
Abstract
Targeted therapies, including small molecule inhibitors directed against aberrant kinase signaling and chromatin regulators, are emerging treatment options for high-grade gliomas (HGG). However, when translating these inhibitors into the clinic, their efficacy is generally limited to partial and transient responses. Recent studies in models of high-grade gliomas reveal a convergence of epigenetic regulators and kinase signaling networks that often cooperate to promote malignant properties and drug resistance. This review examines the interplay between five well-characterized groups of chromatin regulators, including the histone deacetylase (HDAC) family, bromodomain and extraterminal (BET)-containing proteins, protein arginine methyltransferase (PRMT) family, Enhancer of zeste homolog 2 (EZH2), and lysine-specific demethylase 1 (LSD1), and various signaling pathways essential for cancer cell growth and progression. These specific epigenetic regulators were chosen for review due to their targetability via pharmacological intervention and clinical relevance. Several studies have demonstrated improved efficacy from the dual inhibition of the epigenetic regulators and signaling kinases. Overall, the interactions between epigenetic regulators and kinase signaling pathways are likely influenced by several factors, including individual glioma subtypes, preexisting mutations, and overlapping/interdependent functions of the chromatin regulators. The insights gained by understanding how the genome and epigenome cooperate in high-grade gliomas will guide the design of future therapeutic strategies that utilize dual inhibition with improved efficacy and overall survival.
Similar content being viewed by others

Introduction
High-grade gliomas (HGGs) are central nervous system (CNS) tumors that occur in both children and adults, although bear distinct molecular features and neuroanatomy in younger compared with older patients [1,2,3,4]. While these types of cancers are relatively rare, patient prognosis is quite poor, with an average 2-year overall survival of only 20% [5], despite multimodal treatment regimens consisting of surgery, radiation therapy, and chemotherapy [6, 7]. Tumor location or disease progression can further complicate therapeutic interventions; therefore, novel treatment modalities such as targeted therapies, including epigenetically directed therapies, are critical to improve patient outcomes.
The identification of cancer-specific targets supporting tumor growth is a major research objective to develop therapeutic options in HGG. Advances in sequencing technology and single-cell analyses of HGGs and subsets of medulloblastoma have revealed frequent alterations in kinase signaling proteins and proteins which regulate their activity (i.e., tumor suppressors) [8,9,10,11]. For example, several receptor tyrosine kinases (RTKs) are frequently overexpressed or mutated in HGG [8, 11], causing hyperactivity of downstream signaling cascades leading to increased cell proliferation, growth, and survival of cancer cells. To date, there has been limited clinical efficacy from single-agent inhibition of dysregulated signaling pathways in HGG [12].
In addition to aberrant proliferative signaling pathways, disruption of the epigenome has been identified as a contributor to tumorigenesis, cancer progression, and chemotherapy resistance [13, 14]. Epigenetic regulators, termed “writers” and “readers”, catalyze the reversible chemical modifications of histones and DNA [15]. The most predominant epigenetic alterations are post-translational modifications to histones, involving the addition and removal of methyl and acetyl marks, and DNA methylation [16]. The epigenetic “readers” recognize specific modifications and translate their effects on gene expression and other cellular processes. Pharmacological inhibitors have been developed to target various chromatin regulators, such as those directed against the histone deacetylase (HDAC) family, bromodomain and extraterminal (BET)-containing proteins, protein arginine methyltransferase (PRMT) family, Enhancer of zeste homolog 2 (EZH2), and lysine-specific demethylase 1 (LSD1).
Understanding the complex interactions between the cancer genome and epigenome is paramount when designing novel therapeutic strategies. Targeted therapies directed solely at epigenetic regulators or dysregulated kinases have shown limited success in sustaining clinical responses in HGG [17,18,19,20,21,22]. Evidence increasingly shows that epigenetic modulators cooperate with several relevant kinase signaling pathways in gliomas to promote cancer progression and contribute to therapeutic resistance. In this review, we systematically explore the epigenetic regulators and their interactions with kinase signaling networks in HGG and how combination strategies have developed and could be envisioned via existing small molecule inhibitors.
Molecular dysfunction of the genome and epigenome
Kinase signaling pathway alterations
Over the past two decades, substantial effort has been made to sequence and molecularly characterize primary cancer samples across all cancer types, including adult and pediatric HGG. This initiative has identified frequent alterations in several kinase signaling pathways and their regulators (Table 1.) One of the more common alterations detected involve the RTKs, including epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFRA), and KIT, also known as mast/stem cell growth factor receptor. These alterations are composed of gene mutations and/or copy number amplification, which hyperactivate downstream signaling networks involved in cell proliferation, differentiation, cell growth, metabolism, and survival. One convergent activating pathway downstream of these RTKs is phosphatidylinositol-3 kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling. Alterations are found in the catalytic subunit of PI3K, PIK3CA, and the regulatory subunit, PIK3R1, which can activate downstream signaling at AKT and mTOR. Additionally, copy number deletions are found within Phosphatase and Tensin Homolog (PTEN), a tumor suppressor that negatively regulates PI3K/AKT signaling. A common deletion in PTEN includes homozygous deletion, which contributes to hyperactivation of the PI3K/AKT/mTOR pathway.
In addition to the PI3K/AKT pathway, the mitogen-activated protein kinase (MAPK) cascade is downstream of RTKs and involved in HGGs. In this pathway, loss of function mutation in neurofibromin 1 (NF1), a small GTPase activating protein that regulates signal transduction through RAS (Rat sarcoma virus), affects the downstream MAPK signaling leading to activation. Lastly, the cell cycle control gene, cyclin-dependent kinase inhibitor 2 A (CDKN2A), is frequently found to have homozygous deletion. Beyond the aforementioned kinase alterations, several other gene alterations form the HGG genomic landscape. When comparing adult and pediatric gene alterations, there are many overlapping changes within the kinase signaling pathways; however, the frequency at which they occur differs with age.
Epigenetic abnormalities from histone modifiers
Unlike genetic changes, epigenetic dysregulation is not typically the result of mutations and instead occurs through alterations in chromatin accessibility and gene expression [25]. Transcriptional dysregulation can result from overexpression of chromatin modulators and their subsequent hyperactivity. The absence or presence of specific histone modifications, including acetylation and methylation of critical amino acids, governs chromatin structure leading to changes in gene transcription.
Numerous enzymes and protein complexes have been identified to be responsible for regulating the expression of various genes. These epigenetic proteins are over- or under-expressed in tumors, including HGG. One of the most well-studied epigenetic modulators is the HDAC family of enzymes. In gliomas, HDAC activity generally suppresses the expression of regulatory proteins and DNA repair genes as a component of repressive transcriptional complexes. Several HDAC family members have been identified as having altered gene expression in HGG. For example, HDAC1, 2, 3, and 7 have been found to be overexpressed in grade IV gliomas compared to normal brain tissue and low-grade gliomas [26]. Meanwhile, other enzymes that act as readers of these acetylated amino acids are often dysregulated in gliomas. These sets of proteins include the BET proteins. Two BET proteins, BRD2 and BRD4, are significantly overexpressed in gliomas, and the knockout of BRD4 diminishes glioma proliferation [27]. Similarly, PRMT enzymes, such as PRMT1, 2, and 5, function to methylate arginine residues and can promote dysregulation in brain tumors arising from the aberrant expression or activity [28,29,30,31]. Another epigenetic regulator relevant to HGG is the methyltransferase, EZH2, which is overexpressed in gliomas and correlated with high-grade gliomas [32, 33]. EZH2’s activity contributes to glioma progression by silencing tumor-suppressor genes [34]. An additional epigenetic modulator that is commonly dysregulated in gliomas is LSD1, a histone demethylase that is a component of several repressive complexes and is associated with reduced gene transcription. LSD1 is found to be overexpressed in several cancer types, including glioblastoma [35,36,37], and has been associated with poor patient prognosis in certain types of cancer [38,39,40,41,42]. Understanding the dysregulation of epigenetic modulators in HGG can inform the development of targeted therapies aimed at restoring normal gene expression and halting tumor growth.
The aforementioned epigenetic modulators are found in large epigenetic protein complexes, often with several epigenetic proteins. The complexes serve to activate its members and increase their stability. One of the most well-known epigenetic protein complexes is the CoREST (REST corepressor 1), which functions to enhance nucleosome regulation and drive gene repression [43,44,45,46]. The CoREST complex includes both HDAC1/2 proteins and LSD1. Another complex associated with LSD1 is the nucleosome remodeling and deacetylase (NuRD) complex, also including HDAC1/2 proteins [47]. An important role the NuRD complex plays is to maintain the genomic landscape and regulate cell cycle progression [48]. Similarly, the polycomb repressive complex (PRC2) is a repressive chromatin complex relevant to HGG and includes EZH2. This complex functions to regulate normal embryonic development and proper cell identity [49]. Finally, PRMT5 is a part of the methylosome, a protein complex that functions to methylate arginine residues of spliceosomal proteins important for the assembly of small nuclear ribonucleoproteins [50].
Overall, the activity of chromatin modulators and their histone modification can prompt severe changes in gene transcription that can lead to an oncogenic phenotype (Table 2). Their effects on gene transcription can impact various biological processes, from cell division and proliferation to differentiation. Largely, oncogenic conversion via epigenetic regulation can be driven by the actions of tumor suppressor proteins and signaling kinases, highlighting the potential interplay of kinase activity and epigenetic intervention.
Altered DNA methylation patterns
DNA methylation, like histone modifications, is involved in the regulation of gene expression, typically to repress gene transcription [16]. The addition of methyl groups to DNA most commonly occurs at cytosine residues to form 5-methylcytosine in CpG sites, where cytosine is linked to a guanine nucleotide by a phosphate group [16]. The enzymes responsible for generating 5-methylcytosine at the CpG sites are the DNA methyltransferases (DNMTs). The DNMT family includes DNMT1, DNMT3A, and DNMT3B, which establish and maintain the pattern of DNA methylation. The de novo methyltransferases, DNMT3A and DNMT3B, initiate the CpG methylation pattern. Meanwhile, DNMT1 is a maintenance methyltransferase and retains methylation marks throughout DNA replication and cell division.
