tissue, resulting in cerebral edema [104]. Additionally, elevated bicarbonate levels in dialysate and rapid rise in pH may induce paradoxical intracellular acidosis. This phenomenon occurs when bicarbonate-derived carbon dioxide crosses the cell membrane, leading to neuronal swelling and cerebral edema [105]. DDS and cerebral edema could lead to brain herniation or decreased CPP in patients with ABI.
Intradialytic hypotension, defined as a decrease in SBP by ≥ 20 mmHg or a reduction in MAP by ≥ 10 mmHg with associated symptoms [106], might occur during RRT and affect CPP because BP is its key determinant. It occurs when the body's hemodynamic compensation mechanisms fail to respond adequately to the decrease in plasma volume caused by ultrafiltration. This leads to reduced cardiac output, impaired peripheral vasoconstriction and refilling capacity, and decreased MAP [107]. If cerebral autoregulation is impaired, excessive ultrafiltration with decreased MAP might lead to reduced cerebral perfusion and CPP, potentially resulting in decreased ICP.
RRT has the potential to reduce brain tissue oxygenation through multiple pathways. One mechanism involves dialysis-related brain edema, which can impede oxygen diffusion. Additionally, increased ICP associated with a hypermetabolic state in the brain may increase oxygen consumption [108]. Dialysis-induced inflammation could result in pulmonary leukosequestration, leading to reduced arterial oxygen levels. This can impact the brain's respiratory center perfusion and metabolism, ultimately decreasing cerebral oxygenation and causing intermittent short apneic episodes [109]. In a recent study involving 17 adult hemodialysis patients, magnetic resonance imaging, diffusion tensor imaging, and proton magnetic resonance spectroscopy demonstrated that a single hemodialysis session could lead to an increase in brain tissue volume during the session [110]. This change was accompanied by alterations in white matter diffusion metrics and brain metabolite concentrations consistent with ischemic injury [110].
RRT timing
RRT is generally advised for critically ill patients when absolute solute/volume criteria are met and medical treatment proves insufficient [111]. However, RRT initiation should not be delayed in patients with ABI since rapid changes in osmolality could create an osmotic gradient with adverse neurologic consequences. Despite the association between pre-RRT BUN level and subsequent ICP elevation [112], there is no conclusive evidence on the optimal timing of RRT initiation in patients with ABI. Clinical practice suggests that maintaining pre-dialysis BUN under 30–35 mg/dL with optimized RRT could decrease the risk of increasing ICP during treatment [103].
RRT modalities
The RRT modalities commonly used in critically ill patients include intermittent hemodialysis (IHD), continuous RRT (CRRT), and prolonged intermittent RRT (PIRRT); (Table 2). IHD is typically used three times weekly for 3–4 h per session and allows for rapid solute control and fluid removal. CRRT encompasses various modalities developed specifically for critically ill patients to provide slower solute and fluid removal than IHD and maintain better hemodynamic stability. PIRRT combines the advantages of both, using conventional hemodialysis machines with blood-pump speeds and dialysate flow rates between IHD and CRRT. PIRRT improves hemodynamic stability through slow solute and fluid removal while avoiding the need for 24-h therapy (Table 3).
CRRT is less likely to cause DDS and intradialytic hypotension than IHD [112, 115] as its slower blood and dialysate/replacement fluid flow rates and smaller dialyzer surface area result in decreased urea clearance from the plasma (Table 3). Brain computed tomography studies showed increased brain water content after IHD but not after CRRT [116]. Moreover, CRRT slower net ultrafiltration rate facilitates hemodynamic stability, prevents intradialytic hypotension, and maintains CPP and brain tissue oxygenation. Although IHD remains an option in patients with mild brain injury and stable conditions, the 2012 KDIGO guidelines recommend CRRT for patients with ABI requiring RRT [6].
There are several strategies to prevent intradialytic hypotension. Common ones include establishing and adjusting the patient’s dry (target) weight; more frequent and longer RRT sessions to be able to decrease ultrafiltration rates; the avoidance of meals during RRT to mitigate the postprandial drop in blood pressure; the preemptive pharmacological use of midodrine, droxidopa, or sertraline; the use of cool dialysate; sodium profiling; and the use of a high calcium bath [117,118,119].
Conclusive evidence comparing the clinical outcomes after CRRT versus PIRRT in patients with ABI is lacking. A study in patients with brain hemorrhage found similar effects on hemodynamics and ICP [120]. PIRRT is potentially more efficient in resource utilization and offers greater patient care flexibility since it requires a standard IHD device and its administration time is shorter [121]. While the PIRRT effect is intermediate between IHD and CRRT, it could be an alternative to CRRT in patients who might not necessarily require CRRT or with concerns regarding IHD use in ABI and in centers without CRRT availability [68].
Sodium regulation with RRT
Sodium-based osmotherapy is crucial in managing cerebral edema and preventing ICP increase in patients with ABI. However, the standard sodium concentration in CRRT solutions is 140 mmol/L, lower than the recommended serum sodium concentration to maintain the osmotic pressure between the brain and the plasma (145–155 mmol/L). Sodium concentration in CRRT can be adjusted by adding hypertonic saline (NaCl 23.4%) to the CRRT solution bags or by administering hypertonic saline (NaCl 3%) as CRRT post-filter replacement fluid or as a separate infusion (Table 4). In IHD, a 145 mmol/L sodium bath and a separate hypertonic saline infusion could help achieve the desired serum sodium concentration.
