
Abstract
Immunotherapy has emerged as a promising strategy for boosting antitumoral immunity. Blockade of immune checkpoints (ICs), which regulate the activity of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells has proven clinical benefits. Antibodies targeting CTLA-4, PD-1, and PD-L1 are IC-blockade drugs approved for the treatment of various solid and hematological malignancies. However, a large subset of patients does not respond to current anti-IC immunotherapy. An integrative understanding of tumor-immune infiltrate, and IC expression and function in immune cell populations is fundamental to the design of effective therapies. The simultaneous blockade of newly identified ICs, as well as of previously described ICs, could improve antitumor response. We review the potential for novel combinatory blockade strategies as antitumoral therapy, and their effects on immune cells expressing the targeted ICs. Preclinical evidence and clinical trials involving the blockade of the various ICs are reported. We finally discuss the rationale of IC co-blockade strategy with respect to its downstream signaling in order to improve effective antitumoral immunity and prevent an increased risk of immune-related adverse events (irAEs).
Similar content being viewed by others

Background
Tumor growth involves a complex interplay between tumor, immune, and stromal cells, and extracellular matrix components. In the last decade, the relevance of the tumor-immune microenvironment and its direct impact on patients’ clinical outcome has become widely acknowledged [1]. The host immune system is primed to identify and kill malignantly transformed cells to prevent tumorigenesis and tumor growth. Cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells are immune cell populations responsible for immunosurveillance and, when required, for eliminating target cells. Tumor cells can be identified by CTLs as altered cells by the expression of neoantigens displayed by the major histocompatibility complex (MHC) [2]. Tumor cells expressing low levels of MHC molecules can become invisible to T cells and may escape T-cell immune control. In these cases, NK cells can identify and target cancer cells that lack MHC expression. However, tumor immune evasion, defined as the ability of tumor cells to evade the host’s immune response, happens during tumorigenesis and tumor growth. Multiple activating and inhibiting mechanisms tightly regulate the effector function of CTLs and NK cells to prevent autoimmune events and preserve tissue homeostasis. In this regard, immune checkpoints (ICs) are signaling pathways that modulate the immune response. CTLs and NK cells can express IC receptors that, when interacting with IC ligands, activate IC signaling pathways, blocking their cytotoxic activity [3]. These IC ligands can be expressed by immunosuppressive cells, including M2-like macrophages, myeloid-derived suppressor cells (MDSCs), and T-regulatory (Treg) cells, as well as cancer cells. The continuous interaction between IC ligands and their respective IC receptors expressed by CTLs and NK, help produce a dysfunctional state in these immune cells known as exhaustion. Tumors avoid antitumoral immunity by upregulating the expression of IC ligands and recruiting immunosuppressive cells, which give rise to an immunosuppressive tumor microenvironment (TME). Tumors with a strong immunosuppressive TME have been associated with impaired immune cytotoxicity, are more aggressive, and have a poor prognosis [4].
Immunotherapy is based on stimulating the host immune system to elicit a response against cancer cells. Pharmacological blockade of the interaction between ICs and their ligands with IC inhibitors has been identified as a promising strategy for restoring immune cytotoxic activity [5]. In recent years, T cell-mediated cytolysis has been the focus of major efforts to modulate antitumor cytotoxicity as therapy. However, NK cells are another immunotherapy target for boosting antitumor response. Importantly, the IC receptors expressed by NK and T cells can be expressed by other immune cell populations, and their blockade may also modulate the function of those populations. Here, we describe the various IC receptors expressed by T, NK, and other immune cells, and their biological function. We also analyze the antitumoral activity of IC blockade therapies, as single agents or in combination, for cancer treatment.
Main text
Effector cells: cytotoxic T lymphocytes and natural killer cells
CTLs and NK cells are the two major immune populations that are able to eliminate malignant cells. CTLs participate in the adaptive immune response while NK cells are part of the innate immune system. Cytotoxicity arises by two pathways: Perforin/Granzyme B/Granulysin-related lysis, and death receptor-induced apoptosis. Although CTLs and NK cells act in a mechanistically similar fashion, the regulation of the activity of these immune cells, and the recognition of the targets differ. CTL cytotoxicity is acquired after T cell activation upon antigen presentation by antigen presenting cells (APCs) —mainly dendritic cells (DCs) — whereas NK cells lyse target cells without antigen presentation [6]. When activated, CTLs and NK cells both secrete interferon (IFN)-γ and tumor necrosis factor (TNF)-α, which stimulate a pro-inflammatory immune response. Antitumoral effects have been extensively ascribed to these two immune cell populations, highlighting the relevance of comprehensively understanding the activation and inhibition mechanisms that regulate their cytotoxic activity against cancer cells by pharmacological strategies.