In gliomas, DNA methylation patterns are used alongside histopathology for tumor classification, glioma subtyping, and as a prognostic biomarker [53]. For example, methylation profiling can be used as a surrogate to identify the mutation status of isocitrate dehydrogenase (IDH) [53]. IDH mutations, associated with the production of an oncometabolite (2-hydroxyglutarate), leads to global hypermethylation of the CpG islands, thereby causing gene silencing [54, 55]. The presence of this methylation pattern in gliomas is called the glioma – CpG island methylator phenotype (G-CIMP) and occurs frequently in low-grade gliomas [54, 55]. Large cohort studies have shown that G-CIMP is highly associated with the presence of an IDH mutation and correlated with a favorable prognosis [56]. Similarly, histone mutations common to pediatric HGGs lead to global changes in DNA methylation that can be detected through DNA methylation profiling [55, 57]. Methylation profiling can be used to derive copy number profiles inclusive of gene amplifications/deletions and chromosome alterations (7+/10- and 1p/19q codeletion) associated with different glioma subtypes [53]. Finally, the DNA methylation status of O6-methylguanine-DNA methyltransferase (MGMT) is widely used as a predictive biomarker for therapeutic response to the alkylating agent, temozolomide, and as a prognostic marker in glioblastoma patients [55, 58]. Thus, the value of methylation profiling in gliomas is expanding beyond its impact on gene expression, to help determine tumor phenotype and prognosis.
The World Health Organization (WHO) is beginning to adopt DNA methylation profiling in their classification of CNS tumors to provide a robust classification method and identify new tumor types and subtypes [53]. Notably, the emergence of DNA methylation profiling reveals an intriguing overlap with distinct kinase mutations traditionally associated with gliomas. For example, through methylation profiling, HGG is grouped into eight classes, including three classes characterized by RTK alterations, such as PDGFRA and EGFR amplification [59]. Furthermore, in pediatric HGGs with histone alterations, methylation profiling revealed several alterations in kinase signaling pathways, including PDGFRA, EGFR, KIT, MET, KRAS, PTEN, PIK3CA, and CDK4/6 [57]. Additional studies of pediatric HGGs used DNA methylation alongside whole genome sequencing and RNA sequencing in a clinical trial to molecularly characterize tumors and determine a treatment approach based on the identified alterations [60]. Overall, these studies highlight the potential of methylation profiling and its utility in understanding glioma subtypes, their associated kinase alterations, and appropriate therapeutic selection.
Single-agent targeted therapies in clinical development
Kinase inhibitors
Clinical trials have been implemented and are currently underway to assess the safety and efficacy of small molecule inhibitors directed against protein or lipid kinases in HGG patients with kinase dysregulation and aberrant signaling activation (Table 3). Several clinical studies have tested the effects of inhibition of RTKs, including small molecule inhibition of EGFR, PDGFR, FGFR (fibroblast growth factor receptor), c-MET (mesenchymal-epithelial transition factor), KIT (also referred to as CD117), AXL, and VEGFR (vascular endothelial growth factor receptor). Numerous inhibitors have assessed inhibition of EGFR in HGG as single agents and predominantly show a tolerable safety profile without improvements in survival. Likewise, the results from available clinical trials targeting other RTKs, PDGFR and MET, show tolerable safety profiles but limited efficacy. Currently, other RTK inhibitors targeting FGFR and KIT are being investigated in early-phase trials to determine the safety and efficacy of these small molecule inhibitors for adult (FGFR and AXL) and pediatric HGGs (KIT). Studies are also investigating the inhibition of intracellular signaling kinases in HGG downstream of RTKs. For example, clinical studies are ongoing in pediatric HGG patients receiving small molecule inhibitors against MEK and PI3K. Overall, many completed clinical trials conclude that single-agent inhibitors have tolerable toxicities in phase I but limited efficacy when assessed in phase II trials. This lack of clinical efficacy has been extensively reviewed elsewhere [61,62,63,64]. To summarize, resistance can occur from downstream activation of nontargeted kinase pathways or activation of parallel signaling pathways. Other resistance mechanisms, or perhaps a limited tumor penetrance, may be at play, underscoring the lack of response in these clinical trials. Heterogeneity among the brain tumor cells may also prevent the various single-agent strategies utilizing kinase inhibitors from producing efficacious results in reducing tumor burden or prolonging survival.
Epigenetically directed therapies
Targetable epigenetic regulators are being investigated in HGGs as single-agent therapies (Table 4). Similar to kinase inhibition, available results from clinical trials lend tolerable safety profiles. To date, there is limited information on the clinical benefit of these drugs compared to the standard of care. So far, pharmacological HDAC inhibition via vorinostat has been well tolerated and has provided a modest improvement in progression-free survival (PFS) [75]. Other HDAC inhibitors, panobinostat and an aqueous formulation of panobinostat (MTX110), have been assessed in a population of diffuse midline gliomas (DMG, previously referred to as DIPG) and show an acceptable safety profile [19, 76]. However, at tolerable doses panobinostat has no significant clinical benefit likely due to lack of drug exposure at the tumor site [19]. Fortunately, the administration of MTX110 via convection-enhanced delivery provided encouraging results with a benefit on overall survival compared with historical outcomes [76]. The positive results from MTX110 on survival will be assessed further in a multicenter phase II study. In contrast, the clinical studies of birabresib, a BET inhibitor, in HGGs showed a lack of clinical efficacy [18]. Perhaps the insufficient activity is due to intra-tumor heterogeneity and the emergence of subpopulations resistant to treatment. Lack of brain penetrance seems unlikely as preclinical pharmacokinetic analysis indicated biologically active levels of birabresib [77]. Altogether, the outcome of these clinical trials justifies a need to understand further mechanisms of resistance and ways to enhance their efficacy. One potential approach is to investigate the interplay with proliferative kinase signaling pathways.
Crosstalk from epigenetic modulators of histone acetylation with kinase signaling
Histone deacetylases (HDAC)
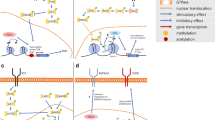
Histone deacetylases (HDACs) are a large family of enzymes that catalyze the removal of acetyl groups from lysine residues on histone tails and non-histone targets; this action generally limits chromatin accessibility and reduces gene transcription. Eighteen human HDACs are separated into four groups based on homology [79]. While HDACs are responsible for deacetylation, they oppose histone acetyltransferase (HATs), which catalyzes reversible lysine acetylation that results in a more permissive gene expression. Therefore, the balance in HDAC and HAT activity is vital for the proper and timely repression or expression of genes. Inhibitors of HDAC have progressed into the clinic, receiving FDA approval for the treatment of several hematological malignancies. To date, the clinically approved HDAC inhibitors (vorinostat, romidepsin, panobinostat, and belinostat) are known as pan-HDAC inhibitors and act on multiple classes of HDACs. In cancer treatment, HDAC inhibitors are used to induce cell cycle arrest and cell death by increasing gene transcription, such genes involved in both intrinsic, BAX, BAK, APAF1, and extrinsic, TRAIL, DR5, FAS, FAS-L, and TNF-α, apoptotic pathways [80]. Currently, HDAC inhibitors are being assessed in glioblastoma, pediatric HGG, and medulloblastoma models. HDACs have been suggested to regulate glioma proliferation via interactions with the PI3K/AKT signaling pathway, MAPK signaling, and upstream at receptor tyrosine kinases (Fig. 1).

Modulation of Kinase Signaling Pathways by HDAC Inhibitors. HDAC inhibitors exhibit synergistic interactions with EGFR and AXL inhibitors, enhancing their therapeutic potential [92]. Additionally, HDAC inhibitors downregulate EGFR expression and attenuate ERK signaling, potentially disrupting downstream cascades [87, 93, 95]. HDAC inhibitors also synergize with PI3K inhibitors, leading to a reduction in phospho-AKT signaling [83]. These observations highlight the intricate regulatory role of HDAC inhibitors in modulating diverse aspects of kinase signaling networks for potential therapeutic applications
Created by BioRender.com
In various cancer models, HDAC antagonists have a synergistic relationship with PI3K inhibitors that is associated with a strong inhibition of the PI3K/AKT signaling pathway compared to single-agent PI3K inhibitor [81, 82]. In GBM cell models, panobinostat has been shown to synergize with a PI3K/mTOR inhibitor to reduce cell viability [83]. In this study, a dual inhibitor of PI3K and mTOR, dactolisib, was used in combination with panobinostat: their effects on cell viability, cell proliferation, and induction of apoptosis were assessed. The combination treatment resulted in a synergistic reduction in cell viability, enhanced antiproliferative effects, and induced apoptosis relative to single-agent treatment [83]. Furthermore, the combination treatment enhanced AKT signaling reduction compared to the single-agent treatment [83].
Similar to adult GBM, a study found that inhibition of HDAC/PI3K via dual inhibitor, CUDC-907, in pediatric HGG models exerts substantial antitumor effects compared to single-target inhibition [84]. CUDC-907 has been shown to reduce the PI3K signaling network and, importantly, blocks compensatory signaling via MAPK and STAT3 (signal transducer and activator of transcription) signaling pathways [85]. Interestingly, this dual inhibitor acts as a radiosensitizer in pediatric HGG models mediated by decreasing NFκB/Forkhead box M1 (FOXM1) expression [84]; this additional effect is particularly important in the pediatric HGG and DMG, as radiation therapy is the current standard of care. Identifying novel strategies to enhance radiotherapy efficacy will be important when progressing into clinical trials. One additional study investigated the HDAC inhibitor, panobinostat, in combination with PI3K inhibition in medulloblastoma. This study showed that HDAC inhibition combined with PI3K inhibition synergistically inhibited growth by activating a tumor suppressor, FOXO1 via two distinct mechanisms [86]. In this case, HDAC inhibition increased FOXO1 protein expression, associated with increased acetylation of H3K9 and H3K27, while PI3K inhibition promoted nuclear localization of FOXO1 through its dephosphorylation [86]. Overall, multiple studies have examined the co-inhibition of HDACs and PI3K/AKT signaling networks and observed enhanced efficacy compared to HDAC inhibition alone.
