
Abstract
Background
The clinicopathological parameters such as residual tumor, grade, the International Federation of Gynecology and Obstetrics (FIGO) score are often used to predict the survival of ovarian cancer patients, but the 5-year survival of high grade serous ovarian cancer (HGSOC) still remains around 30%. Hence, the relentless pursuit of enhanced prognostic tools for HGSOC, this study introduces an unprecedented gene expression-based molecular prognostic score (mPS). Derived from a novel 20-gene signature through Least Absolute Shrinkage and Selection Operator (LASSO)-Cox regression, the mPS stands out for its predictive prowess.
Results
Validation across diverse datasets, including training and test sets (n = 491 each) and a large HGSOC patient cohort from the Ovarian Tumor Tissue Analysis (OTTA) consortium (n = 7542), consistently shows an area-under-curve (AUC) around 0.7 for predicting 5-year overall survival. The mPS’s impact on prognosis resonates profoundly, yielding an adjusted hazard-ratio (HR) of 6.1 (95% CI: 3.65–10.3; p < 0.001), overshadowing conventional parameters—FIGO score, residual disease, and age. Molecular insights gleaned from mPS stratification uncover intriguing pathways, with focal-adhesion, Wnt, and Notch signaling upregulated, and antigen processing and presentation downregulated (p < 0.001) in high-risk HGSOC cohorts.
Conclusion
Positioned as a robust prognostic marker, the 20-gene signature-derived mPS emerges as a potential game-changer in clinical settings. Beyond its role in predicting overall survival, its implications extend to guiding alternative therapies, especially targeting Wnt/Notch signaling pathways and immune evasion—a promising avenue for improving outcomes in high-risk HGSOC patients.
Similar content being viewed by others
Introduction
Epithelial ovarian cancer (EOC) is classified into different categories based on histotypes and grade [1]. Despite the initial responses with cyto-reductive surgery and platinumbased chemotherapy, highgrade serous ovarian cancer (HGSOC) continued to account for 70% of EOC-related cases with more than 75% deaths within 10 years of initial diagnosis. It might be due to the high rate of intra-tumor genetic heterogeneity and chromosome instability within the HGSOCs [2, 3] subsequently supporting clonal evolution [4], resulting in chemo- or therapy- resistance. Therefore, a search for efficient gene signatures or prognostic markers is an urgent unmet clinical need for HGSOC.
Survival prediction takes various factors into account; like age, the International Federation of Gynecology and Obstetrics (FIGO) stage, histology, residual disease, and tumor recurrence [5, 6]. However, prediction based on this orthodox clinical information has limited potential to give rise to a robust prognostic method. It is because of the complex interaction of various molecules as well as immunological factors leading to variable responses within the HGSOCs. Recently the molecular subtypes of HGSOCs based on transcriptome profiles have been identified [7, 8]. The most common and consensus subtypes using various clustering algorithms are mesenchymal, immunoreactive, differentiated, and proliferative. Although these molecular subtypes showed distinct and differential regulation of biological pathways between the groups, but showed relatively less influence on the survival of patients using the Cancer Genome Atlas (TCGA) HGSOC cohort data [9]. It has been reported previously that gene signatures could potentially and significantly play- a role in determining the survival of cancer patients [10] including ovarian cancer [9]. A similar approach has been applied using 101prognostic gene signatures for predicting the survival of HGSOCs [11]. This approach of using molecular gene signatures as prognostic marker has been studied or reported in various cancers: breast [12], colon [13], and prostate [14].
Herein, we proposed to develop a molecular prognostic score (mPS), a machine learning approach for stratifying the prognosis of HGSOCs based on the expression of only 20 predictor genes and the associated coefficients as derived from Least Absolute Shrinkage and Selection Operator (LASSO)-Cox regression [15]. The proposed study design is schematically shown in Fig. 1. In this study, we have considered 1022 subjects/samples and screened or considered only 10,225 genes - found common in TCGA and Gene Expression Omnibus (GEO) databases. The micro-arraybased expression analysis of the same or similar platform (Affymetrix human U133A microarray or Affymetrix Human Genome U133 plus 2.0 Array) has been used here to filter the common genes for subsequent analysis. These common genes across different datasets were further screened to obtain the prognostic gene signature of HGSOCs based on the Cox (proportional hazards) regression model [16]. Finally, further trimming of prognostic genes and feature extraction was done by applying the LASSO-Cox regression model [16] on training datasets of HGSOCs. This resulted in obtaining predictive markers along with derived coefficients that were subsequently used to obtain mPS that eventually determined the prognosis in test or validation datasets (Fig. 1).

Methodology adapted to screen and filter genes for obtaining molecular prognostic score (mPS) based on prognostic signature genes: RNA expression data obtained from TCGA and GEO as indicated were used to find prognostic genes. These prognostic genes were further used in training datasets, 10-fold cross validation to obtain predictor genes and associated coefficients (feature extraction) after applying LASSO regression. These predictor genes and the derived mPS were applied in validation or test datasets. It was also applied in different mRNA expression platforms such as RNA Sequencing by Illumina and NanoString. DEGs; Differentially expressed genes that are significantly (FDR < 0.05) expressed between tumor samples as compared to normal samples. AI; Artificial intelligence. OTTA-SPOT; Ovarian Tumor Tissue Analysis consortium - Stratified Prognosis of Ovarian Tumors
Methods
Datasets
Gene expression raw microarray datasets were downloaded from TCGA viz., TCGA-OV (https://gdac.broadinstitute.org/?cohort=OV) and GEO database managed by the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/geo/). The GEO datasets are GSE18520 (n = 63), GSE26712 (n = 195), GSE26193 (n = 79), GSE63885 (n = 73), GSE14764 (n = 68). Combining TCGA-OV (n = 544) and GEO datasets (n = 478) resulted in a total of 1022 clinical samples with 10,225 as the common gene symbols across all datasets. The datasets underwent preprocessing and normalization using the Robust Microarray Average (RMA) approach. Quantile normalization, followed by batch effect removal, ensured consistent patterns or log ratios across various datasets (Appendix A Suppl Fig. S1). To eliminate outliers, a correlation matrix of mRNA expression (Array-Array Intensity correlation) [17] with a cutoff of 0.7 was employed, resulting in 1016 samples for subsequent analysis.