CTLs are defined as activated effector CD3+ CD8+ T lymphocytes and recognize target cells via the interaction between polyclonally rearranged T-cell receptor (TCR) with a peptide/MHC class I complex. Naïve CD8+ T cells interact with APCs and, upon the correct presentation of the peptide-MHC class I complex, TCR signaling causes the formation of a stabilization complex between T cells and the APC. To become fully activated, the co-stimulatory receptor CD28 must interact with its ligands, CD80 (B7.1) and CD86 (B7.2). The activity of T cells is determined by the balance of positive and negative signals from co-activator and co-inhibitory receptors when they recognize their target. To eliminate target cells, CTLs produce a stabilization complex, after which, lytic granules are secreted. Perforin forms pores in the extracellular membrane of the target cells, allowing Granzyme B and Granulysin to enter the cytosol and induce apoptosis, while membrane-bound FasL binds to its receptor Fas, inducing apoptosis in an independent manner [6].
Human NK cells are phenotypically defined as CD3− CD56+ lymphocytes. Two functionally distinct subsets of NK cells have been defined on the basis of their levels of expression of CD56 and CD16 (also known as Fcγ receptor III). NK cells with high-density expression of CD56 (CD56bright) and CD16− are mostly found in lymph nodes and have a great ability to release immune-modulating cytokines such as IFN-γ and TNF-α. On the other hand, low-density expression of CD56 (CD56dim) CD16+ NK cells mostly occurs in peripheral blood, where it presents a more cytotoxic phenotype characterized by high levels of Perforin and Granzyme B expression [7]. Cytotoxic NK cell activity is independent of foreign antigens presented by MHC molecules. The balance between activation and inhibition signals, which NK cells sense through multiple innate receptors, allows the cells to respond to alterations such as cellular stress, cellular transformation, and malignancy. When activated, NK cells form a stabilization complex similarly to CTLs and release cytotoxic granules.
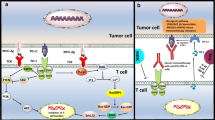
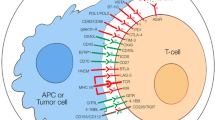
CTL and NK-cell activity is tightly controlled to preserve antigenic self-tolerance. Autoreactive T-cell clones are eliminated in the thymus by a process known as central tolerance. Also, a peripheral regulation of the cytotoxic response is essential to avoid inappropriate responses to self-antigens. The release of immunosuppressive molecules by M2-like macrophages and Treg cells plays a key role in establishing immune self-tolerance [8]. Activated CTLs and NK cells upregulate the expression of multiple coinhibitory receptors, known as ICs receptors, which downregulate their cytotoxic activity when binding to their ligands, ensuring the precise regulation of their effector function (Fig. 1). Although self-tolerance mechanisms are tightly regulated, T-cell exhaustion occurs and is often observed in tumors and chronic infections [9]. NK cells can present a similar exhausted phenotype that is characterized by stronger expression of coinhibitory receptors and weaker expression of activating receptors [10]. Tumor-infiltrating lymphocytes (TILs) and tumor-infiltrating NK cells exhibit enhanced expression of IC receptors [5, 10]. This has boosted interest in understanding how these coinhibitory receptors function in order to therapeutically block them. The best characterized IC receptors are the cytotoxic T-lymphocyte-associated molecule 4 (CTLA-4) and the programmed cell death protein 1 (PD-1), but many other ICs play key central roles in controlling CTL and NK cell effector functions (Table 1).