Apart from the PI3K/AKT signaling pathway, HDAC inhibitors cooperate with several members of the MAPK family proteins. For example, the HDAC inhibitor, sodium butyrate, markedly reduces ERK (extracellular signal-regulated kinase) activation in medulloblastoma with increased H3 acetylation [87]. From this study, the dual inhibition of HDAC and MAPK signaling reduces medulloblastoma proliferation and viability. C-Jun N-terminal kinase (JNK), another member of the MAPK superfamily, can be activated in gliomas and is crucial in the maintenance of stemness [88, 89]. HDAC6 promotes cell growth in glioblastoma through the JNK signaling pathway [90]. Furthermore, inhibition of HDAC6 by ricolinostat, associated with increased H3K9 and H3K27 acetylation, suppresses JNK activity and mediates reduced proliferation and invasion of glioma cells [90]. An additional regulator of MAPK signal transduction is the MAPK phosphatase, MKP1. MKP1 is an inhibitor of ERK1/2, JNK, and MAPK to regulate glioma self-renewal and differentiation. Interestingly, upregulation of MKP1 occurs following treatment with the HDAC inhibitor vorinostat and further sensitizes glioblastoma cells to temozolomide [91]. Finally, the inhibition of RTKs, located upstream of PI3K/AKT and MAPK signaling pathways, has also been shown to improve anti-tumor activity in combination with HDAC inhibitors to target HGG. A study in DMG found that the RTK, AXL, is upregulated in DMG, initiates the mesenchymal transition, and dual AXL/HDAC inhibition caused a synergistic anti-tumor effect [92]. The treatment combination of BGB324 (AXL inhibitor) and panobinostat causes the downregulation of genes associated with mesenchymal transition and DNA damage repair, ultimately leading to decreases in cell viability [92]. Understanding the interplay between HDAC activity and RTK/MAPK signaling pathways is valuable to establish the comprehensive mechanism by which HDAC inhibitors act in the context of HGG.
Beyond AXL, HDAC inhibitors have shown synergistic relationship with other RTKs, including EGFR. Two recent studies assessed the effects of dual inhibition of HDAC and EGFR. In glioblastoma cells, combining EGFR inhibition via AG1478 with sodium butyrate, resulted in decreased cell viability with more activity than either agent alone [93]. Interestingly, the combination mentioned previously increased STAT3 mRNA expression [93]. The authors speculate that the upregulation of STAT3 may mediate the anti-tumor effects of dual HDAC/EGFR inhibition through a tumor-suppressive role, as opposed to its dual action as an oncoprotein [93, 94]. Furthermore, another study also combined EGFR and HDAC inhibitors to study their effects on glioblastoma cells with various models of EGFR alterations [95]. The combined effects of erlotinib and investigational HDAC inhibitor, scriptaid, increased H3K9 acetylation and could overcome erlotinib resistance and re-sensitize glioblastoma cells to EGFR inhibition [95]. In fact, the combination enhanced single-agent efficacy in glioblastoma cells independent of their EGFR status. Further experiments were completed to understand this relationship of enhanced efficacy; HDAC inhibition, via vorinostat or scriptaid, caused a decrease in the mRNA and protein expression of EGFR, both wild-type and EGFRvIII (EGFR variant III extracellular domain mutation) [95]. Due to promising results, the combination of EGFR and HDAC inhibition has been assessed in a phase I/II clinical trial in recurrent glioblastoma (NCT01110876). Unfortunately, this study was terminated due to unanticipated toxicities before there could be any assessment of efficacy in the phase II portion of the trial. While the combination had unacceptable toxicities, their EGFR inhibitor, erlotinib, has limited ability to penetrate the blood-brain-barrier (BBB) and is only active against wild-type EGFR. Future combination studies could include EGFR inhibitors with high penetrance across the BBB as well as a focus on the development of EGFR inhibitors selective for the mutant EGFR in glioblastoma, EGFRvIII. In summary, combining EGFR and HDAC inhibitors in glioblastoma may be able to overcome resistance clinically, but the selection of targeted therapies is important to minimize toxicities.
Bromodomain and extraterminal (BET)
The class of proteins that contain two acetyl-histone reading domains, the bromodomain (BD) and the extraterminal (ET) domain, are collectively known as the bromodomain and extraterminal (BET)-containing proteins. BET proteins are a family of epigenetic readers which recognize specific histone modifications to facilitate the assembly of transcription complexes. This family of epigenetic reader proteins includes BRD2, BRD3, BRD4, and BRDT, and they exert their effects on transcription by binding to acetylated lysine residues on histones [96, 97]. BRD4, perhaps the most studied BET protein, typically recognizes acetylated histone marks on histone 4 lysine 5 and 12 (H4K5 and H4K12) as well as histone 3 lysine 14 (H3K14) [96, 98]. It is proposed that recognizing such acetylated lysine residues can facilitate the activation of transcriptional processes by interacting with cyclinT1 and CDK9 to regulate the positive transcription elongation factor b (P-TEFb) [96, 99, 100]. The BET protein, BRD2, is a protein serine/threonine kinase that promotes the recruitment of transcription factors, such as E2F1, to transcriptional complexes to regulate cell cycle progression [96, 101]. Moreover, BRD2 recognizes acetylated lysine 12 on histone 4 (H4K12) [96, 102]. Several BET proteins, BRD4 and BRD2, have been shown to be overexpressed in gliomas making their inhibition an emerging therapeutic strategy [27]. Unfortunately, resistance to BET inhibition has already been reported secondary to the amplification of RTKs or activation of PI3K/AKT and MAPK signaling pathways [103, 104]. Not only can kinase activation act as a resistance mechanism for BET inhibition, but BET proteins also rewire various kinase transduction pathways to dampen their activity (Fig. 2). A critical understanding of how BET interacts with individual signaling pathways will be important to guide the design and development of novel therapeutic regimens targeting epigenetic readers in HGG.

BET Protein Interactions with Key Signaling Pathways in High-Grade Gliomas. BET proteins influence transcriptional processes by recognizing acetylated lysine residues on histones and recruitment of transcriptional factors, including P-TEFb, to activate RNA polymerase [99, 100]. In models of medulloblastoma and HGG, the inhibition of BET proteins impacts the VEGFR/PI3K/AKT signaling pathway [107] and the transcription factor MYC [111]. Furthermore, synergistic effects are observed upon combining BET inhibitors with kinase inhibitors of MEK, AURKA and CDKs, suggesting potential therapeutic strategies [106, 108, 111]
Created by BioRender.com
Past studies have discovered that inhibition of BET proteins can be used to overcome adaptive resistance associated with inhibition of the kinase signaling protein, MAPK kinase (MEK) [105]. While this study was conducted in triple negative breast cancer models, the relationship between BET and MEK in HGG is poorly understood. In glioblastoma models, hexamethylene bisacetamide (HMBA), a high-affinity BRD2 inhibitor, was shown to block cellular proliferation without leading to cell death [106]. Further in vitro and in vivo screenings revealed a synergistic relationship between the combination of HMBA and MEK inhibition leading to enhanced apoptosis of glioma cells [106]. The study concluded that combined inhibition was more effective relative to monotherapy in glioblastoma models and such evidence provides rationale for a clinical trial in selected patients [106]. Moreover, these synergistic effects underscore the convergence of BET proteins with MAPK signaling among malignant brain tumors.
Similar to BET’s influence on MAPK kinases, BET inhibitors also interact with the PI3K/AKT signaling pathways in HGG models to elicit anti-tumor effects. One study used the bromodomain inhibitor, JQ1, to prevent BET protein binding and activity, allowing for the impact of BRD4 in glioma stem cells to be investigated [107]. In this study, JQ1 inhibited cell proliferation, induced cell cycle arrest, and promoted glioma differentiation. The PI3K/AKT signaling pathway mediated the effect of JQ1, and upon inhibition of BRD4, there was a decrease in phosphorylated AKT [107]. This effect was found to be altered upstream via the RTK, vascular endothelial growth factor receptor (VEGFR), where JQ1 inhibited the expression of VEGF and phosphorylated VEGFR [107]. Importantly, JQ1 is a pan-BET inhibitor, and it is likely that BRD4, or a different BET protein, may regulate the growth and development of gliomas via the VEGF/PI3K/AKT signaling axis. Interestingly, another BET inhibitor, OTX015 (birabresib), a BRD3 selective inhibitor, was found to activate the AKT/mTOR pathway by increasing the level of SESN3, a protein coding gene for stress-inducible protein, sestrin 3 [77]. The study then combined the treatment of birabresib and everolimus producing additive anti-tumor activity. These studies highlight the role of the BET proteins in the regulation of kinases and suggest improved treatment outcomes with the combination of BET and kinase inhibitors.
Not only do BET proteins cooperate with MAPK and PI3K/AKT signaling cascades, but new studies show that BET proteins interact with kinases involved in mitosis. For example, a recent study reported a synergistic relationship between inhibitors of Aurora Kinase A (AURKA) and BET proteins in MYC-driven glioblastoma cells [108]. AURKA plays a vital role in cell division, in particular during mitosis, and the proper functioning of microtubules [108, 109]. Notably, the oncogenic MYC genes are known to be epigenetically regulated by BRD4 via binding at the MYC gene promoter region [108, 110]. As expected, the inhibition of BET with the small molecule inhibitor, JQ1, suppressed the expression of MYCN in the sensitive cell line [108]. Additionally, the expression of MYCN correlated with AURKA levels. Next, JQ1-sensitive and resistant glioblastoma cells displayed a synergistic effect when JQI was combined with an AURKA inhibitor [108]. Ultimately, the discovered relationship between BRD4 and AUKRA inhibitors identified a potential therapeutic approach when translating BET inhibitors into the clinic for trials in HGG with MYC dysregulation.
Cell cycle progression is a fine-tuned biological process with several components essential for its regulation. In addition to kinases like AURKA, cyclin-dependent kinases (CDKs) are required for progression in the cell cycle, and epigenetic interactions may influence these CDKs, creating new opportunities for combination treatments. For example, in MYC-driven models of medulloblastoma, BET inhibition, via JQ1, in combination with CDK inhibitor, milciclib, diminished proliferative markers and induced apoptosis [111]. Importantly, the regulation of MYC via phosphorylation is an essential role of CDK proteins, specifically CDK1 and CDK2 [111,112,113]. While milciclib suppressed phosphorylation of MYC at residues S62 and T58 to destabilize MYC, BET inhibition reduced MYC transcription [111]. The study identified a synergistic relationship between BET and CDK, which reduced medulloblastoma tumor burden and prolonged in vivo survival [111]. In conclusion, combining BET and CDK2 inhibition offers a potential strategic therapy for targeting MYC-dependent medulloblastoma among pediatric patients.