Anticoagulation for RRT
Systemic anticoagulation for RRT should be avoided in patients with ABI. While most intermittent IHD sessions can be conducted without anticoagulation, regional citrate anticoagulation (RCA) is recommended for CRRT [68]. However, some considerations are important. The use of citrate can lead to either metabolic acidosis or alkalosis, depending on the ability of the liver to metabolize citrate. Additionally, due to its calcium-chelating effect, careful calcium replacement is necessary to prevent neurotoxicity stemming from low ionized calcium levels in the patient. RCA should not be used in cases of shock liver or lactic acidosis exceeding 4 mmol/L due to citrate accumulation. Therefore, careful monitoring of acid–base status, calcium, and lactic acid is recommended. If RCA is unavailable or contraindicated, CRRT can be performed without anticoagulation. Although nafamostat could be an alternative anticoagulant [123,124,125,126], its efficacy in multiple CRRT clinical settings, including ABI, requires further evaluation.
Conclusions
AKI, common in neurocritical patients, is associated with increased morbidity and mortality and has significant implications for managing ABI and its sequelae. Prompt identification of the cause of AKI, with a focus on reversible factors, and the adoption of preventive measures are crucial. The use of RRT in ABI patients is challenging due to potential negative impacts on ICP, CPP, brain oxygenation, and more. CRRT is preferred in ABI cases for gradual solute, electrolyte, and volume adjustments. Unless contraindicated, RCA should be considered for anticoagulation during CRRT. When CRRT is unavailable, intermittent dialysis methods with careful adjustments might be used to minimize complications. Given the magnitude of the problem, future research should focus on better understanding the mechanisms leading to AKI during ABI, and optimizing AKI management in neurocritical care.
Availability of data and materials
Not applicable.
Abbreviations
- ABI:
-
Acute brain injury
- AKI:
-
Acute kidney injury
- ATACH-II:
-
Antihypertensive treatment of acute cerebral hemorrhage II
- BP:
-
Blood pressure
- CKD:
-
Chronic kidney disease
- COBI:
-
Continuous hyperosmolar therapy for traumatic brain-injured patients
- CPP:
-
Cerebral perfusion pressure
- CRRT:
-
Continuous renal replacement therapy
- DDS:
-
Dialysis disequilibrium syndrome
- EPO-TBI:
-
Erythropoetin in traumatic brain injury
- ESICM:
-
European Society of Intensive Care Medicine
- GFR:
-
Glomerular filtration rate
- ICH:
-
Intracerebral hemorrhage
- ICP:
-
Intracranial pressure
- ICU:
-
Intensive care unit
- IHD:
-
Intermittent hemodialysis
- KDIGO:
-
Kidney Disease: Improving Global Outcomes
- MAP:
-
Mean arterial pressure
- NCS:
-
Neurocritical Care Society
- PIRRT:
-
Prolonged intermittent renal replacement therapy
- RCA:
-
Regional citrate anticoagulation
- RRT:
-
Renal replacement therapy
- SAH:
-
Subarachnoid hemorrhage
- SBP:
-
Systolic blood pressure
- SCr:
-
Serum creatinine
- TBI:
-
Traumatic brain injury
- UOP:
-
Urinary output
References
Griffin BR, Liu KD, Teixeira JP. Critical care nephrology: core curriculum 2020. Am J Kidney Dis. 2020;75(3):435–52.
Kellum JA, Ronco C, Bellomo R. Conceptual advances and evolving terminology in acute kidney disease. Nat Rev Nephrol. 2021;17(7):493–502.
James MT, Bhatt M, Pannu N, Tonelli M. Long-term outcomes of acute kidney injury and strategies for improved care. Nat Rev Nephrol. 2020;16(4):193–205.
Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P, Acute Dialysis Quality Initiative workgroup. Acute renal failure–definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8(4):R204-212.
Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A, Acute Kidney Injury Network. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31.
Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2(1):1–138.
Vanmassenhove J, Steen J, Vansteelandt S, Morzywolek P, Hoste E, Decruyenaere J, Benoit D, Van Biesen W. The importance of the urinary output criterion for the detection and prognostic meaning of AKI. Sci Rep. 2021;11(1):11089.
Hoste EA, Bagshaw SM, Bellomo R, Cely CM, Colman R, Cruz DN, Edipidis K, Forni LG, Gomersall CD, Govil D, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. 2015;41(8):1411–23.
Ramirez-Guerrero G, Lucero C, Villagran-Cortes F, Hauway E, Torres-Cifuentes V, Baghetti-Hernandez R, Vera-Calzaretta A, Ronco C, Garay O. Acute kidney injury in neurocritical patients: a retrospective cohort study. Int Urol Nephrol. 2023;55(7):1875–83.
Robba C, Banzato E, Rebora P, Iaquaniello C, Huang CY, Wiegers EJA, Meyfroidt G, Citerio G, Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (TBI) ICU Participants and Investigators. Acute kidney injury in traumatic brain injury patients: results from the collaborative European neurotrauma effectiveness research in traumatic brain injury study. Crit Care Med. 2021;49(1):112–26.
Skrifvars MB, Bailey M, Moore E, Martensson J, French C, Presneill J, Nichol A, Little L, Duranteau J, Huet O, et al. A post hoc analysis of osmotherapy use in the erythropoietin in traumatic brain injury study-associations with acute kidney injury and mortality. Crit Care Med. 2021;49(4):e394–403.