Coinhibitory receptors expressed by cytotoxic T lymphocytes (A) and natural killer cells (B) and their ligands. Cytotoxic T lymphocytes and natural killer cells can express multiple coinhibitory receptors, known as immune checkpoints, that produce downstream inhibitory signals when activated upon binding to their ligands. Note that not all ICs are expressed simultaneously by cytotoxic T lymphocytes or NK cells
Approved IC inhibitors for cancer treatment
IC signaling inhibition can be exploited as a therapeutic strategy for treating cancer [11, 12]. CTLA-4 and PD-1 were the first ICs to be blocked in preclinical models addressing cancer therapy. Several IC blockade antibodies were approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), and are currently used to treat a variety of tumor types (Table 2). However, we are far from fully exploiting the potential of this therapeutic strategy. CTLs and NK cells may be modulated by anti-IC drugs by direct mechanisms when the effector cells express the targeted IC, and/or by indirect mechanisms, when blockade of the IC alters the immunomodulatory functions of other immune cell populations. While the currently approved IC blockade drugs produce a good clinical response in some patients, a percentage of patients show short-duration response or even no response at all (Table 3). Furthermore, late relapses occur in subsets of patients who initially responded to IC blockade therapy [13], which has sparked interest in targeting other coinhibitory receptors to find new therapeutic combinations to treat non-responsive tumors.
CTLA-4 blockade
CTLA-4, which binds CD80 and CD86 ligands, was the first IC to be described in T cells [11]. CD80 and CD86 are also ligands of the T cell co-activator receptor CD28. CTLA-4 has a high sequence similarity to that of CD28 and competes, with higher affinity, to bind CD80 and CD86. CTLA-4 regulates early stages of CD8+ T cell activation, ensuring tolerance of self-antigens in the lymph nodes. Mechanistically, CTLA-4 indirectly limits CD28 signaling by capturing and sequestering CD80 and CD86 from ligand-expressing cells through a process named trans-endocytosis [14]. Genetic ablation of CTLA-4 increases early lethality in mice due to lymphoproliferation [5]. Blockade experiments demonstrated that in vitro and in vivo anti-CTLA-4 treatment enhance T-cell proliferation [15]. CTLA-4 is also expressed by Treg cells, and its signaling enhances their suppressive functions [16]. In addition, CD4+ T cells expressing CTLA-4 exhibit lower rates of proliferation [17]. The role of CTLA-4 in NK cells remains to be elucidated. While some studies indicated that CTLA-4 is expressed in mouse and human NK cells, and that it regulates their effector functions, others reported that human NK cells are not affected by the CD28/CTLA-4 axis [18].
In vivo administration of anti-CTLA-4 antibody increases antitumor immunity, causing pre-established tumors to be rejected [11]. Further experiments demonstrated that blockade of CTLA-4 stimulates the CD8+ T cell cytotoxic response against tumor cells [5]. Importantly, the complete antitumoral response provoked by CTLA-4 blockade requires Treg cells. CTLA-4 conditional knockout (KO) in Treg cells reduces tumor growth [15]. Blockade of CTLA-4 accelerates Th1 proliferation and increases interleukin (IL)-2 production. While no direct function of anti-CTLA-4 in NK cells has yet been clearly identified, in vivo blockade of CTLA-4 might enhance NK cell activity indirectly by inhibiting Treg immunosuppressive activity and enhancing Th1 pro-inflammatory function [18]. Finally, the concomitant blockade of CTLA-4 and PD-1 signaling enhances antitumor response and increases the cytotoxic activity of CD8+ T cells in preclinical models [19].
CTLA-4 was the first IC receptor to be clinically targeted. In 2011, ipilimumab, an anti-CTLA-4 monoclonal antibody (mAb), was approved by the FDA and the EMA for treating patients with advanced melanoma. At present, ipilimumab is the only anti-CTLA-4 mAb approved for clinical use. It has value as a monotherapy for melanoma in adjuvant and metastatic settings, or combined with nivolumab, an anti-PD-1 antibody, for patients suffering several advanced cancer types (Table 2). Several clinical trials are testing the efficacy of anti-CTLA-4 combined with other anti-ICs in a variety of tumor types (Table 4).