Differential gene expression
Differential gene expression analysis was conducted between HGSOC cases (n = 988) and control samples (n = 28) using R (version 4.1.0)/ Bioconductor, limma, and associated packages. Genes with a fold-change (FC) ≥ 1.5 and a false discovery rate (FDR) < 0.05 were considered as differentially expressed. The methodology is illustrated schematically in Fig. 1.
Univariate analyses on differential gene expression
The significantly (FDR < 0.05) differentially expressed genes (DEGs) between HGSOC tumors and controls, as explained above, were selected. To investigate prognostic genes, we utilized univariate Cox proportional hazards regression analyses [16] on these DEGs (HGSOC vs. Control) along with the corresponding survival data from HGSOC cohorts. The genes influencing the survival of HGSOC patients were filtered based on log-rank p-value < 0.05 and hazard ratio (HR) either greater than 1.1 or less than 0.9. This pre-filtering strategy, akin to approaches used in previous studies [13, 18], aims to mitigate noise (number of genes > > number of samples) [12, 19] prior to applying multivariate analyses.
Regularized Cox Regression on selected genes
The genes identified through univariate analyses underwent further scrutiny in a multivariate regression analysis. This involved employing the LASSO estimation, implemented using R/Rstudio with the “glmnet” package [15, 20]. The HGSOC samples were divided randomly into training (n = 491) and test (n = 491) datasets. The predictor-gene signatures (predictor variables; genei) and the corresponding coefficients (coefi) were then utilized to formulate the mPS or risk score, using the training dataset, as illustrated in the equation below.
Utilizing the predictor variables (e.g., genes) and their associated coefficients, we proceeded to predict the test datasets. Receiver operating characteristic (RoC) curve analyses were then carried out at different time points (in years) using survival data [21] to evaluate the predictive capacity.
The molecular prognostic score (mPS) determines the risk score for overall survival
The mPS or risk scores obtained, as mentioned above, were used to divide or partition the samples (HGSOC patients) into high (values above the median) and low-risk groups, determined by the median values of mPS. The HGSOC samples were also divided into quartiles, forming four equal parts based on associated mPS values. Subsequently, a Kaplan-Meier survival plot was generated using R with ‘survival’ and ‘survminer’ packages.
Gene enrichment analysis using GO and KEGG databases
The gene enrichment analysis [22] was conducted by applying the Bioconductor package ‘limma’ [23]. This analysis aimed to uncover the roles of various pathways associated with different groups in HGSOC cohorts. The functions ‘goana’ and ‘kegga’ conducted over-representation analyses for Gene Ontology (GO) terms or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, respectively. The list of differentially expressed genes (FDR < 0.05) with the associated Entrez Gene IDs was used as a gene set for over-representation or pathway enrichment analysis. The MArrayLM method automatically extracted the gene sets from a linear model fit object [23]. The top 20-dysregulated pathways based on p-values were shown.
Data mining and analyses
Data retrieval from repositories (TCGA, GEO) and subsequent analysis were performed using R/Rstudio, specifically R version 4.1.0. The analysis code, detailed packages, and approach are available at the following link: https://rpubs.com/siddik/mPS. The various packages and other associated base packages are described briefly in Appendix A Supplementary file.
Results
Differential gene expression and gene set enrichment analysis (GSEA) between ovarian carcinoma and normal ovarian tissue
A subset of data encompassing 10,225 genes and 1,016 samples with a minimum gene expression matrix correlation of 0.7 was selected. The data utilized in this study spans five different datasets as mentioned in the Methodology section. Appendix A Suppl Table S1 provides detailed information about the samples, including 988 ovarian cancer samples and 28 normal samples representing ovarian surface epithelial cells without indications of ovarian tumors.
Multidimensional scaling plots were generated to visualize distances between gene expression profiles of the samples. Pairwise distances between samples were calculated using the top 500 variable genes, revealing separation or clustering patterns. Notably, the samples within the same dataset were clustered together (Appendix A Suppl Fig. S1) rather than the types of samples (normal vs. cancer) indicating the requirement for the removal of batch effect prior to further analysis. This observation emphasized the necessity to address batch effects before further analysis. To mitigate the batch effect attributed to different datasets, a corrective step was implemented (Appendix A Suppl Fig. S1). This adjustment aimed to establish a uniform pattern of log2-expression ratios across subjects and samples, independent of the originating datasets. This process ensured a more robust and consistent foundation for subsequent analyses, facilitating a more accurate exploration of gene expression dynamics.