Epigenetic modulators of histone methylation interplay with kinase signaling cascades
Protein arginine methyltransferases (PRMT)
The protein arginine methyltransferase (PRMT) family consists of nine members that act as a component of complexes that epigenetically regulate transcription, translation, splicing, and cell signaling [114, 115]. PRMTs catalyze the transfer of a methyl group to the guanidine nitrogen atoms of arginine, mainly those present on the histone tail. The methylation pattern on arginine by PRMTs can occur in three forms: monomethylarginines, asymmetric dimethylarginines, and symmetric dimethylarginines. Based on the methylation pattern, PRMT members are separated into one of three types. Type I PRMTs form the monomethylarginine and asymmetric dimethylarginine. The type II isoforms produce monomethylarginine and symmetric dimethylarginine. In contrast, type III arginine methyltransferase forms only the monomethylarginine. While mutations in PRMT are uncommon in cancer, high protein expression levels have been associated with poor outcomes [116, 117]. Additionally, PRMT1 and PRMT5 expression has been associated with the development of glioblastoma and medulloblastoma [30, 118]. Overexpression of PRMT3 promotes tumor growth in GBM while also conflicting poor survival with heightened expression; PRMT3 promotes tumorigenesis in GBM by regulating glycolysis, specifically, HIF1A [51]. PRMT2, another overexpressed PRMT protein, confers poor patient prognosis, and knockout of PRMT2 causes reductions of phosphorylated STAT3, AKT, and MAPK in glioma cell lines [31]. Meanwhile, PRMT6 is overexpressed in GBM, causing increased self-renewal, and PRMT6 expression is correlated to poor patient prognosis [52]. PRMT6 is postulated to methylate RCC1 (regulator of chromosome condensation 1) for chromatin binding, thereby modulating mitosis [51, 52]. Therefore, several family members of PRMT have recently become of interest as cancer targets based on the association of PRMT expression with poor patient outcomes and brain development and tumorigenesis (Fig. 3). Numerous PRMT inhibitors have been developed and are largely separated into Type I PRMT and Type II PRMT inhibitors which have been investigated in various cancer models [119]. Drug discovery efforts are ongoing to design new agents with selective activity against different isoforms of PRMT and considerations for strategies to improve efficacy through combination treatments.

Patterns of PRMT Methylation and Kinase Signaling Crosstalk. The PRMT family initiates distinct methylation patterns, including monomethylarginines, asymmetric dimethylarginines, and symmetric dimethylarginines, to epigenetically regulate cell processes. Highlighted are PRMT1, 2, 3, 5, and 6 which are associated with glioblastoma and medulloblastoma progression and/or tumorigenesis [30, 31, 51, 52, 118]. Functional genetic studies of PRMTs emphasize their modulation of kinase networks. For example, gene depletion of either PRMT2 or 5 is shown to decrease kinase signaling of both PI3K/AKT and MAPK pathways [31, 120]. Moreover, PRMT1 activates JAK/STAT3 pathway to promote cell differentiation, offering insights into potential PRMT-directed therapies [121]
Created by BioRender.com
The PRMT-PTEN signaling axis is one potential intersection for consideration when designing combination treatments with PRMT inhibitors. As a tumor suppressor, PTEN attenuates the kinase signaling cascade of the PI3K/AKT/mTOR pathway. In glioblastoma models, one study found PTEN as a downstream target of the type II arginine methyltransferase, PRMT5 [120]. Here, PRMT5 expression was enriched at the promotor region of PTEN in glioblastoma neurospheres but not in the differentiated glioblastoma counterpart. Depletion of PRMT5 caused the expression of PTEN transcript and protein expression to significantly increase in the glioblastoma neurospheres [120]. In parallel, the expression of phosphorylated AKT and ERK was reduced with gene silencing of PRMT5. Furthermore, reduced proliferative and self-renewal capacity among the neurospheres was observed following PRMT5 depletion, partially restored upon PTEN knockdown. Overall, this study demonstrates PTEN as a target of PRMT5 methylation, which regulates important processes involving cell proliferation, cell growth, and self-renewal of neurospheres.
Like the previously described histone targets, PRMTs can also have non-histone targets. The type I arginine methyltransferase, PRMT1, influences the JAK/STAT3 pathway in neural stem precursor cells. Activated STAT3 signaling can impact many cellular processes, including cell differentiation. Activation of STAT3 can be achieved via phosphorylation or methylation. A recent study found STAT3 as a non-histone target of PRMT1, and its methylation resulted in the enhanced activation of STAT3 [121]. The methylated STAT3 promoted astrocytic differentiation of neural stem precursor cells [121]. Taken together, this study demonstrates STAT3 regulation via PRMT1 to promote cell differentiation. This is relevant to HGG when constructing treatment strategies to target PRMT1 pharmacologically, leading to therapies that will promote differentiation of stem-like cancer cells.
Enhancer of zeste homolog 2 (EZH2)
Enhancer of zeste homolog 2 (EZH2) is an epigenetic writer that catalyzes the methylation of lysine residues on histone and non-histone targets through its involvement with Polycomb-group (PcG) proteins. EZH2 performs a critical role in transcription processes, particularly transcriptional repression, by acting as the functional subunit of the PcG protein complex, Polycomb Repressive Complex 2 (PCR2) [122, 123]. The methyltransferase activity of EZH2 arises from its SET domain and uses S-adenosyl-L-methionine (SAM) as a cofactor [124]. The PRC2 houses the core proteins: EZH1/2, Suz12, Eed, and Rbbp4 [125]. Collectively, the primary target of PRC2 is histone H3 lysine 27 (H3K27), which recruits PRC1 to exert alterations in gene expression via chromatin remodeling [124]. Interestingly, EZH2 has demonstrated a potential tumor-suppressive role in subsets of diffuse midline gliomas by inducing oxidative phosphorylation [126]. However, a different study identified EZH2 inhibition as a therapeutic target among gliomas harboring H3K27M mutations, which inhibits the PRC2 complex [127]. In glioblastoma, high expression of EZH2 has been associated with worse survival and high tumor grade [33]. Based on the proposed role of EZH2 in glioblastoma, inhibition of this writer has triggered interest as a therapeutic target [128]. While preclinical studies are ongoing, caution is necessary when considering using EZH2 inhibitors in other glioma models, as EZH2 may have context-specific functions and various downstream effects such as regulation of tumor suppressors or other signaling pathways.
In an effort to develop EZH2 inhibitors, several small molecules have been designed to prevent its activity with varying mechanisms of action [122]. Current EZH2 inhibitors that are in development include SAM-competitive inhibitors, inhibitors that disrupt EZH2 protein interactions, and those that promote EZH2 degradation [122]. One of the largest groups of inhibitors is those that bind to the SET domain and compete with SAM to inhibit the methyltransferase activity of EZH2, such as tazemetostat and GSK126. Similarly, there is a drug in development, 3-Deazaneplanocin A (DZNep), which inhibits global histone methylation by targeting S-adenosyl homocysteine (SAH) hydrolase. Another group of EHZ2 inhibitors work by disrupting EZH2s’ interaction with the PCR2 complex. These inhibitors target the PCR2 scaffolding proteins, Suz12 and Eed, and include A769662 and astemizole, respectively. Lastly, there is a class of EZH2 inhibitors that promote its degradation, including ANCR, GNA002, and FBW7. In addition to direct inhibition of EZH2, it is important to consider other means of vulnerability, perhaps through its interactions with kinase signaling pathways including the PI3K/AKT and JAK/STAT pathways (Fig. 4).

Multifaceted Role of EZH2 in Kinase-Mediated Regulation. As a methyltransferase, EZH2 facilitates the compaction of chromatin by adding methyl groups to histone protein, H3K27. This epigenetic modification condenses the chromatin structure, affecting gene expression patterns. In this manner, EZH2 can exert its influence on kinase signaling pathways. It downregulates PTEN expression, leading to activation of the PI3K/AKT pathway [129]. Beyond its chromatin-related functions, EZH2 acts as a methyltransferase for non-histone protein, STAT3, increasing its activity in gene regulation [134]. Moreover, the kinase MELK plays a crucial role, as it phosphorylates EZH2, enhancing its methyltransferase activity and amplifying its effect [131, 133]. TF – transcription factor
Created by BioRender.com
Evaluation of EZH2 and its relationship with cell signaling processes may provide insights into kinase signaling dysfunction or mechanisms of therapeutic resistance. EZH2’s cellular interplay with E2F transcription factors in glioblastoma models is one mechanism that offers tumor cell proliferation and growth. The clinically relevant E2F transcription factor, E2F7, was overexpressed among high-grade glioma patients compared with low-grade gliomas or normal tissues. Furthermore, high expression of E2F7 was associated with poor prognosis [129]. Moreover, E2F7 acts upstream of EZH2 as a transcriptional activator in glioblastoma by binding to its promoter [129]. Kinase signaling pathways are affected by the activity of E2F7 and its action on EZH2. The tumor suppressor protein, PTEN, is a negative regulator of the PI3K/AKT signaling. PTEN, an established target of EZH2, is downregulated in glioblastoma and associated with poor survival [129]. Overall, E2F7 is responsible, in part, for the proliferative advantages in glioblastoma mediated by EZH2 inhibition of PTEN and leading to activation of the PI3K/AKT/mTOR pathway [129]. The relationship between EZH2 and PTEN highlights critical insights into the origins of aberrant signaling in glioblastoma and considerations for therapy.
Interestingly, the PTEN/PI3K/AKT signaling network is not the only kinase signaling cascade EZH2 interacts with among brain tumor models. EZH2 interacts with maternal embryonic leucine-zipper kinases (MELK) in glioblastoma and medulloblastoma. MELK is an important kinase signaling protein involved in cell growth, cell cycle regulation, DNA repair, migration, invasion, and apoptosis [130,131,132]. In glioblastoma, poorer overall survival was noted among patients with higher expression levels of MELK and EZH2 [131]. Similar to glioblastoma, medulloblastoma patients had poor overall survival with increased MELK expression postoperatively [133]. Beyond its association with poor prognosis, MELK activates EZH2 through phosphorylation to promote glioma stem-like cells to proliferate and self-renew [131]. EZH2 and MELK also have a similar relationship in medulloblastoma. In medulloblastoma models, phosphorylated EZH2, and its H3K27 methylation activity, were reduced upon MELK gene silencing [133]. Importantly, in vivo models of medulloblastoma displayed a significant survival benefit from the knockdown and pharmacological inhibition of MELK and EZH2, highlighting the relevance of these two proteins in brain tumor proliferation [133]. Once EZH2 is activated via phosphorylation in glioblastoma, it proceeds to methylate a downstream target, NFĸB. NFĸB methylation mediates the effect of EZH2 to induce glioblastoma proliferation and maintenance of stem-like characteristics [131]. Nonetheless, EZH2 and MELK cooperate in glioblastoma and medulloblastoma to promote cell proliferation and this interplay offers a potential combination therapeutic strategy.