PD-1 and PD-L1 blockade
PD-1 promotes inhibitory signals upon binding to its ligands, programmed death-ligand 1 (PD-L1) and programmed cell death 1 ligand 2 (PD-L2), in T cells [12]. PD-1 is expressed in distinct immune cell populations, including those of B, Treg, and myeloid cells. While PD-L1 may be expressed in hematopoietic and non-hematopoietic cells, physiological expression of PD-L2 is restricted to DCs, macrophages, and B cells. Notably, PD-L1 and PD-L2 expression is upregulated in many tumor types and is usually associated with poor patient outcome. PD-1 regulates activated T cell function in the later stages of the immune response in peripheral tissues [15]. Activated T cells induce PD-1 expression, and its signaling, upon binding to PD-L1/−L2, decreases T cell proliferation and cytokine secretion. PD-1 cytoplasmic tail contains classic inhibitory motifs ITIM and ITSM which recruit SHP-2 phosphatases resulting in reduced TCR downstream phosphorylation and signaling [20] (Fig. 2B). High PD-1 levels, concomitantly with other ICs, can be detected in TILs and in association with an exhausted phenotype [21]. PD-1 signaling in Treg cells enhances their proliferative and suppressive functions [5]. In NK cells, PD-1 expression has only been identified under pathological conditions [22]. PD-1 is upregulated in tumor-infiltrating NK cells and PD-1+ NK cells are known to be functionally exhausted [23]. Although PD-1 downstream mechanisms leading to NK exhaustion have not been completely elucidated, SHP-2 recruitment might participate in the process, given its role damping NK cell function [24]. An interaction between PD-L1 and CD80 expressed in T cells was also characterized, which led to inhibition of T cell function in vitro [25]. However, other studies suggest that the PD-L1 interaction with CD80 could cause T cell expansion without promoting exhaustion. Specifically, it has been reported that in bone marrow transplantations, the interaction of CD80 from donor CD8+ T cells with PD-L1 in lymphoid tissues from recipient patients, promotes T cell expansion, resulting in increased graft-versus-leukemia activity [26]. According to these findings, the blockade of PD-L1 signaling could reduce the antitumoral activity of T cells in specific tissue environments.

Immune checkpoint downstream inhibitory signaling in CD8+ T cells. Immune checkpoint pathways initiated after binding of ligands to their respective IC receptors (blue boxes) interfere with TCR signaling by a variety of mechanisms. ICs have inhibitory motifs in their cytoplasmic tail that can recruit (blue arrows) protein tyrosine phosphatases SHP1 and/or SHP2, which are responsible for dephosphorylating (red inhibitory arrows) TCR downstream signaling proteins. This is the case for PVRIG, 2B4, Siglec-7/−9, ILT2, BTLA, KIR-L, NKG2A, TIGIT, PD-1, and KLRG1. However, some ICs, such as CTLA-4, TIM-3, CD47, and CD200R1, present alternative downstream mechanisms, while other IC downstream signaling, such as that involving LAG-3, VISTA, CD96, CD160, and B7-H3, remains to be fully elucidated. Schematic representation of (A) SHP1-dependent inhibition of TCR signaling, (B) SHP2-dependent inhibition of TCR signaling, and (C) non-dependent SHP1 and SHP2 inhibition of TCR signaling. Dotted lines indicate indirect mechanisms (created with BioRender.com)
PD-1 signaling blockade increases antitumor immunity and decreases tumor growth. Specifically, PD-L1 transgenic expression in tumor cells enhances tumorigenesis, which can be reversed with anti-PD-L1 antibodies. PD-L1 mAb blockade enhances DC-mediated T cell activation and antitumor function. Tumor-infiltrating exhausted NK cells express PD-1 and its blockade partially restores NK antitumoral activity [23]. Although PD-1 signaling in Treg cells enhances immunosuppressive functions, in vivo PD-1 blockade in Treg cells leads to different tumor-dependent responses [27]. Moreover, a complete anti-PD-1 antitumoral effect requires DC stimulation and function [28]. A bispecific anti-PD-1/PD-L1 antibody gave rise to greater antitumoral efficacy relative to monospecific therapies in a high-grade serous ovarian cancer model by inducing a superior cytotoxicity in both T and NK cells [29]. This suggests that PD-L1 could be involved in interactions other than those binding to PD-1. PD-1 blockade alongside other ICs has shown itself to increase T cell response [30] and to bestow therapeutic benefits, which will be further discussed in the corresponding IC blockade section.
Inhibition of the PD-1/PD-L1 axis is the most commonly applied IC blockade therapy. Nivolumab, pembrolizumab, and cemiplimab are PD-1-blocking mAbs that have proven therapeutic efficacy in treating patients suffering from different tumor types (Table 2). Multiple anti-PD-1 antibodies, including these three, are being tested either alone (Table S1) or in combination with other anti-IC antibodies in multiple cancers (Table 4). The anti-PD-L1 antibodies, atezolizumab, durvalumab, and avelumab, also have a therapeutic value in treating certain tumor types and have been approved for clinical use (Table 2). These three antibodies, among others, are being tested as treatments for a wide range of solid tumors in clinical trials, either as monotherapy (Table S1) or in combination with other IC blockade antibodies (Table 4).