The differential gene expression was performed between normal (n = 28) and primary HGSOC (n = 973) samples. Among the analyzed genes, 649 were downregulated and 473 were upregulated in the primary HGSOC tumors compared to normal samples of ovarian surface epithelial tissues without any indications of tumors (Fig. 2A, and Appendix A Suppl Table S2). The top ten dysregulated genes based on adjusted p-value (FDR < 0.05) and fold change, were illustrated in Fig. 2B. Noteworthy, the upregulated genes included CP (Ceruloplasmin Ferroxidase), FOLR1, TOP2A, CRABP2, MAL, SOX17, CKS2, TPX2, S100A2, and UBE2C, while the top downregulated genes were ABCA8, ALDH1A2, BCHE, EFEMP1, NELL2, HBB, TCEAL2, SFRP1, HBA2 and FLRT2.

Differential gene expression and pathways involved in HGSOC A; Mean-difference plot (aka MA plot) with color coding for highlighted points (genes) that are differentially expressed in primary tumor (TP) as compared to normal samples (NT). B; Box plot showing the top ten dysregulated genes in primary tumor (TP) vs. normal samples (NT). C; The key biological/ molecular pathways that are upregulated (Up; red color) or downregulated (Down; blue) are shown by bubble-plot. The number denotes the number of genes that are significantly up/down regulated. The pathways indicated are curated from KEGG pathway database. D; The key molecules involve in cell cycle (hsa04110) regulation with FDR < 0.05 and the indicated log2-fold change are shown by gradient color scale
To elucidate pathways involved in HGSOC, gene enrichment analyses were performed on these 1,122(649 + 473) differentially expressed genes. According to the Gene Ontology (GO) database, upregulated genes were predominantly associated with cell cycle processes, cell cycle transition, cell/nuclear division, chromatin organization, chromatid segregation, and DNA replication (Appendix A Suppl Table S3). KEGG pathway analysis revealed dysregulation in cell cycle, Complement and coagulation cascades, DNA replication, Oxidative phosphorylation, ECM-receptor interaction, and Drug metabolism-cytochrome P450 (Fig. 2C, Appendix A Suppl Table S4).
Given the importance of cell cycle-related molecules in cancer, a detailed analysis focused on the cell cycle pathway was conducted. Using the Bioconductor package ‘Pathview’: a tool set for pathway based data integration and visualization [24], differential expressed genes of primary tumors were mapped onto the cell cycle pathway (hsa04110). Among the 25 differentially upregulated molecules, 23 were significantly associated with cell proliferation and tumor mass in HGSOC. The cell cycle-related molecules are ARF, Ink4a (CDKN2A), CycD, CycA, CycB, Cdc7, ChK1, MCM (minichromosome maintenance complex component), and others (Fig. 2D).
Construction of risk model
In the analysis, 1,062 differentially expressed genes were identified in tumors compared to normal samples, with an adjusted p-value (FDR) < 0.05 (Appendix A Suppl Table S2). Subsequently, these genes were subjected to univariate Cox regression analyses, further refined by log-rank testing (p-value < 0.05) and restricting the hazard ratio (HR) either greater than 1.1 or less than 0.9. A total of 122 genes emerged, with 63 associated with worse overall survival (HR > 1.1, p-value < 0.05) and 59 linked to improved survival (HR < 0.9, p-value < 0.05) for HGSOC patients (Appendix A Suppl Table S5).
The selected 122 genes were then employed to construct a LASSO estimation using a training dataset comprising 491 randomly chosen samples from both TCGA and GEO data cohorts. A log(Λ) vs. partial likelihood deviance plot [25] was generated with different alpha (α) values, revealing that the optimal fit occurred with α = 1, signifying the LASSO regression model (Fig. 3). The 10-fold cross-validation, also with α = 1, was employed for LASSO estimation in further analyses. Dotted vertical lines on the plot denote corresponding Λ values (primary x-scale) and gene numbers (secondary x-scale) with minimal deviance on the left. The right vertical line signifies the most regularized model with a cross-validation error within 1 standard deviation of the minimum.

LASSO regression and selection of various parameters: LASSO model fitting on 122 prognostic genes affecting overall survival: The plots with alpha (α) = 1, i.e., LASSO (top left), alpha (α) = 0.5; elastic net (top right) and alpha (α) = 0; ridge regression (bottom left) are shown. The combined/ merged plot (bottom right) with regression curves for LASSO (α = 1), elastic net (α = 0.5) and ridge (α = 0) regression are shown for comparison
From this analysis, 20 predictor genes were identified, and their associated coefficients were determined using LASSO regression (Table 1). Detailed information on these 20 predictor genes, including relative expression, fold change (tumor vs. normal), and hazard ratio (HR), is provided in Appendix A Suppl Table S6. The 20 predictor genes and their coefficients were then used to calculate the mPS score. This mPS score serves as a predictive metric for the survival of HGSOC patients.
Survival analysis based on molecular prognostic score (mPS)
In this study, mPS or risk score was meticulously developed based on the expression levels of 20 predictor genes. The resulting score was stratified into high and low-risk groups using median values, forming the basis for subsequent analyses.
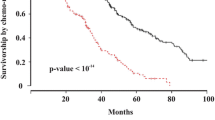
To validate the robustness of the mPS, a two-fold approach was adopted. First, the training dataset comprising 491 samples was utilized, followed by the application of the mPS to the remaining 491 samples as the test dataset. The outcomes of this validation process were compelling, revealing significant differences in survival curves between the identified high and low-risk groups (Fig. 4). The log-rank p-values, both for the training and test datasets, were less than 0.0001, reinforcing the prognostic efficacy of the mPS.