The interactions between EZH2 and its non-histone targets are also important in HGG, and this relationship could offer further understanding of oncogenic signaling pathways. One non-histone target of EZH2 is the STAT3 in the JAK/STAT pathway [134]. Following JAK activation by interleukin, interferons, or growth factors, STAT3 is phosphorylated and acts as a transcription factor to promote various processes such as immune cell response, cell division, metastasis, and cell differentiation [135]. Post-translational modifications of STAT3, such as methylation, can affect its activity. Upon methylation of STAT3 via EZH2 in glioblastoma stem-like cells, STAT3 activity was enhanced [134]. This positive regulation promoted the self-renewal capacity of stem-like glioblastoma cells. As expected, EZH2 inhibition via gene knockdown and pharmacological inhibition, via DZNep, reduced the methylation of STAT3 and dampened its activity [134]. Ultimately, EZH2 directly regulates STAT3, and its inhibition may impair key signaling pathways that promote tumor growth and maintain a stem-like tumor cell population in glioblastoma.
Lysine-specific demethylase 1 (LSD1)
Histone demethylases, including lysine-specific demethylase 1 (LSD1/ KDM1A), regulate gene transcription and chromatin structure via the demethylation of lysine residues. LSD1 was the first discovered lysine demethylase, and it belongs to a family of two histone lysine demethylases (LSD1 and LSD2) [136]. LSD1 is a flavin-dependent monoamine oxidase that catalyzes the demethylation of mono- and dimethyl groups from histone 3 on lysine residues 4 and 9 (H3K4 and H3K9) [136]. Overexpression of LSD1 is found in many cancer types, and the resulting increase in its activity can lead to gene dysregulation and support cancer progression [137]. For example, LSD1 plays a role in cancer by maintaining cancer stemness, regulating differentiation, promoting EMT, and regulating hypoxia [138]. Therefore, LSD1 is regarded as a cancer drug target with several small molecule inhibitors already in various stages of clinical development. In HGG, LSD1-directed agents can induce tumor regression when assessed in vivo [36, 139, 140].
Pharmacological LSD1 inhibitors have been developed and are separated into two categories, reversible and irreversible inhibitors. The first identified irreversible inhibitor was tranylcypromine. Tranylcypromine covalently binds to the FAD domain within the active site of LSD1, rendering LSD1 inactive [141,142,143,144,145]. Since the identification of tranylcypromine, multiple irreversible LSD1 inhibitors have been developed and have been investigated in various other tumor models, mostly hematological and small-cell lung cancer; these molecules include GSK-LSD1, ORY-1001, RN-1, IMG-7289, INCB059872, and ORY-2001 [142, 143, 145,146,147,148]. In contrast, reversible LSD1 inhibitors, such as SP-2509, are proposed to bind to the allosteric site and have effects independent of LSD1 demethylase activity [149]. Meanwhile, another reversible inhibitor, CC-990,011 binds at the amine oxidase pocket of LSD1 while having anti-tumor activity in small-cell lung cancer [150].
Part of the mechanism by which LSD1 inhibitors induce an anti-tumor response is through its interaction with kinase signaling pathways involving cell metabolism and cell cycle progression (Fig. 5). One study found that LSD1 inhibition via tranylcypromine impaired mitochondrial respiration in glioblastoma cells by reducing their oxidative capacity [151]. This impairment was accompanied by a reduction in mitochondrial proteins Tom20, PDH, and SDH. Furthermore, tranylcypromine treatment caused a decrease in the kinase activity of mTOR that reduced the downstream activation of two key players to regulate cell growth and cell cycle progression: ribosomal protein S6 kinase beta-1 (S6K1) and eukaryotic translation initiation factor 4E-binding protein 1 (4-EBP1) [151]. Overall, this study concluded that LSD1 inhibition impairs mitochondrial respiration with subsequent effects on cell cycle progression.

LSD1 Modulation of Cell Growth and Kinase Signaling Pathways. As a histone demethylase, LSD1 targets H3K4 demethylation, influencing crucial cellular processes. Inhibition of LSD1 leads to reduced mTOR signaling and subsequent attenuation of mitochondrial respiration, impacting cell metabolism and growth [151]. Moreover, LSD1 inhibition upregulates the expression of p21, a negative regulator of the cell cycle, culminating in cell cycle arrest [151, 152]. In contrast, cells without LSD1 inhibition demonstrate heightened cell growth and progression through the cell cycle
Created by BioRender.com
The effects of LSD1 inhibition on cell cycle progression extend beyond its influence on mTOR signaling. An additional role for LSD1 inhibitors in glioblastoma is to induce cell senescence through its involvement in the retinoblastoma (RB)/E2F/CDK-Cyclin pathway. Both gene silencing and pharmacological inhibition of LSD1 increased the expression of cyclin-dependent kinase inhibitor, p21 [151]. Accordingly, the upregulation of p21 led to the reduced phosphorylation of RB and a negative cell cycle regulation to induce cell senescence. A different study in glioblastoma also found that LSD1 activity/inhibition regulates the expression of genes involved in cell cycle progression. Following inhibition of LSD1, the gene encoding p21 was upregulated, and its downstream targets, cyclin-dependent kinase (CDK4/6/2), had reduced activity [152]. Interestingly, this study combined LSD1 inhibition, GSK-LSD1, with a small molecule inhibitor that disrupts EZH2 interactions, AC1Q3QWB, with an oncogenic long noncoding RNA (lnRNA), HOTAIR (Hox transcript antisense intergenic RNA). The combination treatment yielded synergistic cell cycle inhibition via upregulation of CDKN1A encoding p21 and resulted in improved survival of orthotopic glioblastoma models [152]. In summary, LSD1 expression is implicated in promoting HGG, and its successful inhibition disrupts cell cycle progression through the interplay with kinase signaling networks.
Discussion
The review of recent studies in HGG and medulloblastoma shows that the interplay between epigenetics and kinase signaling pathways is a multifactorial mechanism. Most likely, the precise relationship has tumor-specific and drug-specific implications. Overall, the cooperation between epigenetic and kinase signaling networks highlights new multimodal treatment strategies to build upon and enhance the standard of care in HGG. As epigenetically directed therapies and kinase inhibitors continue to translate into the clinic, sustaining partial and complete responses to single target agents will likely be challenging. Studies should continue to look for opportunities to understand the interplay of chromatin regulators and kinase signaling networks to overcome single-agent barriers. In addition, studies should evaluate the treatment regimens for kinase/chromatin inhibitors including the timing of treatment, sequence of drug administration, and dosing to maximize efficacy and limit toxicities. Understanding these parameters will be necessary to build novel therapeutic strategies with greater efficacy against HGGs.
Targeted therapies have become a significant tool in treating many cancer types, but their utility in HGG has not yet been established. A major constraint for targeted therapy in HGG is dose-limiting toxicities (DLTs). The DLTs from targeted therapy are a result of off-target and off-tumor effects. For example, most epigenetically targeted inhibitors are pan-inhibitors, which target all or several proteins within that class. Developing more selective agents, including both epigenetic and kinase inhibitors, will aid in reducing off-target effects and improve safety. Off-tumor effects are a consequence of target inhibition outside of the tumor site; this can be opposed by using small molecule inhibitors with high brain penetrance to reach adequate concentrations within the tumor. Another factor to consider is the tumor-specific mechanism and individual genomic landscape that predict response to targeted therapy. This measure is already relevant as EZH2 can act in a tumor-specific manner with oncogenic activity in most tumors, but a tumor suppressive role in a small subset of tumors, including cases of DMGs [126, 153]. Thus, this idea of identifying predictive biomarkers, or precision medicine, by analyzing a patient’s genomic landscape will improve patient selection and spare non-responders from toxicity. Inhibitors need to be more effective at lower doses and given to patients with a high likelihood of a response to improve the toxicity profile of targeted therapy.
Beyond safety, resistance is another obstacle to targeting chromatin modifiers and kinase signaling proteins. Due to the redundancy in kinase signaling pathways, inhibiting a single kinase is often unsuccessful, as compensatory pathways mitigate the effects of single-agent kinase inhibition. An additional mechanism of resistance to kinase inhibition is through epigenetic changes. Furthermore, epigenetic regulation is highly interdependent between the writers, readers, and erasers and can produce unexpected effects that may limit therapeutic efficacy. For example, inhibition of HDAC via vorinostat can increase H3K4 methylation, making it vulnerable to LSD1 demethylation [35]. One solution to overcome resistance and improve drug toxicity profile is to design combination treatment strategies inclusive of kinase inhibition and epigenetically directed inhibition.
Several in vitro and in vivo models have identified synergistic relationships between kinase and epigenetic inhibition. However, there is still a need to explore the interplay between epigenetic regulators and kinase signaling pathways and understand their specific mechanisms. Identifying safe drug combinations with synergistic or additive effects may afford improvements in efficacy and create an opportunity in the clinical trial setting for HGGs. In the clinic, treatment combinations could allow for dose reductions that exert a clinical effect and decrease unwanted side effects. Certain drug combinations may also circumvent resistance mechanisms associated with single-agent inhibition to extend a drug response. Additionally, other combination treatment strategies to overcome the lack of single-agent success may include introducing immunotherapies to either kinase inhibition or epigenetically directed agents. Previous studies have shown that epigenetic alterations change the tumor microenvironment to contribute to the immune suppressive niche for tumor cells. More recently, studies in several cancer types have shown inhibition of epigenetic regulators (LSD1, EZH2, BET proteins) can enhance the anti-tumor immune response of anti-PD1 therapy [13, 154,155,156]. Interestingly, studies in other cancer models highlight that kinase inhibition can enhance anti-tumor effects with immunotherapies [157,158,159]. Investigating triple therapy targeting chromatin modulators and kinase pathways and using immunotherapy in the ongoing efforts to attenuate tumor cell proliferation to improve patient outcomes and increase overall survivability would be worthwhile. In conclusion, enhancing our understanding of the cooperation across the HGG epigenome and genome will guide the development of new therapeutic strategies.
Data Availability
Not applicable.