IC inhibitors under clinical development
LAG-3 blockade
The coinhibitory receptor lymphocyte activation gene-3 (LAG-3) is an IC receptor expressed by CD8+ T cells and NK cells that regulates peripheral tolerance. LAG-3 binds to HLA class II as well as to fibrinogen-like protein 1 (FGL-1), Galectin-3 (Gal-3), and LSECTin. LAG-3 ligands are expressed by tumor cells, and, notably, FGL-1 participates in immune evasion mechanisms that reduce the T cell response [31]. Mice deficient in LAG-3 have altered T cell proliferation. LAG-3 is expressed in Treg cells and its blockade disrupts Treg suppressor functions [32]. LAG-3 is also expressed in NK cells, but its function is not yet fully understood. Blockade of LAG-3 in human NK cells does not induce NK cytotoxicity against different tumor types [18]. Recent findings indicate that LAG-3 blockade in vitro increases cytokine production by NK cells without affecting their cytotoxicity [33]. The mechanism of action of LAG-3 remains largely unknown. LAG-3 cytoplasmic tail does not contain classical inhibitory motifs but presents a KIEELE motif that may mediate LAG-3 inhibitory functions [34] (Fig. 2C).
Anti-LAG-3 mAb administration was found to increase the proliferation and effector function of CD8+ T cells and delay tumor growth in a prostate cancer mouse model. LAG-3 is co-expressed with PD-1 in CD4+ and CD8+ TILs in many mouse and human tumors [35]. Simultaneous blockade of PD-1 and LAG-3 synergizes to enhance anti-tumor CTLs activity and reduces tumor growth in a colon adenocarcinoma model [36], a chronic lymphocytic leukemia (CLL) model [37], and a malignant pleural mesothelioma model [38]. LAG-3 expression has been linked to a stronger suppressive function of Treg cells [39]. The role of NK cells in LAG-3 blockade therapy response, and the possible contribution of NK cells to the observed antitumoral effects remain largely unknown. A unique study determined that treatment of the 4T1 mouse model of metastatic breast cancer with IL-12, combined with anti-LAG-3 or anti-PD-1, recovered NK cell cytotoxicity and proliferation, which resulted in a reduced presence of pulmonary metastases [40]. LAG-3 blockade is currently being investigated in clinical trials, either as monotherapy or combined with the inhibition of other ICs, to treat multiple tumor types (Table 4).
TIM-3 blockade
T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) acts as a coinhibitory receptor of T cells and is also expressed in Tregs, NK cells, DCs, and macrophages. TIM-3 expression is known to be increased in exhausted TILs [41]. The first TIM-3 ligand to be described was Galectin-9 (Gal-9), but TIM-3 also binds to phosphatidylserine (PS), carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), and high-mobility group protein 1 (HMGB1). The expression of TIM-3 ligands is upregulated by APCs, endothelial cells, and neutrophils, among other immune cells, and has been linked to carcinogenesis and tumor progression. Gal-9 interaction with TIM-3 negatively regulates T-helper function and can induce T-cell death [42]. TIM-3 blockade and gene depletion downregulate Th1 cell function and increase CTL proliferation and cytokine production [31]. TIM-3+ Treg cells increase suppressor functions in vitro [43]. Blockade of TIM-3 reverses the exhausted phenotype of CTLs from patients with advanced tumors. TIM-3 interaction with its ligands leads to the phosphorylation of conserved tyrosine residues in its cytoplasmic tail by the tyrosine ITK [44]. Phosphorylation of the TIM-3 cytoplasmic tail leads to the release of BAT3 protein and the recruitment of the tyrosine kinase FYN, ultimately resulting in TCR downstream kinase LCK inhibition [45] (Fig. 2C). In NK cells, TIM-3 acts as a negative regulator of NK effector functions. Its expression is upregulated in cancer, and has been associated with NK exhaustion [18]. Additionally, TIM-3 ligand Gal-9 can also interact with PD-1 expressed by T cells, which dampens the CD4+ and CD8+ T cell response [42, 46]. Specifically, Gal-9 interacts with PD-1 through glycans without affecting the PD-1/PD-L1 interaction. Likewise, the Gal-9/PD-1 interaction enables the formation of PD-1 and TIM-3 heterodimers, which promotes T cell exhaustion but dampens Gal-9/TIM-3-induced T cell apoptosis [47]. It is of note that, human Treg cells express high levels of Gal-9, which induces Treg death upon interacting with TIM-3 expressed by tumor resident cells [47]. Hence, anti-Gal-9 therapy might promote Treg function, thereby limiting the antitumoral function of CD8+ T cells. In this sense, the combination of anti-Gal-9 with Treg cell treatment depletion promotes synergistic antitumor activity in a breast tumor model by activating specific subsets of TILs [47].