Wang R, Zhang J, Xu J, He M, Xu J. Incidence and burden of acute kidney injury among traumatic brain-injury patients. Risk Manag Healthc Policy. 2021;14:4571–80.
Liu Z, Wang R, He M, Kang Y. Hypomagnesemia is associated with the acute kidney injury in traumatic brain injury patients: a pilot study. Brain Sci. 2023;13(4):593.
Huet O, Chapalain X, Vermeersch V, Moyer JD, Lasocki S, Cohen B, Dahyot-Fizelier C, Chalard K, Seguin P, Hourmant Y, et al. Impact of continuous hypertonic (NaCl 20%) saline solution on renal outcomes after traumatic brain injury (TBI): a post hoc analysis of the COBI trial. Crit Care. 2023;27(1):42.
Huang ZY, Liu Y, Huang HF, Huang SH, Wang JX, Tian JF, Zeng WX, Lv RG, Jiang S, Gao JL, et al. Acute kidney injury in traumatic brain injury intensive care unit patients. World J Clin Cases. 2022;10(9):2751–63.
Wang D, Guo Y, Zhang Y, Li Z, Li A, Luo Y. Epidemiology of acute kidney injury in patients with stroke: a retrospective analysis from the neurology ICU. Intern Emerg Med. 2018;13(1):17–25.
Gwak DS, Chung I, Kim BK, Lee S, Jeong HG, Kim YS, Chae H, Park CY, Han MK. High chloride burden and clinical outcomes in critically ill patients with large hemispheric infarction. Front Neurol. 2021;12:604686.
Sadan O, Singbartl K, Kandiah PA, Martin KS, Samuels OB. Hyperchloremia is associated with acute kidney injury in patients with subarachnoid hemorrhage. Crit Care Med. 2017;45(8):1382–8.
Sadan O, Singbartl K, Kraft J, Plancher JM, Greven ACM, Kandiah P, Pimentel C, Hall CL, Papangelou A, Asbury WH, et al. Low-chloride- versus high-chloride-containing hypertonic solution for the treatment of subarachnoid hemorrhage-related complications: the ACETatE (A low ChloriE hyperTonic solution for brain Edema) randomized trial. J Intensive Care. 2020;8:32.
Riha HM, Erdman MJ, Vandigo JE, Kimmons LA, Goyal N, Davidson KE, Pandhi A, Jones GM. Impact of moderate hyperchloremia on clinical outcomes in intracerebral hemorrhage patients treated with continuous infusion hypertonic saline: a pilot study. Crit Care Med. 2017;45(9):e947–53.
Burgess LG, Goyal N, Jones GM, Khorchid Y, Kerro A, Chapple K, Tsivgoulis G, Alexandrov AV, Chang JJ. Evaluation of acute kidney injury and mortality after intensive blood pressure control in patients with intracerebral hemorrhage. J Am Heart Assoc. 2018;7(8):e008439.
Tujjar O, Belloni I, Hougardy JM, Scolletta S, Vincent JL, Creteur J, Taccone FS. Acute kidney injury after subarachnoid hemorrhage. J Neurosurg Anesthesiol. 2017;29(2):140–9.
Kumar AB, Shi Y, Shotwell MS, Richards J, Ehrenfeld JM. Hypernatremia is a significant risk factor for acute kidney injury after subarachnoid hemorrhage: a retrospective analysis. Neurocrit Care. 2015;22(2):184–91.
Luu D, Komisarow J, Mills BM, Vavilala MS, Laskowitz DT, Mathew J, James ML, Hernandez A, Sampson J, Fuller M, et al. Association of severe acute kidney injury with mortality and healthcare utilization following isolated traumatic brain injury. Neurocrit Care. 2021;35(2):434–40.
Zacharia BE, Ducruet AF, Hickman ZL, Grobelny BT, Fernandez L, Schmidt JM, Narula R, Ko LN, Cohen ME, Mayer SA, et al. Renal dysfunction as an independent predictor of outcome after aneurysmal subarachnoid hemorrhage: a single-center cohort study. Stroke. 2009;40(7):2375–81.
Buttner S, Stadler A, Mayer C, Patyna S, Betz C, Senft C, Geiger H, Jung O, Finkelmeier F. Incidence, risk factors, and outcome of acute kidney injury in neurocritical care. J Intensive Care Med. 2020;35(4):338–46.
Nadkarni GN, Patel AA, Konstantinidis I, Mahajan A, Agarwal SK, Kamat S, Annapureddy N, Benjo A, Thakar CV. Dialysis requiring acute kidney injury in acute cerebrovascular accident hospitalizations. Stroke. 2015;46(11):3226–31.
Murray AM. Cognitive impairment in the aging dialysis and chronic kidney disease populations: an occult burden. Adv Chronic Kidney Dis. 2008;15(2):123–32.
Lee M, Saver JL, Chang KH, Liao HW, Chang SC, Ovbiagele B. Low glomerular filtration rate and risk of stroke: meta-analysis. BMJ. 2010;341:c4249.
Kumai Y, Kamouchi M, Hata J, Ago T, Kitayama J, Nakane H, Sugimori H, Kitazono T, FSR Investigators. FSR investigators proteinuria and clinical outcomes after ischemic stroke. Neurology. 2012;78(24):1909–15.