Abbreviations
- 4-EBP1:
-
Eukaryotic translation initiation factor 4E-binding protein 1
- AMP:
-
Amplification
- AURKA:
-
Aurora kinase A
- BBB:
-
Blood-brain-barrier
- BET:
-
Bromodomain and extraterminal
- BRD) c-Jun N-terminal kinase (JNK:
-
Bromodomain
- c-MET:
-
Mesenchymal-epithelial transition factor
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2 A
- CDKs:
-
Cyclin-dependent kinases
- CNA:
-
Copy number alterations
- CNS:
-
Central nervous system
- CoREST:
-
REST corepressor 1
- DCR:
-
Disease control rate
- DIPG:
-
Diffuse intrinsic pontine glioma
- DLT:
-
Dose-limiting toxicity
- DMG:
-
Diffuse midline gliomas
- DNMT:
-
DNA methyltransferase
- DZNep:
-
3-Deazaneplanocin A
- EGFR:
-
Epidermal growth factor receptor
- ERK:
-
Extracellular signal-regulated kinase
- EZH2:
-
Enhancer of zeste homolog 2
- FGFR:
-
Fibroblast growth factor receptor
- FOXM1:
-
Forkhead box M1
- G-CIMP:
-
Glioma – CpG island methylator phenotype
- GBM:
-
Glioblastoma
- H3K4/9/27:
-
Histone 3 lysine 4/9/27
- H4K5/8/12:
-
Histone 4 lysine 5/8/12
- HATs:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- HGGs:
-
High-grade gliomas
- HMBA:
-
Hexamethylene bisacetamide
- HOMDEL:
-
Homozygous deletion
- HOTAIR:
-
Hox transcript antisense intergenic RNA HOTAIR
- IDH:
-
Isocitrate dehydrogenase
- JAK:
-
Janus kinase
- KIT:
-
Mast/stem cell growth factor receptor
- lnRNA:
-
Long noncoding RNA
- LSD1/KDM1A:
-
Lysine-specific demethylase 1
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
Mitogen-activated protein kinase kinase
- MELK:
-
Maternal embryonic leucine-zipper kinases
- MGMT:
-
O6-methylguanine-DNA methyltransferase
- MKP1:
-
MAPK phosphatase 1
- MTD:
-
Maximum tolerated dose
- mTOR:
-
Mammalian target of rapamycin
- NF1:
-
Neurofibromin 1
- NuRD:
-
Nucleosome remodeling and deacetylase
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- P-TEFb:
-
Positive transcription elongation factor b
- PcG:
-
Polycomb-group
- PD:
-
Pharmacodynamic
- PDGFRA:
-
Platelet-derived growth factor receptor
- PFS:
-
Progression-free survival
- pHGG:
-
Pediatric high-grade glioma
- PI3K:
-
Phosphatidylinositol-3 kinase
- PK:
-
Pharmacokinetic
- PRC2:
-
Polycomb repressive complex
- PRMT:
-
Protein arginine methyltransferase
- PTEN:
-
Phosphatase and tensin homolog
- RAS:
-
Rat sarcoma virus
- RB:
-
Retinoblastoma
- RCC1:
-
Regulator of chromosome condensation 1
- RT:
-
Radiation therapy
- RTKs:
-
Receptor tyrosine kinases
- S6K1:
-
S6 kinase beta-1
- SAH:
-
S-adenosyl homocysteine
- SAM:
-
S-adenosyl-L-methionine
- STAT:
-
Signal transducer and activator of transcription
- VEGFR:
-
Vascular endothelial growth factor receptor
- WHO:
-
World Health Organization
References
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–63.
Aggarwal P, Luo W, Pehlivan KC, Hoang H, Rajappa P, Cripe TP, et al. Pediatric versus adult high grade glioma: immunotherapeutic and genomic considerations. Front Immunol. 2022;13:1038096.
Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014;14(2):92–107.
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–37.
Marra JS, Mendes GP, Yoshinari GH, da Silva Guimarães F, Mazin SC, de Oliveira HF. Survival after radiation therapy for high-grade glioma. Rep Pract Oncol Radiother. 2019;24(1):35–40.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96.
Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: state of the art and future directions. CA Cancer J Clin. 2020;70(4):299–312.
Sakthikumar S, Roy A, Haseeb L, Pettersson ME, Sundström E, Marinescu VD, et al. Whole-genome sequencing of glioblastoma reveals enrichment of non-coding constraint mutations in known and novel genes. Genome Biol. 2020;21(1):127.
Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547(7663):311–7.
Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014;46(5):451–6.
Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol. 2011;29(30):3999–4006.
Wang H, Xu T, Jiang Y, Xu H, Yan Y, Fu D, et al. The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia. 2015;17(3):239–55.
Lu Y, Chan YT, Tan HY, Li S, Wang N, Feng Y. Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol Cancer. 2020;19(1):79.
Muntean AG, Hess JL. Epigenetic dysregulation in cancer. Am J Pathol. 2009;175(4):1353–61.
Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019;4:62.
Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity (Edinb). 2010;105(1):4–13.
Pan PC, Magge RS. Mechanisms of EGFR resistance in glioblastoma. Int J Mol Sci. 2020;21(22).
Hottinger A, Sanson M, Moyal E, Delord J-P, Micheli RD, Rezai K, et al. Dose optimization of MK-8628 (OTX015), a small molecule inhibitor of bromodomain and extra-terminal (BET) proteins, in patients (pts) with recurrent glioblastoma (GB). J Clin Oncol. 2016;34(15):e14123.
Monje M, Cooney T, Glod J, Huang J, Peer CJ, Faury D et al. A phase I trial of panobinostat in children with diffuse intrinsic pontine glioma: a report from the Pediatric Brain Tumor Consortium (PBTC-047). Neuro Oncol. 2023.
Lassman AB, Sepúlveda-Sánchez JM, Cloughesy TF, Gil-Gil MJ, Puduvalli VK, Raizer JJ, et al. Infigratinib in patients with recurrent gliomas and FGFR alterations: a multicenter phase II study. Clin Cancer Res. 2022;28(11):2270–7.
Tinkle CL, Broniscer A, Chiang J, Campagne O, Huang J, Orr BA, et al. Phase I study using crenolanib to target PDGFR kinase in children and young adults with newly diagnosed DIPG or recurrent high-grade glioma, including DIPG. Neurooncol Adv. 2021;3(1):vdab179.
Hu H, Mu Q, Bao Z, Chen Y, Liu Y, Chen J, et al. Mutational landscape of secondary glioblastoma guides MET-targeted trial in brain tumor. Cell. 2018;175(6):1665–78e18.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
Zoghbi HY, Beaudet AL. Epigenetics and human disease. Cold Spring Harb Perspect Biol. 2016;8(2):a019497.
Was H, Krol SK, Rotili D, Mai A, Wojtas B, Kaminska B, et al. Histone deacetylase inhibitors exert anti-tumor effects on human adherent and stem-like glioma cells. Clin Epigenetics. 2019;11(1):11.
Pastori C, Daniel M, Penas C, Volmar CH, Johnstone AL, Brothers SP, et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics. 2014;9(4):611–20.
Yan F, Alinari L, Lustberg ME, Martin LK, Cordero-Nieves HM, Banasavadi-Siddegowda Y, et al. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 2014;74(6):1752–65.
Gu X, He M, Lebedev T, Lin CH, Hua ZY, Zheng YG, et al. PRMT1 is an important factor for medulloblastoma cell proliferation and survival. Biochem Biophys Rep. 2022;32:101364.
Wang S, Tan X, Yang B, Yin B, Yuan J, Qiang B, et al. The role of protein arginine-methyltransferase 1 in gliomagenesis. BMB Rep. 2012;45(8):470–5.
Dong F, Li Q, Yang C, Huo D, Wang X, Ai C, et al. PRMT2 links histone H3R8 asymmetric dimethylation to oncogenic activation and tumorigenesis of glioblastoma. Nat Commun. 2018;9(1):4552.
Chen YN, Hou SQ, Jiang R, Sun JL, Cheng CD, Qian ZR. EZH2 is a potential prognostic predictor of glioma. J Cell Mol Med. 2021;25(2):925–36.
Zhang J, Chen L, Han L, Shi Z, Pu P, Kang C. EZH2 is a negative prognostic factor and exhibits pro-oncogenic activity in glioblastoma. Cancer Lett. 2015;356(2 Pt B):929–36.
Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer. 2012;106(2):243–7.
Singh MM, Manton CA, Bhat KP, Tsai WW, Aldape K, Barton MC, et al. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro Oncol. 2011;13(8):894–903.
Sareddy GR, Viswanadhapalli S, Surapaneni P, Suzuki T, Brenner A, Vadlamudi RK. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene. 2017;36(17):2423–34.
Stitzlein LM, Gangadharan A, Walsh LM, Nam D, Espejo AB, Singh MM, et al. Comparison of pharmacological inhibitors of lysine-specific demethylase 1 in glioblastoma stem cells reveals inhibitor-specific efficacy profiles. Front Neurol. 2023;14:1112207.
Ding J, Zhang ZM, Xia Y, Liao GQ, Pan Y, Liu S, et al. LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br J Cancer. 2013;109(4):994–1003.
Nagasawa S, Sedukhina AS, Nakagawa Y, Maeda I, Kubota M, Ohnuma S, et al. LSD1 overexpression is associated with poor prognosis in basal-like breast cancer, and sensitivity to PARP inhibition. PLoS ONE. 2015;10(2):e0118002.
Zhao ZK, Yu HF, Wang DR, Dong P, Chen L, Wu WG, et al. Overexpression of lysine specific demethylase 1 predicts worse prognosis in primary hepatocellular carcinoma patients. World J Gastroenterol. 2012;18(45):6651–6.
Jie D, Zhongmin Z, Guoqing L, Sheng L, Yi Z, Jing W, et al. Positive expression of LSD1 and negative expression of E-cadherin correlate with metastasis and poor prognosis of colon cancer. Dig Dis Sci. 2013;58(6):1581–9.
Yu Y, Wang B, Zhang K, Lei Z, Guo Y, Xiao H, et al. High expression of lysine-specific demethylase 1 correlates with poor prognosis of patients with esophageal squamous cell carcinoma. Biochem Biophys Res Commun. 2013;437(2):192–8.
Lee MG, Wynder C, Bochar DA, Hakimi MA, Cooch N, Shiekhattar R. Functional interplay between histone demethylase and deacetylase enzymes. Mol Cell Biol. 2006;26(17):6395–402.
Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19(6):857–64.
You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc Natl Acad Sci U S A. 2001;98(4):1454–8.
Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci U S A. 2002;99(11):7420–5.
Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138(4):660–72.
Basta J, Rauchman M. The nucleosome remodeling and deacetylase complex in development and disease. Transl Res. 2015;165(1):36–47.
Yang Y, Li G. Post-translational modifications of PRC2: signals directing its activity. Epigenetics Chromatin. 2020;13(1):47.
Friesen WJ, Paushkin S, Wyce A, Massenet S, Pesiridis GS, Van Duyne G, et al. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified sm proteins. Mol Cell Biol. 2001;21(24):8289–300.
Liao Y, Luo Z, Lin Y, Chen H, Chen T, Xu L, et al. PRMT3 drives glioblastoma progression by enhancing HIF1A and glycolytic metabolism. Cell Death Dis. 2022;13(11):943.
Huang T, Yang Y, Song X, Wan X, Wu B, Sastry N, et al. PRMT6 methylation of RCC1 regulates mitosis, tumorigenicity, and radiation response of glioblastoma stem cells. Mol Cell. 2021;81(6):1276–91e9.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231–51.
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–22.
Wenger A, Carén H. Methylation profiling in diffuse gliomas: diagnostic value and considerations. Cancers (Basel). 2022;14(22).
Malta TM, de Souza CF, Sabedot TS, Silva TC, Mosella MS, Kalkanis SN, et al. Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol. 2018;20(5):608–20.
Chai RC, Yan H, An SY, Pang B, Chen HY, Mu QH, et al. Genomic profiling and prognostic factors of H3 K27M-mutant spinal cord diffuse glioma. Brain Pathol. 2023;33(4):e13153.
Della Monica R, Cuomo M, Buonaiuto M, Costabile D, Franca RA, Del Caro B. M, MGMT and whole-genome DNA methylation impacts on diagnosis, prognosis and therapy of Glioblastoma multiforme. Int J Mol Sci. 2022;23(13).
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–74.
Kline C, Jain P, Kilburn L, Bonner ER, Gupta N, Crawford JR, et al. Upfront biology-guided therapy in diffuse intrinsic pontine glioma: therapeutic, molecular, and biomarker outcomes from PNOC003. Clin Cancer Res. 2022;28(18):3965–78.
Aldaz P, Arozarena I. Tyrosine kinase inhibitors in adult glioblastoma: an (un)closed chapter? Cancers (Basel). 2021;13(22).
Qin A, Musket A, Musich PR, Schweitzer JB, Xie Q. Receptor tyrosine kinases as druggable targets in glioblastoma: do signaling pathways matter? Neurooncol Adv. 2021;3(1):vdab133.
Yang K, Wu Z, Zhang H, Zhang N, Wu W, Wang Z, et al. Glioma targeted therapy: insight into future of molecular approaches. Mol Cancer. 2022;21(1):39.
Fabro F, Lamfers MLM, Leenstra S. Advancements, challenges, and future directions in tackling glioblastoma resistance to small kinase inhibitors. Cancers (Basel). 2022;14(3).
Geoerger B, Hargrave D, Thomas F, Ndiaye A, Frappaz D, Andreiuolo F, et al. Innovative therapies for children with cancer pediatric phase I study of erlotinib in brainstem glioma and relapsing/refractory brain tumors. Neuro Oncol. 2011;13(1):109–18.
Kesavabhotla K, Schlaff CD, Shin B, Mubita L, Kaplan R, Tsiouris AJ, et al. Phase I/II study of oral erlotinib for treatment of relapsed/refractory glioblastoma multiforme and anaplastic astrocytoma. J Exp Ther Oncol. 2012;10(1):71–81.
Qaddoumi I, Kocak M, Pai Panandiker AS, Armstrong GT, Wetmore C, Crawford JR, et al. Phase II trial of erlotinib during and after radiotherapy in children with newly diagnosed high-grade gliomas. Front Oncol. 2014;4:67.
van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27(8):1268–74.
Chakravarti A, Wang M, Robins HI, Lautenschlaeger T, Curran WJ, Brachman DG, et al. RTOG 0211: a phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phys. 2013;85(5):1206–11.
Geyer JR, Stewart CF, Kocak M, Broniscer A, Phillips P, Douglas JG, et al. A phase I and biology study of gefitinib and radiation in children with newly diagnosed brain stem gliomas or supratentorial malignant gliomas. Eur J Cancer. 2010;46(18):3287–93.
Uhm JH, Ballman KV, Wu W, Giannini C, Krauss JC, Buckner JC, et al. Phase II evaluation of gefitinib in patients with newly diagnosed grade 4 astrocytoma: Mayo/North central cancer treatment group study N0074. Int J Radiat Oncol Biol Phys. 2011;80(2):347–53.
Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MC, Delorenzi M, et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib–a phase II trial. Mol Cancer Ther. 2011;10(6):1102–12.
Sepúlveda-Sánchez JM, Vaz M, Balañá C, Gil-Gil M, Reynés G, Gallego Ó, et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol. 2017;19(11):1522–31.
Wang Y, Liang D, Chen J, Chen H, Fan R, Gao Y, et al. Targeted therapy with anlotinib for a patient with an oncogenic FGFR3-TACC3 fusion and recurrent glioblastoma. Oncologist. 2021;26(3):173–7.
Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27(12):2052–8.
Mueller S, Kline C, Stoller S, Lundy S, Christopher L, Reddy AT et al. PNOC015: repeated convection enhanced delivery (CED) of MTX110 (aqueous panobinostat) in children with newly diagnosed diffuse intrinsic pontine glioma (DIPG). Neuro Oncol. 2023.
Berenguer-Daizé C, Astorgues-Xerri L, Odore E, Cayol M, Cvitkovic E, Noel K, et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int J Cancer. 2016;139(9):2047–55.
Bukowinski A, Chang B, Reid JM, Liu X, Minard CG, Trepel JB, et al. A phase 1 study of entinostat in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: trial ADVL1513, Pediatric Early Phase-Clinical Trial Network (PEP-CTN). Pediatr Blood Cancer. 2021;68(4):e28892.
Park SY, Kim JS. A short guide to histone deacetylases including recent progress on class II enzymes. Exp Mol Med. 2020;52(2):204–12.
Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18(7).
Piao J, Chen L, Quan T, Li L, Quan C, Piao Y, et al. Superior efficacy of co-treatment with the dual PI3K/mTOR inhibitor BEZ235 and histone deacetylase inhibitor trichostatin A against NSCLC. Oncotarget. 2016;7(37):60169–80.
Rahmani M, Aust MM, Benson EC, Wallace L, Friedberg J, Grant S. PI3K/mTOR inhibition markedly potentiates HDAC inhibitor activity in NHL cells through BIM- and MCL-1-dependent mechanisms in vitro and in vivo. Clin Cancer Res. 2014;20(18):4849–60.
Meng W, Wang B, Mao W, Wang J, Zhao Y, Li Q, et al. Enhanced efficacy of histone deacetylase inhibitor panobinostat combined with dual PI3K/mTOR inhibitor BEZ235 against glioblastoma. Nagoya J Med Sci. 2019;81(1):93–102.
Pal S, Kozono D, Yang X, Fendler W, Fitts W, Ni J, et al. Dual HDAC and PI3K inhibition abrogates NFκB- and FOXM1-mediated DNA damage response to radiosensitize pediatric high-grade gliomas. Cancer Res. 2018;78(14):4007–21.
Qian C, Lai CJ, Bao R, Wang DG, Wang J, Xu GX, et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin Cancer Res. 2012;18(15):4104–13.
Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA, et al. HDAC and PI3K antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell. 2016;29(3):311–23.
da Cunha Jaeger M, Ghisleni EC, Cardoso PS, Siniglaglia M, Falcon T, Brunetto AT, et al. HDAC and MAPK/ERK inhibitors cooperate to reduce viability and stemness in medulloblastoma. J Mol Neurosci. 2020;70(6):981–92.
Li JY, Wang H, May S, Song X, Fueyo J, Fuller GN. Constitutive activation of c-Jun N-terminal kinase correlates with histologic grade and EGFR expression in diffuse gliomas. J Neurooncol. 2008;88(1):11–7.
Yoon CH, Kim MJ, Kim RK, Lim EJ, Choi KS, An S, et al. c-Jun N-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene. 2012;31(44):4655–66.
Huang Z, Xia Y, Hu K, Zeng S, Wu L, Liu S, et al. Histone deacetylase 6 promotes growth of glioblastoma through the MKK7/JNK/c-Jun signaling pathway. J Neurochem. 2020;152(2):221–34.
Arrizabalaga O, Moreno-Cugnon L, Auzmendi-Iriarte J, Aldaz P, Ibanez de Caceres I, Garros-Regulez L, et al. High expression of MKP1/DUSP1 counteracts glioma stem cell activity and mediates HDAC inhibitor response. Oncogenesis. 2017;6(12):401.
Meel MH, de Gooijer MC, Metselaar DS, Sewing ACP, Zwaan K, Waranecki P, et al. Combined therapy of AXL and HDAC inhibition reverses mesenchymal transition in diffuse intrinsic pontine glioma. Clin Cancer Res. 2020;26(13):3319–32.
Buendia Duque M, Pinheiro KV, Thomaz A, da Silva CA, Freire NH, Brunetto AT, et al. Combined inhibition of HDAC and EGFR reduces viability and proliferation and enhances STAT3 mRNA expression in glioblastoma cells. J Mol Neurosci. 2019;68(1):49–57.
Luwor RB, Stylli SS, Kaye AH. The role of Stat3 in glioblastoma multiforme. J Clin Neurosci. 2013;20(7):907–11.
Liffers K, Kolbe K, Westphal M, Lamszus K, Schulte A. Histone deacetylase inhibitors resensitize EGFR/EGFRvIII-overexpressing, erlotinib-resistant glioblastoma cells to tyrosine kinase inhibition. Target Oncol. 2016;11(1):29–40.
Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282(18):13141–5.
Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399(6735):491–6.
Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci U S A. 2003;100(15):8758–63.
Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19(4):535–45.
Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19(4):523–34.
Denis GV, Vaziri C, Guo N, Faller DV. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ. 2000;11(8):417–24.
Kanno T, Kanno Y, Siegel RM, Jang MK, Lenardo MJ, Ozato K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol Cell. 2004;13(1):33–43.
Shorstova T, Foulkes WD, Witcher M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br J Cancer. 2021;124(9):1478–90.