Survival curve and prediction based on mPS score: The cut-off set at median value of mPS (High vs. Low mPS) indicates that higher mPS is associated with poor OS time whereas lower mPS is associated with higher OS time (in days) both in training A and test B datasets. The groups based on quartiles (Q1; 0-25th percentiles, Q2; 25-50th percentiles; Q3; 50-75th percentiles and Q4; 75-100th percentiles) showed mPS with higher Q value showed poor OS as opposed to lower Q values both in training C and test datasets D. The RoC of sensitivity/specificity of test data E and training data F for indicated time (in year)is also plotted. TP; True positive, FP; False positive, AUC; Area under curve
Survival analyses further unveiled noteworthy insights. In the training dataset, the median overall survival (OS) time for the high-risk group was 1024 days (95% Confidence Interval (CI): 914–1168), while the low-risk group exhibited a median OS time of 1699 days (95% CI: 1446–2012) (Fig. 4A; Table 2). Similarly, in the test dataset, the high-risk group displayed a median OS time of 1091 days (95% CI: 1006–1234), whereas the low-risk group demonstrated a more favorable median OS time of 1976 days (95% CI: 1764–2279) (Fig. 4C; Table 2).
Furthermore, a granular exploration of the training samples involved their division into quartiles (Q1, Q2, Q3,and Q4) based on the mPS score. Q4 bearing the highest with Q1 bearing the lowest mPS score. The survival curves were then generated for these equally divided quartiles (Q1, Q2, Q3 and Q4) to obtain a median overall survival (OS) time. The median OS time in days were 2621, 1354, 1203, and 914 for Q1, Q2, Q3,and Q4 subgroups, respectively (Fig. 4B, and Table 2). The median mPS of respective quartiles were also obtained, and then the Pearson correlation between the median mPS of the quartiles and the respective median OS time was calculated. In the training data, an inverse relationship (r2= -0.902, p = 0.049, Pearson correlation) between mPS (risk score) and median OS time was observed. Similarly, a strong inverse correlation was also observed in test data (r2=-0.954, p = 0.02) between the mPS score and OS time. This indicates that the mPS score can not only qualitatively indicate survival time but can also quantitatively measure or predict survival time (Fig. 4B and D; Table 2). The heat map generated using the relative expression of poor predictor (n = 9 genes) vs. good predictor (n = 11) genes (Appendix A Suppl Fig. S2) could potentially cluster both the training and test datasets based on mPS.
Prediction based on risk score obtained using 20-gene signature
The 20-gene signature was derived based on values plotted in the graph (Fig. 3) using 10-fold cross-validation on both training and test datasets containing HGSOC samples from different datasets. The derived mPS, based on these 20 genes, was further applied to study sensitivity and specificity using receiver operating characteristic (RoC) curve for survival data. The Area under curve (AUC) values of RoC curves indicated the predictive capacity of the prognostic model. Our prognostic model for predicting ovarian cancer OS appeared to be efficient, with AUC values were around 0.70 (± 0.03) and 0.68 (± 0.03) across the span of 5 years for training (Fig. 4E) and test HGSOC samples (Fig. 4F), respectively. This suggests that our model is a very effective predictor for determining the risk or OS time in HGSOC patients (Fig. 4).
Commonly used clinical parameters include FIGO stage, tumor grade, residual disease, along with age and ethnicity, to study OS time or prognosis in HGSOC. These parameters were converted or scaled into numeric values as shown in Appendix A Suppl Tables S7-S9. Univariate analysis using Cox regression on the survival data of HGSOCs revealed that the age, FIGO stage and residual disease at the largest nodule exhibited a positive correlation (β coefficient > 1; HR > 1.2, p-value < 0.05) indicating that higher values of these parameters were associated with worse survival or a poor prognosis (Table 3).
Multivariate Cox regression analysis was performed to adjust for the impact of other significant parameters (Table 3) for deriving the adjusted HR. The forest plot for the multivariate Cox-proportional hazards regression model of these parameters is shown in Fig. 5A. The residual disease at the largest nodule showed a significant effect (p < 0.001) in determining prognosis with an adjusted HR of 1.3 (95% CI = 1.13–1.40). This infers that the larger residual disease after primary cytoreduction surgery was associated with decreased survival of HGSOC patients. Interestingly, the mPS score was the most significant parameter (p-value < 0.001) with the adjusted HR (adjusted to age, residual disease and FIGO) of 6.1 (95% CI = 3.65–10.30). Further analysis was conducted to assess whether adding parameters such as age and residual disease of the largest nodule could increase the additional prognostic value or sensitivity to mPS. It was found that the AUC for the 5-year OS of HGSOC using the mPS score alone was 0.71 as compared to 0.60 contributed by the residual disease of the largest nodule. Moreover, the addition of parameters such as residual disease and age to the mPS showed only a nominal improvement in the predictive capacity (AUC = 0.72) for HGSOC patients (Fig. 5B). Thus, the mPS outperformed various traditional parameters, such as age and residual disease of the largest nodule, in predicting the OS of HGSOC. In conclusion, the mPS alone could serve as a pivotal prognostic factor in predicting the outcome of the severity of HGSOC in terms of OS.

Prognosis of HGSOC using clinicopathological and mPS: A; The ggforest plot of Cox proportional Hazard regression fitting of various parameters as indicated. The HR and p-value obtained are adjusted values with respect to other shown parameters. B; Area under curve (AUC) using mPS, age and residual disease of the largest nodule (RD) alone or combination as indicated. C; Gene enrichment score as shown by barcode plot of indicated KEGG pathways. D; Molecules/ genes involved in upregulation of Wnt signaling and E; downregulation of Antigen processing and presentation signaling. The log2-fold change of the molecules between high vs. low risk group involved are shown with gradient scale
Gene enrichment study/pathway analysis using high (poor) vs. low (good) risk group
To compare gene expression between the high-risk (higher mPS) and low-risk groups, we conducted a differential gene expression analysis. We identified 1988 significantly upregulated and 2453 significantly downregulated genes (FDR < 0.05) in the high-risk group compared to the low-risk group (Appendix A Suppl Table S10).