Beuscher VD, Sprugel MI, Gerner ST, Sembill JA, Madzar D, Reindl C, Lucking H, Lang S, Hoelter P, Kuramatsu JB, et al. Chronic kidney disease and clinical outcomes in patients with intracerebral hemorrhage. J Stroke Cerebrovasc Dis. 2020;29(8):104802.
Ng L, Wang J, Altaweel L, Athar MK. Neurologic aspects of cardiac emergencies. Crit Care Clin. 2014;30(3):557–84.
Prunet B, Basely M, D’Aranda E, Cambefort P, Pons F, Cimarelli S, Dagain A, Desse N, Veyrieres JB, Jego C, et al. Impairment of cardiac metabolism and sympathetic innervation after aneurysmal subarachnoid hemorrhage: a nuclear medicine imaging study. Crit Care. 2014;18(3):R131.
Nguyen H, Zaroff JG. Neurogenic stunned myocardium. Curr Neurol Neurosci Rep. 2009;9(6):486–91.
Kerro A, Woods T, Chang JJ. Neurogenic stunned myocardium in subarachnoid hemorrhage. J Crit Care. 2017;38:27–34.
Messerer DAC, Halbgebauer R, Nilsson B, Pavenstadt H, Radermacher P, Huber-Lang M. Immunopathophysiology of trauma-related acute kidney injury. Nat Rev Nephrol. 2021;17(2):91–111.
Cook AM, Morgan Jones G, Hawryluk GWJ, Mailloux P, McLaughlin D, Papangelou A, Samuel S, Tokumaru S, Venkatasubramanian C, Zacko C, et al. Guidelines for the acute treatment of cerebral edema in neurocritical care patients. Neurocrit Care. 2020;32(3):647–66.
Lin SY, Tang SC, Tsai LK, Yeh SJ, Shen LJ, Wu FL, Jeng JS. Incidence and risk factors for acute kidney injury following mannitol infusion in patients with acute stroke: a retrospective cohort study. Medicine (Baltimore). 2015;94(47):e2032.
Kim MY, Park JH, Kang NR, Jang HR, Lee JE, Huh W, Kim YG, Kim DJ, Hong SC, Kim JS, et al. Increased risk of acute kidney injury associated with higher infusion rate of mannitol in patients with intracranial hemorrhage. J Neurosurg. 2014;120(6):1340–8.
Fang L, You H, Chen B, Xu Z, Gao L, Liu J, Xie Q, Zhou Y, Gu Y, Lin S, et al. Mannitol is an independent risk factor of acute kidney injury after cerebral trauma: a case–control study. Ren Fail. 2010;32(6):673–9.
Gondim Fde A, Aiyagari V, Shackleford A, Diringer MN. Osmolality not predictive of mannitol-induced acute renal insufficiency. J Neurosurg. 2005;103(3):444–7.
Fink ME. Osmotherapy for intracranial hypertension: mannitol versus hypertonic saline. Continuum (Minneap Minn). 2012;18(3):640–54.
Erdman MJ, Riha H, Bode L, Chang JJ, Jones GM. Predictors of acute kidney injury in neurocritical care patients receiving continuous hypertonic saline. Neurohospitalist. 2017;7(1):9–14.
Sigmon J, May CC, Bryant A, Humanez J, Singh V. Assessment of acute kidney injury in neurologically injured patients receiving hypertonic sodium chloride: Does chloride load matter? Ann Pharmacother. 2020;54(6):541–6.
Semler MW, Kellum JA. Balanced crystalloid solutions. Am J Respir Crit Care Med. 2019;199(8):952–60.
Swenson ER. Balanced crystalloid versus saline solution in critically ill patients: Is chloride the villain? Am J Respir Crit Care Med. 2019;200(3):398.
Hansen PB, Jensen BL, Skott O. Chloride regulates afferent arteriolar contraction in response to depolarization. Hypertension. 1998;32(6):1066–70.
Wilcox CS. Regulation of renal blood flow by plasma chloride. J Clin Invest. 1983;71(3):726–35.
Chowdhury AH, Cox EF, Francis ST, Lobo DN. A randomized, controlled, double-blind crossover study on the effects of 2-L infusions of 0.9% saline and plasma-lyte(R) 148 on renal blood flow velocity and renal cortical tissue perfusion in healthy volunteers. Ann Surg. 2012;256(1):18–24.
Davenport MS, Perazella MA, Yee J, Dillman JR, Fine D, McDonald RJ, Rodby RA, Wang CL, Weinreb JC. Use of intravenous iodinated contrast media in patients with kidney disease: consensus statements from the American College of Radiology and the National Kidney Foundation. Radiology. 2020;294(3):660–8.
McDonald JS, McDonald RJ, Williamson EE, Kallmes DF, Kashani K. Post-contrast acute kidney injury in intensive care unit patients: a propensity score-adjusted study. Intensive Care Med. 2017;43(6):774–84.
Schonenberger E, Martus P, Bosserdt M, Zimmermann E, Tauber R, Laule M, Dewey M. Kidney injury after intravenous versus intra-arterial contrast agent in patients suspected of having coronary artery disease: a randomized trial. Radiology. 2019;292(3):664–72.
Corral L, Javierre CF, Ventura JL, Marcos P, Herrero JI, Manez R. Impact of non-neurological complications in severe traumatic brain injury outcome. Crit Care. 2012;16(2):R44.