Iniguez AB, Alexe G, Wang EJ, Roti G, Patel S, Chen L, et al. Resistance to epigenetic-targeted therapy engenders tumor cell vulnerabilities associated with enhancer remodeling. Cancer Cell. 2018;34(6):922–38e7.
Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, et al. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P-TEFb complex. Cancer Discov. 2017;7(3):302–21.
Funck-Brentano E, Vizlin-Hodzic D, Nilsson JA, Nilsson LM. BET bromodomain inhibitor HMBA synergizes with MEK inhibition in treatment of malignant glioma. Epigenetics. 2021;16(1):54–63.
Wen N, Guo B, Zheng H, Xu L, Liang H, Wang Q, et al. Bromodomain inhibitor jq1 induces cell cycle arrest and apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling pathway. Int J Oncol. 2019;55(4):879–95.
Čančer M, Drews LF, Bengtsson J, Bolin S, Rosén G, Westermark B, et al. BET and aurora kinase A inhibitors synergize against MYCN-positive human glioblastoma cells. Cell Death Dis. 2019;10(12):881.
Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013;70(4):661–87.
Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–17.
Bolin S, Borgenvik A, Persson CU, Sundström A, Qi J, Bradner JE, et al. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene. 2018;37(21):2850–62.
Hydbring P, Bahram F, Su Y, Tronnersjö S, Högstrand K, von der Lehr N, et al. Phosphorylation by Cdk2 is required for Myc to repress ras-induced senescence in cotransformation. Proc Natl Acad Sci U S A. 2010;107(1):58–63.
Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The Cdk1 complex plays a prime role in regulating N-myc phosphorylation and turnover in neural precursors. Dev Cell. 2005;9(3):327–38.
Tewary SK, Zheng YG, Ho MC. Protein arginine methyltransferases: insights into the enzyme structure and mechanism at the atomic level. Cell Mol Life Sci. 2019;76(15):2917–32.
Guccione E, Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol. 2019;20(10):642–57.
Chen Z, Gan J, Wei Z, Zhang M, Du Y, Xu C, et al. The emerging role of PRMT6 in cancer. Front Oncol. 2022;12:841381.
Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci. 2015;72(11):2041–59.
Favia A, Salvatori L, Nanni S, Iwamoto-Stohl LK, Valente S, Mai A, et al. The protein arginine methyltransferases 1 and 5 affect Myc properties in glioblastoma stem cells. Sci Rep. 2019;9(1):15925.
Dong J, Duan J, Hui Z, Garrido C, Deng Z, Xie T, et al. An updated patent review of protein arginine N-methyltransferase inhibitors (2019–2022). Expert Opin Ther Pat. 2022;32(12):1185–205.
Banasavadi-Siddegowda YK, Russell L, Frair E, Karkhanis VA, Relation T, Yoo JY, et al. PRMT5-PTEN molecular pathway regulates senescence and self-renewal of primary glioblastoma neurosphere cells. Oncogene. 2017;36(2):263–74.
Honda M, Nakashima K, Katada S. PRMT1 regulates astrocytic differentiation of embryonic neural stem/precursor cells. J Neurochem. 2017;142(6):901–7.
Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. 2020;13(1):104.
Satijn DP, Gunster MJ, van der Vlag J, Hamer KM, Schul W, Alkema MJ, et al. RING1 is associated with the polycomb group protein complex and acts as a transcriptional repressor. Mol Cell Biol. 1997;17(7):4105–13.
Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of Zeste protein. Genes Dev. 2002;16(22):2893–905.
Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–9.
Dhar S, Gadd S, Patel P, Vaynshteyn J, Raju GP, Hashizume R, et al. A tumor suppressor role for EZH2 in diffuse midline glioma pathogenesis. Acta Neuropathol Commun. 2022;10(1):47.
Mohammad F, Weissmann S, Leblanc B, Pandey DP, Højfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. 2017;23(4):483–92.
Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647(1–2):21–9.
Yang R, Wang M, Zhang G, Bao Y, Wu Y, Li X, et al. E2F7-EZH2 axis regulates PTEN/AKT/mTOR signalling and glioblastoma progression. Br J Cancer. 2020;123(9):1445–55.
Ganguly R, Mohyeldin A, Thiel J, Kornblum HI, Beullens M, Nakano I. MELK-a conserved kinase: functions, signaling, cancer, and controversy. Clin Transl Med. 2015;4:11.
Liu H, Sun Y, Qi X, Gordon RE, O’Brien JA, Yuan H, et al. EZH2 phosphorylation promotes self-renewal of glioma stem-like cells through NF-κB methylation. Front Oncol. 2019;9:641.
Speers C, Zhao SG, Kothari V, Santola A, Liu M, Wilder-Romans K, et al. Maternal embryonic leucine zipper kinase (MELK) as a novel mediator and biomarker of radioresistance in human breast cancer. Clin Cancer Res. 2016;22(23):5864–75.
Liu H, Sun Q, Sun Y, Zhang J, Yuan H, Pang S, et al. MELK and EZH2 cooperate to regulate medulloblastoma cancer stem-like cell proliferation and differentiation. Mol Cancer Res. 2017;15(9):1275–86.
Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23(6):839–52.
Jin W. Role of JAK/STAT3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial-mesenchymal transition. Cells. 2020;9(1).
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–53.
Karakaidos P, Verigos J, Magklara A. LSD1/KDM1A, a gate-keeper of cancer stemness and a promising therapeutic target. Cancers (Basel). 2019;11(12).
Kim D, Kim KI, Baek SH. Roles of lysine-specific demethylase 1 (LSD1) in homeostasis and diseases. J Biomed Sci. 2021;28(1):41.
Bailey CP, Figueroa M, Gangadharan A, Yang Y, Romero MM, Kennis BA, et al. Pharmacologic inhibition of lysine-specific demethylase 1 as a therapeutic and immune-sensitization strategy in pediatric high-grade glioma. Neuro Oncol. 2020;22(9):1302–14.
Alejo S, Palacios BE, Venkata PP, He Y, Li W, Johnson JD, et al. Lysine-specific histone demethylase 1A (KDM1A/LSD1) inhibition attenuates DNA double-strand break repair and augments the efficacy of temozolomide in glioblastoma. Neuro Oncol. 2023;25(7):1249–61.
Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, et al. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry. 2007;46(27):8058–65.
Fang Y, Liao G, Yu B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J Hematol Oncol. 2019;12(1):129.
Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015;28(1):57–69.
Binda C, Valente S, Romanenghi M, Pilotto S, Cirilli R, Karytinos A, et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J Am Chem Soc. 2010;132(19):6827–33.
Bailey CP, Figueroa M, Gangadharan A, Lee DA, Chandra J. Scaffolding LSD1 inhibitors impair NK cell metabolism and cytotoxic function through depletion of glutathione. Front Immunol. 2020;11:2196.
Maes T, Mascaró C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. 2018;33(3):495–511e12.
Johnston G, Ramsey HE, Liu Q, Wang J, Stengel KR, Sampathi S, et al. Nascent transcript and single-cell RNA-seq analysis defines the mechanism of action of the LSD1 inhibitor INCB059872 in myeloid leukemia. Gene. 2020;752:144758.
Sacilotto N, Dessanti P, Lufino MMP, Ortega A, Rodríguez-Gimeno A, Salas J, et al. Comprehensive in vitro characterization of the LSD1 small molecule inhibitor class in oncology. ACS Pharmacol Transl Sci. 2021;4(6):1818–34.
Sehrawat A, Gao L, Wang Y, Bankhead A, McWeeney SK, King CJ, et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc Natl Acad Sci U S A. 2018;115(18):E4179–E88.
Kanouni T, Severin C, Cho RW, Yuen NY, Xu J, Shi L, et al. Discovery of CC-90011: a potent and selective reversible inhibitor of Lysine Specific Demethylase 1 (LSD1). J Med Chem. 2020;63(23):14522–9.
Saccà CD, Gorini F, Ambrosio S, Amente S, Faicchia D, Matarese G, et al. Inhibition of lysine-specific demethylase LSD1 induces senescence in glioblastoma cells through a HIF-1α-dependent pathway. Biochim Biophys Acta Gene Regul Mech. 2019;1862(5):535–46.
Zhao J, Jin W, Yi K, Wang Q, Zhou J, Tan Y, et al. Combination LSD1 and HOTAIR-EZH2 inhibition disrupts cell cycle processes and induces apoptosis in glioblastoma cells. Pharmacol Res. 2021;171:105764.
Eich ML, Athar M, Ferguson JE, Varambally S. EZH2-targeted therapies in cancer: hype or a reality. Cancer Res. 2020;80(24):5449–58.
Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell. 2018;174(3):549–63e19.
Wang H, Liu G, Jin X, Song S, Chen S, Zhou P, et al. BET inhibitor JQ1 enhances anti-tumor immunity and synergizes with PD-1 blockade in CRC. J Cancer. 2022;13(7):2126–37.
Zhou L, Mudianto T, Ma X, Riley R, Uppaluri R. Targeting EZH2 enhances antigen presentation, antitumor immunity, and circumvents Anti-PD-1 resistance in head and neck cancer. Clin Cancer Res. 2020;26(1):290–300.
Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015;21(7):1639–51.
Galbán S, Apfelbaum AA, Espinoza C, Heist K, Haley H, Bedi K, et al. A bifunctional MAPK/PI3K antagonist for inhibition of tumor growth and metastasis. Mol Cancer Ther. 2017;16(11):2340–50.
Yan C, Saleh N, Yang J, Nebhan CA, Vilgelm AE, Reddy EP, et al. Novel induction of CD40 expression by tumor cells with RAS/RAF/PI3K pathway inhibition augments response to checkpoint blockade. Mol Cancer. 2021;20(1):85.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Institutes of Health (R21 NS093387, P50 CA127001, TL1 TR003169, R61/R33NS111058 and P30 CA016672), by the Cancer Prevention and Research Institute of Texas (DP160014) and by the Department of Defense (W81XWH2110728).
Author information
Authors and Affiliations
Contributions
Conception and design of the review article: L.S., R.D., and J.C. Interpretation of relevant literature and manuscript writing: L.S., J.A., E.S., R.D., and J.C.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare none.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Stitzlein, L.M., Adams, J.T., Stitzlein, E.N. et al. Current and future therapeutic strategies for high-grade gliomas leveraging the interplay between epigenetic regulators and kinase signaling networks. J Exp Clin Cancer Res 43, 12 (2024). https://doi.org/10.1186/s13046-023-02923-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-023-02923-7