To investigate pathways or events associated with these dysregulated genes involved in risk stratification, we performed gene enrichment studies. Gene enrichment using GO-related terms indicated a significant (p < 0.05) downregulation of pathways related to DNA repair, respiratory electron transport chain, cell cycle, and DNA replication. Conversely, pathways related to cell migration, extracellular matrix interactions, vasculature and blood vessel development were upregulated (p < 0.05) (Appendix A Suppl Table S11). Similar results were obtained from pathway analysis using the KEGG pathway database, where pathways such as Focal adhesion, Notch signaling, Wnt signaling, PI3-Akt signaling, and signaling pathways regulating the pluripotency of stem cells were upregulated, while pathways involving antigen processing and presentation, cell cycle, DNA replication, and base excision repair were downregulated (Fig. 5C; Table 4).
Since molecules involved in Wnt Signaling [26] as well as the antigen processing and presentation [27] have been reported previously for their prognostic values, we further investigated or deciphered the molecules regulating these two pathways (Fig. 5D and E). We found that several key molecules involved in the canonical Wnt signaling pathway, including the Frizzled related family of proteins (FRP), Wnt family members, pigment epithelium-derived factor (PEDF), serpin family F member 1 (SERPINF1), Frizzled (FZD) proteins, BMP and activin membrane-bound inhibitor (BAMBI), segment polarity protein dishevelled (Dvl), protein kinase A (PKA), β-catenin, and transcription factor-like (TCF)/lymphoid enhancer-binding factor (LEF), were upregulated in the high-risk group of HGSOC. This activation of canonical Wnt signaling could lead to increased cell movement and proliferation (Fig. 5D). Considering the favorable outcome of immunoreactive subtypes [27] in ovarian cancer, we analyzed the molecules involved in antigen processing and presentation signaling. This pathway was found downregulated (p < 1.27E-10) in high risk HGSOC patients. There were almost 37 molecules significantly (FDR < 0.05) downregulated in this pathway, including the key mediators such as IFN-γ, TNF-α, immuno proteasome activator PA28, TAP1/2, TAPBP, MHC-I (HLA-A, HLA-B, HLA-C), affecting MHC-I pathway mediated killing of cancer cells. MHC-II pathway via HLA-DMA, HLA-DMB, HLA-DOA, CLIP (CD74), cathepsin S (CTSS) was also downregulated, leading to decreased antitumor cytokine production and activation of other immune cells. Hence, immune evasion and escape were associated with high- risk group of HGSOC patients based on our findings.
Discussion
The 20-gene signature used to develop a mPS score, has shown potential in determining the overall survival of HGSOC patients. The AUC (~ 0.7) of mPS-based stratification in both training and test data encompassing both TCGA and GEO datasets indicates its influence on patient survival. Previous studies [11, 12] have demonstrated an inverse correlation between mPS score and patient survival, with recent work [11] showing that a 101 predictor gene-based mPS outperformed age and stage in predicting survival in advanced HGSOC cases. Interestingly, our study achieved similar results using only 20 genes (Fig. 5A and B), a smaller set compared to the 101 predictor genes used in previous work [11] for ovarian cancer risk stratification.
Our approach involved pre-filtering common genes across TCGA and GEO datasets, followed by filtering for differential genes in HGSOC tumor vs. normal samples and further refinement using Cox proportional hazards regression (univariate) prior to LASSO-Cox fitting (multivariate) (Fig. 1). This methodological difference may explain the deviation in predictor genes between our study and previous reports [11]. Furthermore, the selection of the 101-prognostic genes [11] from a total set of 513 genes, also used for the molecular classification of HGSOC [28], might have omitted many crucial prognostic genes. Hence, the deviation is expected. To compare the predictive capacity of set of 101prognostic genes previously discussed, we have done across-platform or dataset validation. Among the 101prognostic genes [11], 85 genes were common in the integrated TCGA and GEO datasets encompassing 982 ovarian cancer serous patients with mRNA expression and the associated survival data that we have used in our study. This set of 85 predictor genes was used to calculate the mPS based on their associated coefficients, as previously reported [11], and the expression data of ovarian cancer cohorts of the afore mentioned TCGA and GEO repositories. The AUC for these 85 predictor genes in determining OS time ranged from 0.61 to 0.68 over a 1 to 5-year period (Fig. 6A). Similarly, our derived mPS using our 20 predictor genes (Table 1) in the same integrated TCGA and GEO datasets showed an AUC range of 0.67 to 0.72 over 1 to 5 years (Fig. 6B).