Barea-Mendoza JA, Chico-Fernandez M, Quintana-Diaz M, Servia-Goixart L, Fernandez-Cuervo A, Bringas-Bollada M, Ballesteros-Sanz MA, Garcia-Saez I, Perez-Barcena J, Llompart-Pou JA, et al. Traumatic brain injury and acute kidney injury-outcomes and associated risk factors. J Clin Med. 2022;11(23):7216.
Adachi N, Lei B, Deshpande G, Seyfried FJ, Shimizu I, Nagaro T, Arai T. Uraemia suppresses central dopaminergic metabolism and impairs motor activity in rats. Intensive Care Med. 2001;27(10):1655–60.
Jeppsson B, Freund HR, Gimmon Z, James JH, von Meyenfeldt MF, Fischer JE. Blood-brain barrier derangement in uremic encephalopathy. Surgery. 1982;92(1):30–5.
Trachtman H, Futterweit S, Tonidandel W, Gullans SR. The role of organic osmolytes in the cerebral cell volume regulatory response to acute and chronic renal failure. J Am Soc Nephrol. 1993;3(12):1913–9.
Silver SM. Cerebral edema after rapid dialysis is not caused by an increase in brain organic osmolytes. J Am Soc Nephrol. 1995;6(6):1600–6.
Liu M, Liang Y, Chigurupati S, Lathia JD, Pletnikov M, Sun Z, Crow M, Ross CA, Mattson MP, Rabb H. Acute kidney injury leads to inflammation and functional changes in the brain. J Am Soc Nephrol. 2008;19(7):1360–70.
Palkovits M, Sebekova K, Gallatz K, Boor P, Sebekova K Jr, Klassen A, Bahner U, Heidland A. Neuronal activation in the CNS during different forms of acute renal failure in rats. Neuroscience. 2009;159(2):862–82.
Salama M, Farrag SM, abulasrar S, Amin MM, Ali AA, Sheashaa H, Sobh M, Arias-Carrion O. Up-regulation of TLR-4 in the brain after ischemic kidney-induced encephalopathy in the rat. CNS Neurol Disord Drug Targets. 2013;12(5):583–586.
Chou AH, Lee CM, Chen CY, Liou JT, Liu FC, Chen YL, Day YJ. Hippocampal transcriptional dysregulation after renal ischemia and reperfusion. Brain Res. 2014;1582:197–210.
Andres-Hernando A, Dursun B, Altmann C, Ahuja N, He Z, Bhargava R, Edelstein CE, Jani A, Hoke TS, Klein C, et al. Cytokine production increases and cytokine clearance decreases in mice with bilateral nephrectomy. Nephrol Dial Transplant. 2012;27(12):4339–47.
Chen J, Hartono JR, John R, Bennett M, Zhou XJ, Wang Y, Wu Q, Winterberg PD, Nagami GT, Lu CY. Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int. 2011;80(5):504–15.
Lu R, Kiernan MC, Murray A, Rosner MH, Ronco C. Kidney-brain crosstalk in the acute and chronic setting. Nat Rev Nephrol. 2015;11(12):707–19.
Nongnuch A, Panorchan K, Davenport A. Brain-kidney crosstalk. Crit Care. 2014;18(3):225.
Ramirez-Guerrero G, Baghetti-Hernandez R, Ronco C. Acute kidney injury at the neurocritical care unit. Neurocrit Care. 2021;36:640–9.
Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. 2012;120(4):c179-184.
Bouma GJ, Muizelaar JP, Bandoh K, Marmarou A. Blood pressure and intracranial pressure-volume dynamics in severe head injury: relationship with cerebral blood flow. J Neurosurg. 1992;77(1):15–9.
Asfar P, Meziani F, Hamel JF, Grelon F, Megarbane B, Anguel N, Mira JP, Dequin PF, Gergaud S, Weiss N, et al. High versus low blood-pressure target in patients with septic shock. New Engl J Med. 2014;370(17):1583–93.
Chesnut R, Aguilera S, Buki A, Bulger E, Citerio G, Cooper DJ, Arrastia RD, Diringer M, Figaji A, Gao G, et al. A management algorithm for adult patients with both brain oxygen and intracranial pressure monitoring: the Seattle International Severe Traumatic Brain Injury Consensus Conference (SIBICC). Intensive Care Med. 2020;46(5):919–29.
Carney N, Totten AM, O’Reilly C, Ullman JS, Hawryluk GW, Bell MJ, Bratton SL, Chesnut R, Harris OA, Kissoon N, et al. Guidelines for the management of severe traumatic brain injury, Fourth edition. Neurosurgery. 2017;80(1):6–15.
Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, Machado FR, McIntyre L, Ostermann M, Prescott HC, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021;47(11):1181–247.
Huijben JA, Volovici V, Cnossen MC, Haitsma IK, Stocchetti N, Maas AIR, Menon DK, Ercole A, Citerio G, Nelson D, et al. Variation in general supportive and preventive intensive care management of traumatic brain injury: a survey in 66 neurotrauma centers participating in the Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (CENTER-TBI) study. Crit Care. 2018;22(1):90.
Steiner LA, Siegemund M. Vasoactive agents to improve brain perfusion: pathophysiology and clinical utilization. Curr Opin Crit Care. 2019;25(2):110–6.
Bhatraju PK, Zelnick LR, Herting J, Katz R, Mikacenic C, Kosamo S, Morrell ED, Robinson-Cohen C, Calfee CS, Christie JD, et al. Identification of acute kidney injury subphenotypes with differing molecular signatures and responses to vasopressin therapy. Am J Respir Crit Care Ced. 2019;199(7):863–72.
Greenberg SM, Ziai WC, Cordonnier C, Dowlatshahi D, Francis B, Goldstein JN, Hemphill JC 3rd, Johnson R, Keigher KM, Mack WJ, et al. 2022 Guideline for the management of patients with spontaneous intracerebral hemorrhage: a guideline from the American Heart Association/American Stroke Association. Stroke. 2022;53(7):e282–361.
Ravera M, Re M, Deferrari L, Vettoretti S, Deferrari G. Importance of blood pressure control in chronic kidney disease. J Am Soc Nephrol. 2006;17(4 Suppl 2):S98-103.
Qureshi AI, Huang W, Lobanova I, Barsan WG, Hanley DF, Hsu CY, Lin CL, Silbergleit R, Steiner T, Suarez JI, et al. Outcomes of intensive systolic blood pressure reduction in patients with intracerebral hemorrhage and excessively high initial systolic blood pressure: post hoc analysis of a randomized clinical trial. JAMA Neurol. 2020;77(11):1355–65.
Qureshi AI, Huang W, Lobanova I, Hanley DF, Hsu CY, Malhotra K, Steiner T, Suarez JI, Toyoda K, Yamamoto H, et al. Systolic blood pressure reduction and acute kidney injury in intracerebral hemorrhage. Stroke. 2020;51(10):3030–8.
Clifton GL, Miller ER, Choi SC, Levin HS. Fluid thresholds and outcome from severe brain injury. Crit Care Med. 2002;30(4):739–45.
Clifton GL, Valadka A, Zygun D, Coffey CS, Drever P, Fourwinds S, Janis LS, Wilde E, Taylor P, Harshman K, et al. Very early hypothermia induction in patients with severe brain injury (the National Acute Brain Injury Study: Hypothermia II): a randomised trial. Lancet Neurol. 2011;10(2):131–9.
Wiegers EJA, Lingsma HF, Huijben JA, Cooper DJ, Citerio G, Frisvold S, Helbok R, Maas AIR, Menon DK, Moore EM, et al. Fluid balance and outcome in critically ill patients with traumatic brain injury (CENTER-TBI and OzENTER-TBI): a prospective, multicentre, comparative effectiveness study. Lancet Neurol. 2021;20(8):627–38.
Husain-Syed F, Grone HJ, Assmus B, Bauer P, Gall H, Seeger W, Ghofrani A, Ronco C, Birk HW. Congestive nephropathy: a neglected entity? Proposal for diagnostic criteria and future perspectives. ESC Heart Fail. 2021;8(1):183–203.
Oddo M, Poole D, Helbok R, Meyfroidt G, Stocchetti N, Bouzat P, Cecconi M, Geeraerts T, Martin-Loeches I, Quintard H, et al. Fluid therapy in neurointensive care patients: ESICM consensus and clinical practice recommendations. Intensive Care Med. 2018;44(4):449–63.
Romero-Gonzalez G, Manrique J, Slon-Roblero MF, Husain-Syed F, De la Espriella R, Ferrari F, Bover J, Ortiz A, Ronco C. PoCUS in nephrology: a new tool to improve our diagnostic skills. Clin Kidney J. 2023;16(2):218–29.
Hammond NE, Zampieri FG, Di Tanna GL, Garside T, Adigbli D, Cavalcanti AB, Machado FR, Micallef S, Myburgh J, Ramanan M, et al. Balanced crystalloids versus saline in critically ill adults—a systematic review with meta-analysis. NEJM Evid. 2022. https://doi.org/10.1056/EVIDoa2100010.
Lehmann L, Bendel S, Uehlinger DE, Takala J, Schafer M, Reinert M, Jakob SM. Randomized, double-blind trial of the effect of fluid composition on electrolyte, acid-base, and fluid homeostasis in patients early after subarachnoid hemorrhage. Neurocrit Care. 2013;18(1):5–12.
Roquilly A, Loutrel O, Cinotti R, Rosenczweig E, Flet L, Mahe PJ, Dumont R, Marie Chupin A, Peneau C, Lejus C, et al. Balanced versus chloride-rich solutions for fluid resuscitation in brain-injured patients: a randomised double-blind pilot study. Crit Care. 2013;17(2):R77.
Reinhart K, Perner A, Sprung CL, Jaeschke R, Schortgen F, Johan Groeneveld AB, Beale R, Hartog CS, European Society of Intensive Care Medicine. Consensus statement of the ESICM task force on colloid volume therapy in critically ill patients. Intensive Care Med. 2012;38(3):368–83.
Maguigan KL, Dennis BM, Hamblin SE, Guillamondegui OD. Method of hypertonic saline administration: effects on osmolality in traumatic brain injury patients. J Clin Neurosci. 2017;39:147–50.
Chau K, Yong J, Ismail K, Griffith N, Liu M, Makris A. Levetiracetam-induced severe acute granulomatous interstitial nephritis. Clin Kidney J. 2012;5(3):234–6.
Rossert J. Drug-induced acute interstitial nephritis. Kidney Int. 2001;60(2):804–17.