Prognostic performance of molecular prognostic score (mPS)across sequencing platforms and datasets: RoC curves for prognostic performance of mPS derived from the 101-predictor genes A as described previously [11] along with our (Table 1)20-predictor genes B. Prediction was studied using AUC for the period of 1 to 5 years’ duration in the integrated datasets (TCGA-OV and GSE14764, GSE18520, GSE26193, GSE26712 and GSE63855) spanning 982 samples. Cross validation across sequencing platforms is done with our 20-predictor genes. There are only 3 out of 20 genes found to be included in the gene expression based NanoString platforms as indicated. Quartiles divides the HGSOC patients into four equal parts based on mPS derived from 3 predictor genes: Q1 bearing the lowest where as Q4 bearing the highest mPS score. Kaplan–Meier curves and the associated risk table of overall survival for HGSOC patients in the GSE135820 (n = 3773) C, D and GSE132342 (n = 3769) E, F datasets
Similarly, we applied our derived 20-gene signature to NanoString-based mRNA expression datasets (GSE132342, n = 3769; GSE135820, n = 3773), which were previously used in other studies [11, 28], for cross-dataset and cross-platform validation. Only three genes (GFPT2, WT1, RASA1) were common between our 20-predictor gene signature and the mRNA expression datasets from the OTTA consortium (GSE132342, GSE135820) (Appendix A Suppl Table S12). Interestingly, the derived molecular prognostic score (mPS), based on the linear addition of coefficients along with the expression of these three genes in the OTTA dataset (GSE132342, GSE135820), potentially predicted the overall survival of HGSOC patients (Fig. 6C-F). To study the association between median overall survival (OS) time and mPS, we further partitioned these cross data HGSOC samples into four equal parts, with Q1 having the lowest mPS value and Q4 having the highest mPS value. The median survival time was found to be the shortest for samples in the group with the highest mPS value (Appendix A Suppl Table S13). The median survival time in the groups stratified based on mPS value were significantly different (Fig. 6C-F).
These findings suggest that although only three genes overlapped with the OTTA datasets, the derived mPS score shows promise in predicting overall survival in HGSOC patients. This could have important implications for prognosis and potentially guide treatment decisions.
Next, to study the dynamic and robustness application of our 20-gene-based mPS in predicting the OS, we have derived the mPS value by a similar approach using coefficients of 20-predictor genes and their respective expression data of RNASequencing platform (Illumina). It efficiently predicted the OS time. The lowest mPS value is associated with the shortest OS time whereas the highest is associated with the least OS time (Appendix A Suppl Fig. S3).
Hence, our 20-gene expression-based mPS value, derived from Affymetrix Human U133A/U133 Plus 2.0 microarray data in TCGA and GEO repositories, is a robust and dynamic prognostic indicator. This mPS efficiently predicted the outcome of HGSOC across various datasets and was applicable regardless of the mRNA expression data platform used. It effectively predicted ovarian cancer outcomes using data from both the NanoString platform (Fig. 6) and the Illumina RNASeq platform (Appendix A Suppl Fig. S3).
In addition to the prognostic index of the 20-gene signature-based mPS score, which acts as a risk classifier, we conducted an in-depth study of the key regulatory pathways responsible for the poor prognosis of HGSOCs. We found that the poor prognostic group or high-risk group of HGSOCs exhibited altered pathways regulating TGF-β [29], PI3K-Akt [30], and Wnt/Notch [31] signaling, which are often associated with poor survival in cancer patients. Immune evasion or escape was also observed in the high-risk group and found to be associated with poor outcomes [27]. Thus, targeting these dysregulated pathways [32] might prove beneficial for the high-risk group, which was anticipated to have poor survival outcomes with current prevailing treatments. Interestingly, we found that the molecules involved in DNA replication and repair, as well as antigen processing and presentation, were downregulated in the high-risk group. This suggests a role for immune evasion or antigenic escape [33] and defective DNA repair pathways [34] in therapy resistance in high-risk HGSOC. Further investigation into their role in therapy resistance is needed to identify target molecules and reprogram HGSOC towards an immune-reactive state. Therefore, it would be noteworthy to conduct combination therapy using immunotherapies/agents that overcome immune suppression and PARP inhibitors [35, 36] in HGSOC patients in anticipation of improving overall survival time.
Conclusion
The conventional parameters like age, clinicopathological parameters: stage/ FIGO score, histology, and residual disease shows a trend in prognosis, yet the 5-year survival of ovarian cancer remains unchanged. Currently, a molecular gene-based prognostic score derived from the predictor genes and associated coefficients was found to be a robust prognostic marker/ classifier applied in various cancers including breast, prostate and colon cancer. A similar approach was used in ovarian cancer using 101prognostic genes. We have applied only 20 prognostic genes to predict the over-all survival of HGSOCs. Our system was found to be universal and robust as it was applicable and reproducible in various gene expression platforms including microarray, RNASeq, or NanoString. Our 20-gene signature based mPS for the prognosis of survival of HGSOC significantly outperformed the conventional parameters: age, residual disease, and FIGO score. The high-risk group with lower survival time could benefit from targeted therapies focusing on dysregulated pathways such as TGF-β, Notch signaling, DNA repair, and antigen processing and presentation pathways. Further investigations into the functional roles of identified prognostic genes and pathways are warranted. Prospective clinical studies are essential to validate the clinical utility of the 20-gene signature as a prognostic tool and to explore potential targeted therapies based on identified dysregulated pathways.
In conclusion, this study provides a comprehensive molecular understanding of HGSOC, offering a 20-gene signature-based mPS as a promising prognostic indicator. The identified dysregulated pathways open avenues for targeted interventions, bringing us one step closer to personalized and effective management of high-grade serous ovarian cancer.
Data availability
The relevant data and the supplementary files are shared in the manuscript. The raw data used and/or analysed during the study are available in the TCGA data repository (https://gdac.broadinstitute.org/), and GEO accession numbers: GSE18520, GSE26712, GSE26193, GSE63885 GSE14764, and GSE135820. The code for R/ Rstudio used for data analysis can be found at the link: https://rpubs.com/siddik/mPS.
Change history
11 August 2024
Added the supplementary figure file.
References
Peres LC, Cushing-Haugen KL, Kobel M, Harris HR, Berchuck A, Rossing MA, et al. Invasive epithelial ovarian Cancer survival by Histotype and Disease Stage. J Natl Cancer Inst. 2019;111(1):60–8. https://doi.org/10.1093/jnci/djy071. Epub 2018/05/03.