Hefny F, Stuart A, Kung JY, Mahmoud SH. Prevalence and risk factors of augmented renal clearance: a systematic review and meta-analysis. Pharmaceutics. 2022;14(2):445.
Udy AA, Jarrett P, Lassig-Smith M, Stuart J, Starr T, Dunlop R, Deans R, Roberts JA, Senthuran S, Boots R, et al. Augmented renal clearance in traumatic brain injury: a single-center observational study of atrial natriuretic peptide, cardiac output, and creatinine clearance. J Neurotrauma. 2017;34(1):137–44.
Chen Y, Liu L, Zhu M. Effect of augmented renal clearance on the therapeutic drug monitoring of vancomycin in patients after neurosurgery. J Int Med Res. 2020;48(10):300060520949076.
Ong CLJ, Goh PSJ, Teo MM, Lim TP, Goh KKK, Ang XY, Lim LJK, Jamaludin NHB, Ang BT, Kwa LHA. Pharmacokinetics of levetiracetam in neurosurgical ICU patients. J Crit Care. 2021;64:255–61.
Ostermann M, Zarbock A, Goldstein S, Kashani K, Macedo E, Murugan R, Bell M, Forni L, Guzzi L, Joannidis M, et al. Recommendations on acute kidney injury biomarkers from the acute disease quality initiative consensus conference: a consensus statement. JAMA Netw Open. 2020;3(10):e2019209.
Li N, Zhao WG, Xu FL, Zhang WF, Gu WT. Neutrophil gelatinase-associated lipocalin as an early marker of acute kidney injury in patients with traumatic brain injury. J Nephrol. 2013;26(6):1083–8.
Shimoyama T, Sato T, Sakamoto Y, Nagai K, Aoki J, Suda S, Nishiyama Y, Kimura K. Urinary biomarkers of kidney tubule injury, risk of acute kidney injury, and mortality in patients with acute ischaemic stroke treated at a stroke care unit. Eur J Neurol. 2020;27(12):2463–72.
Wang S, Xie L, Xu J, Hu Y, Wu Y, Lin Z, Pan S. Predictive value of serum creatinine/cystatin C in neurocritically ill patients. Brain Behav. 2019;9(12):e01462.
Jiang F, Su L, Xiang H, Zhang X, Xu D, Zhang Z, Peng Z. Incidence, risk factors, and biomarkers predicting ischemic or hemorrhagic stroke associated acute kidney injury and outcome: a retrospective study in a general intensive care unit. Blood Purif. 2019;47(4):317–26.
Davenport A. Renal replacement therapy in the patient with acute brain injury. Am J Kidney Dis. 2001;37(3):457–66.
Trinh-Trang-Tan MM, Cartron JP, Bankir L. Molecular basis for the dialysis disequilibrium syndrome: altered aquaporin and urea transporter expression in the brain. Nephrol Dial Transplant. 2005;20(9):1984–8.
Shapiro JI, Whalen M, Kucera R, Kindig N, Filley G, Chan L. Brain pH responses to sodium bicarbonate and Carbicarb during systemic acidosis. Am J Physiol. 1989;256(5 Pt 2):H1316-1321.
Workgroup KD. K/DOQI clinical practice guidelines for cardiovascular disease in dialysis patients. Am J Kidney Dis. 2005;45(4 Suppl 3):S1-153.
Daugirdas JT. Pathophysiology of dialysis hypotension: an update. Am J Kidney Dis. 2001;38(4 Suppl 4):S11-17.
Ko SB, Choi HA, Gilmore E, Schmidt JM, Claassen J, Lee K, Mayer SA, Badjatia N. Pearls & Oysters: the effects of renal replacement therapy on cerebral autoregulation. Neurology. 2012;78(6):e36-38.
Ross EA, Tashkin D, Chenoweth D, Webber MM, Nissenson AR. Pulmonary leukosequestration without hypoxemia during hemodialysis. Int J Artif Organs. 1987;10(6):367–74.
Anazodo UC, Wong DY, Theberge J, Dacey M, Gomes J, Penny JD, Van Ginkel M, Poirier SE, McIntyre CW. Hemodialysis-Related acute brain injury demonstrated by application of intradialytic magnetic resonance imaging and spectroscopy. J Am Soc Nephrol. 2023;34(6):1090–104.
STARRT-AKI Investigators; Canadian Critical Care Trials Group; Australian and New Zealand Intensive Care Society Clinical Trials Group; United Kingdom Critical Care Research Group; Canadian Nephrology Trials Network; Irish Critical Care Trials Group; Bagshaw SM, Wald R, Adhikari NKJ, et al: Timing of initiation of renal-replacement therapy in acute kidney injury. N Engl J Med. 2020;383(3):240–51.
Lund A, Damholt MB, Wiis J, Kelsen J, Strange DG, Moller K. Intracranial pressure during hemodialysis in patients with acute brain injury. Acta Anaesthesiol Scand. 2019;63(4):493–9.
Beaubien-Souligny W, Trott T, Neyra JA. How to determine fluid management goals during continuous kidney replacement therapy in patients with AKI: focus on POCUS. Kidney360. 2022;3(10):1795–806.
Tsujimoto Y, Tsujimoto H, Nakata Y, Kataoka Y, Kimachi M, Shimizu S, Ikenoue T, Fukuma S, Yamamoto Y, Fukuhara S. Dialysate temperature reduction for intradialytic hypotension for people with chronic kidney disease requiring haemodialysis. Cochrane Database Syst Rev. 2019;7(7):CD012598.
Schneider AG, Bellomo R, Bagshaw SM, Glassford NJ, Lo S, Jun M, Cass A, Gallagher M. Choice of renal replacement therapy modality and dialysis dependence after acute kidney injury: a systematic review and meta-analysis. Intensive Care Med. 2013;39(6):987–97.
Ronco C, Bellomo R, Brendolan A, Pinna V, La Greca G. Brain density changes during renal replacement in critically ill patients with acute renal failure. Continuous hemofiltration versus intermittent hemodialysis. J Nephrol. 1999;12(3):173–8.
Douvris A, Malhi G, Hiremath S, McIntyre L, Silver SA, Bagshaw SM, Wald R, Ronco C, Sikora L, Weber C, et al. Interventions to prevent hemodynamic instability during renal replacement therapy in critically ill patients: a systematic review. Crit Care. 2018;22(1):41.
Flythe JE, Chang TI, Gallagher MP, Lindley E, Madero M, Sarafidis PA, Unruh ML, Wang AY, Weiner DE, Cheung M, et al. Blood pressure and volume management in dialysis: conclusions from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. 2020;97(5):861–76.
Chou JA, Kalantar-Zadeh K, Mathew AT. A brief review of intradialytic hypotension with a focus on survival. Sem Dial. 2017;30(6):473–80.
Wu VC, Huang TM, Shiao CC, Lai CF, Tsai PR, Wang WJ, Huang HY, Wang KC, Ko WJ, Wu KD, et al. The hemodynamic effects during sustained low-efficiency dialysis versus continuous veno-venous hemofiltration for uremic patients with brain hemorrhage: a crossover study. J Neurosurg. 2013;119(5):1288–95.
Schwenger V, Weigand MA, Hoffmann O, Dikow R, Kihm LP, Seckinger J, Miftari N, Schaier M, Hofer S, Haar C, et al. Sustained low efficiency dialysis using a single-pass batch system in acute kidney injury—a randomized interventional trial: the REnal Replacement Therapy Study in Intensive Care Unit PatiEnts. Crit Care. 2012;16(4):R140.
Yessayan LT, Szamosfalvi B, Rosner MH. Management of dysnatremias with continuous renal replacement therapy. Sem Dial. 2021;34(6):472–9.
Choi JY, Kang YJ, Jang HM, Jung HY, Cho JH, Park SH, Kim YL, Kim CD. Nafamostat mesilate as an anticoagulant during continuous renal replacement therapy in patients with high bleeding risk: a randomized clinical trial. Medicine (Baltimore). 2015;94(52):e2392.
Chen T, Wang J, Li C, Zhang W, Zhang L, An L, Pang T, Shi X, Liao H. Nafamostat mesilate attenuates neuronal damage in a rat model of transient focal cerebral ischemia through thrombin inhibition. Sci Rep. 2014;4:5531.
Matsubara H, Imai T, Tsuji S, Oka N, Egashira Y, Enomoto Y, Nakayama N, Nakamura S, Shimazawa M, Iwama T, et al. Nafamostat protects against early brain injury after subarachnoid hemorrhage in mice. J Pharmacol Sci. 2022;148(1):65–72.
Yang JW, Han BG, Kim BR, Lee YH, Kim YS, Yu JM, Choi SO. Superior outcome of nafamostat mesilate as an anticoagulant in patients undergoing maintenance hemodialysis with intracerebral hemorrhage. Ren Fail. 2009;31(8):668–75.
Acknowledgements
None.
Funding
This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
FH-S, TT, JAN, GR-G, and AT prepared all manuscript drafts and were involved in reviewing and editing, including the tables and figures. MHR and CR conceived the concept underlying the manuscript and were involved in editing of the manuscript, including the tables and figures. AT is the paper's senior author. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
JAN reports consultancy agreements with Baxter Healthcare, Outset Medical, Vifor Pharma, and Leadiant Biosciences, all unrelated to this work. AT reports consultancy for Baxter, receiving honoraria from UptoDate, having a patent on 0.5% trisodium citrate solution for CRRT anti- coagulation, the license for which has been bought by Baxter, and serving on a speakers bureau for Baxter, all unrelated to this work. MHR has received consultant fees from Baxter Healthcare, serves as on the Data Safety Monitoring Boards of clinical trials sponsored by Reata, Travere and Astra Zeneca, all unrelated to this work. CR has received funding for lectures been consultant or advisory board member for Asahi, Astute, B. Braun, Baxter, bioMérieux, Bioporto, CytoSorbents, Estor, Fresenius Medical Care, General Electric (GE), Jafron, Medtronic, Toray. FH-S, TT, and GR-G declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Appendix Table 1.
Common causes of AKI in the ICU setting.
Additional file 2.
Appendix Literature Search Strategy.
Additional file 3: Appendix Table 2.
AKI and RRT rates and associated outcomes in neurocritically ill patients.
Additional file 4: Appendix Table 3.
Current and proposed definition and staging of AKI.
Additional file 5: Appendix Table 4.
Characteristics of novel AKI biomarkers.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Husain-Syed, F., Takeuchi, T., Neyra, J.A. et al. Acute kidney injury in neurocritical care. Crit Care 27, 341 (2023). https://doi.org/10.1186/s13054-023-04632-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-023-04632-1