Lambrechts S, Smeets D, Moisse M, Braicu EI, Vanderstichele A, Zhao H, et al. Genetic heterogeneity after first-line chemotherapy in high-grade serous ovarian cancer. Eur J Cancer. 2016;53:51–64. PubMed PMID: 26693899.
Morden CR, Farrell AC, Sliwowski M, Lichtensztejn Z, Altman AD, Nachtigal MW, et al. Chromosome instability is prevalent and dynamic in high-grade serous ovarian cancer patient samples. Gynecol Oncol. 2021;161(3):769–78. https://doi.org/10.1016/j.ygyno.2021.02.038. Epub 2021/03/15.
Cooke SL, Ng CK, Melnyk N, Garcia MJ, Hardcastle T, Temple J, et al. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene. 2010;29(35):4905–13. https://doi.org/10.1038/onc.2010.245. Epub 2010/06/29.
Bruix J, Reig M, Sherman M, Evidence-Based Diagnosis. Staging, and treatment of patients with Hepatocellular Carcinoma. Gastroenterology. 2016;150(4):835–53. https://doi.org/10.1053/j.gastro.2015.12.041. Epub 2016/01/23.
Dao F, Schlappe BA, Tseng J, Lester J, Nick AM, Lutgendorf SK, et al. Characteristics of 10-year survivors of high-grade serous ovarian carcinoma. Gynecol Oncol. 2016;141(2):260–3. https://doi.org/10.1016/j.ygyno.2016.03.010. Epub 2016/03/13.
Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14(16):5198–208. https://doi.org/10.1158/1078-0432.CCR-08-0196. Epub 2008/08/14.
Chen GM, Kannan L, Geistlinger L, Kofia V, Safikhani Z, Gendoo DMA, et al. Consensus on Molecular subtypes of High-Grade Serous Ovarian Carcinoma. Clin Cancer Res. 2018;24(20):5037–47. https://doi.org/10.1158/1078-0432.CCR-18-0784. Epub 2018/08/08.
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. https://doi.org/10.1038/nature10166. Epub 2011/07/02.
Long J, Zhang L, Wan X, Lin J, Bai Y, Xu W, et al. A four-gene-based prognostic model predicts overall survival in patients with hepatocellular carcinoma. J Cell Mol Med. 2018;22(12):5928–38. https://doi.org/10.1111/jcmm.13863. Epub 2018/09/25.
Millstein J, Budden T, Goode EL, Anglesio MS, Talhouk A, Intermaggio MP, et al. Prognostic gene expression signature for high-grade serous ovarian cancer. Ann Oncol. 2020;31(9):1240–50. https://doi.org/10.1016/j.annonc.2020.05.019. Epub 2020/05/31.
Shimizu H, Nakayama KI. A 23 gene-based molecular prognostic score precisely predicts overall survival of breast cancer patients. EBioMedicine. 2019;46:150–9. https://doi.org/10.1016/j.ebiom.2019.07.046. Epub 2019/07/31.
Shimizu H, Nakayama KI. A universal molecular prognostic score for gastrointestinal tumors. NPJ Genom Med. 2021;6(1):6. https://doi.org/10.1038/s41525-021-00172-1. Epub 2021/02/06.
Wu CL, Schroeder BE, Ma XJ, Cutie CJ, Wu S, Salunga R, Development and validation of a 32-gene prognostic index for prostate cancer progression. Proc Natl Acad Sci U S A., M.W et al. K. have served on an advisory board for bioTheranostics, Inc. C.J.C. has served as a consultant for bioMerieux and bioTheranostics. B.E.S, R.S., Y.Z., C.A.S., and M.G.E. are employees and stockholders of bioTheranostics, Inc.
Tibshirani R. The lasso method for variable selection in the Cox model. Statistics in Medicine. 1997;16(4):385–95. https://doi.org/10.1002/(SICI)1097-0258(19970228)16:4%3C385::AID-SIM380%3E3.0.CO;2-3. PubMed PMID: 9044528.
Cox DR. Regression Models and Life-Tables. Journal of the Royal Statistical Society:, Series B, Methodological.). 2018;34(2):187–202. https://doi.org/10.1111/j.2517-6161.1972.tb00899.x.
Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8):e71. https://doi.org/10.1093/nar/gkv1507. Epub 2015/12/26.
Liu GM, Xie WX, Zhang CY, Xu JW. Identification of a four-gene metabolic signature predicting overall survival for hepatocellular carcinoma. J Cell Physiol. 2020;235(2):1624–36. https://doi.org/10.1002/jcp.29081. Epub 2019/07/17.
Jardillier R, Koca D, Chatelain F, Guyon L. Prognosis of lasso-like penalized Cox models with tumor profiling improves prediction over clinical data alone and benefits from bi-dimensional pre-screening. BMC Cancer. 2022;22(1):1045. https://doi.org/10.1186/s12885-022-10117-1. Epub 2022/10/06.
Tibshirani R, Bien J, Friedman J, Hastie T, Simon N, Taylor J, et al. Strong rules for discarding predictors in lasso-type problems. J R Stat Soc Ser B Stat Methodol. 2012;74(2):245–66. https://doi.org/10.1111/j.1467-9868.2011.01004.x. Epub 2012/03/01.
Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56(2):337–44. https://doi.org/10.1111/j.0006-341x.2000.00337.x. Epub 2000/07/06. PubMed PMID: 10877287.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. https://doi.org/10.1186/gb-2010-11-2-r14. Epub 2010/02/06.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. https://doi.org/10.1093/nar/gkv007. Epub 2015/01/22.
Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29(14):1830–1. https://doi.org/10.1093/bioinformatics/btt285. Epub 2013/06/07.
Friedman JH, Hastie T, Tibshirani R. Regularization paths for generalized Linear models via Coordinate Descent. J Stat Softw. 2010;33(1):1–22. https://doi.org/10.18637/jss.v033.i01.
Arend RC, Londono-Joshi AI, Straughn JM Jr., Buchsbaum DJ. The Wnt/beta-catenin pathway in ovarian cancer: a review. Gynecol Oncol. 2013;131(3):772–9. https://doi.org/10.1016/j.ygyno.2013.09.034. Epub 2013/10/16.
Konecny GE, Wang C, Hamidi H, Winterhoff B, Kalli KR, Dering J, et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J Natl Cancer Inst. 2014;106(10). https://doi.org/10.1093/jnci/dju249. PubMed PMID: 25269487; PubMed Central PMCID: PMCPMC4271115. Epub 2014/10/02.
Talhouk A, George J, Wang C, Budden T, Tan TZ, Chiu DS, et al. Development and validation of the Gene expression predictor of high-grade Serous Ovarian Carcinoma Molecular SubTYPE (PrOTYPE). Clin Cancer Res. 2020;26(20):5411–23. https://doi.org/10.1158/1078-0432.CCR-20-0103. Epub 2020/06/20.
de Kruijf EM, Dekker TJA, Hawinkels L, Putter H, Smit V, Kroep JR, et al. The prognostic role of TGF-beta signaling pathway in breast cancer patients. Ann Oncol. 2013;24(2):384–90. https://doi.org/10.1093/annonc/mds333. Epub 2012/10/02.
Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18(1):26. https://doi.org/10.1186/s12943-019-0954-x. Epub 2019/02/21.
Bocchicchio S, Tesone M, Irusta G. Convergence of wnt and notch signaling controls ovarian cancer cell survival. J Cell Physiol. 2019;234(12):22130–43. https://doi.org/10.1002/jcp.28775. Epub 2019/05/16.
Sow HS, Ren J, Camps M, Ossendorp F, Ten Dijke P. Combined inhibition of TGF-beta signaling and the PD-L1 Immune Checkpoint is differentially effective in Tumor models. Cells. 2019;8(4). https://doi.org/10.3390/cells8040320. PubMed PMID: 30959852; PubMed Central PMCID: PMCPMC6523576. Epub 2019/04/10.
Preston CC, Goode EL, Hartmann LC, Kalli KR, Knutson KL. Immunity and immune suppression in human ovarian cancer. Immunotherapy. 2011;3(4):539–56. https://doi.org/10.2217/imt.11.20. Epub 2011/04/06.
Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801 – 17. Epub 2012/11/24. https://doi.org/10.1038/nrc3399. PubMed PMID: 23175119.
Zimmer AS, Nichols E, Cimino-Mathews A, Peer C, Cao L, Lee MJ, et al. A phase I study of the PD-L1 inhibitor, durvalumab, in combination with a PARP inhibitor, olaparib, and a VEGFR1-3 inhibitor, cediranib, in recurrent women’s cancers with biomarker analyses. J Immunother Cancer. 2019;7(1):197. https://doi.org/10.1186/s40425-019-0680-3. Epub 2019/07/28.
Li A, Yi M, Qin S, Chu Q, Luo S, Wu K. Prospects for combining immune checkpoint blockade with PARP inhibition. J Hematol Oncol. 2019;12(1):98. https://doi.org/10.1186/s13045-019-0784-8. Epub 2019/09/16.
Acknowledgements
The authors would like to thank CSIR for providing structural and financial support and acknowledged PAN CSIR cancer research program: Making cancer care affordable (HCP-40). Sarbar Ali Saha is the recipient of Council of Scientific and Industrial Research (CSIR)-Senior Research Fellowship (SRF). Siddik Sarkar is the recipient of Start-up Research Grant (SRG/2019/001880) from Science and Engineering Research Board (SERB), INDIA, and also acknowledged the funding agency, WellcomeTrust UK (Ref: 223547/Z/21/Z). Abhishek Swarnakar is the recipient of Department of Biotechnology (DBT)- Junior Research Fellowship (JRF). Arnab Chakrabarty is the recipient of University Grants Commission (UGC)-JRF. The results shown here are in part based upon the data (raw file) generated by the TCGA Research Network: https://www.cancer.gov/tcga. The Gene Expression Omnibus (GEO maintained by NCBI): http://www.ncbi.nlm.nih.gov/geo. The authors would like to acknowledge all the contributing laboratories and authors for data repository files associated with following accession numbers: GSE18520, GSE26712, GSE26193, GSE63885 GSE14764, and GSE135820 along with TCGA-OV cancer project.
Funding
This work was supported by the Council of Scientific & Industrial Research (CSIR), Science and Engineering Research Board (SERB), and Laboratory Reserve Fund (LRF) from CSIR-Indian Institute of Chemical Biology, INDIA.
Author information
Authors and Affiliations
Contributions
S., Sarkar designed the study. S. A., Saha, P., Sarkar, S., Banerjee download the data from data repositories.S., Sarkar analyzed and interpreted the data. S., Sarkar, S.A., Saha, P., Sarkar, S., Banerjee, and P., Mitra wrote and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sarkar, S., Saha, S.A., Swarnakar, A. et al. The molecular prognostic score, a classifier for risk stratification of high-grade serous ovarian cancer. J Ovarian Res 17, 159 (2024). https://doi.org/10.1186/s13048-024-01482-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-024-01482-5