In a TIM-3-overexpressing mouse model, anti-TIM-3 antibody reduces tumor growth by restoring T-cell activity [48]. Concomitant blockade of PD-1 and TIM-3 further improves T cell anti-tumor function and reduces tumor growth more effectively. Simultaneously targeting TIM-3 and PD-1 increases LAG-3 expression in TILs, suggesting a cross-regulation between IC receptors, and triple blockade of TIM-3, PD-1, and LAG-3 results in reduced tumor growth in a colon adenocarcinoma model [49]. Simultaneous blockade of TIM-3 and PD-L1 significantly reduces tumor growth in orthotopic models of HNSCC [50] whereas treatment with anti-TIM-3 concurrently with anti-PD-1 causes greater regression of murine glioma than is produced by a single checkpoint blockade [51]. TIM-3 blockade reverses exhausted NK cells isolated from lung adenocarcinoma patients [52], and advanced MHC class I-deficient tumors treated with IL-21 combined with anti-TIM-3 and anti-PD-1 reduce tumor progression by enhancing NK cell antitumoral immunity [53]. Hence, TIM-3 blockade also boosts NK cytotoxic activity in specific tumor settings. The presence of TIM-3+ Treg cells is associated with poor prognosis in lung cancer, and pharmacological blockade of TIM-3 reduces the suppressive function of intratumoral Treg cells [41]. In vitro experiments indicated that TIM-3 signaling in DCs blocks DC activation and maturation [54]. A recent study concluded that the accumulation of TIM-3+ CD4+ T cells in tumoral regions favors TIM-3-mediated immunosuppressive functions in hepatocellular carcinoma (HCC) patients. Depletion of CD4+ T cells abrogates the antitumoral effects of anti-TIM-3 therapy, indicating that CD4+ T cells might be responsible for TIM-3-mediated immunosuppression [55]. The response of TIM-3 blockade-based therapy is currently being analyzed in clinical trials as monotherapy or in combination with anti-PD-1 antibodies (Table 4).
TIGIT blockade
The T cell immunoglobulin and ITIM domain (TIGIT) is a member of the immunoglobulin superfamily of paired receptors expressed by T cells and NK cells that interact with ligands of the nectin and nectin-like family. TIGIT acts as an inhibitor receptor when binding to its ligands PVR, nectin-2, and nectin-3 (CD113), all of which are expressed in APCs and a variety of non-hematopoietic cells, including tumor cells [56]. Specifically, TIGIT interacts with PVR with a higher affinity than with nectin-2, while the nectin-3/TIGIT interaction has a weak binding affinity. In addition, DNAM-1 is another co-stimulatory receptor expressed by NK and T cells that competes with TIGIT to bind PVR and nectin-2, which enables it to regulate T cell inhibition precisely [57]. In NK cells, TIGIT signaling reduces NK cytotoxicity and cytokine release, while in T cells it reduces T cell activation, proliferation, and effector functions. Specifically, the TIGIT intracellular cytoplasmic tail contains an ITIM and an ITT-like domain that recruit SHP-1 phosphatases, leading to the blockade of PI3K and MAPK pathways in NK cells [58], and decreased TCR downstream signaling [59] (Fig. 2A). TIGIT+ Treg cells have greater suppressive capacities in vitro and selectively suppress the Th1 pro-inflammatory response in vivo [31]. TIGIT interaction with PRV expressed by DCs forces the DCs to be tolerogenic by increasing their production of immunosuppressive IL-10 cytokine [60].
TIGIT ligands are expressed by cancer cells and exhausted TIGIT+ T and NK cells are detected in various human cancers. Antibody blockade of TIGIT enhances antitumor CD8+ T cell response and prompts tumor regression in a colorectal cancer (CRC) mouse model. TIGIT dual blockade with PD-1, PD-L1, or TIM-3 has a synergistic action that produces enhanced CD8+ T cell activity and tumor regression in a colorectal carcinoma model [61]. Dual blockade of PD-1 and TIGIT enhances antitumor response and increases survival in a mouse glioblastoma model [62]. Conditional TIGIT KO in Treg cells reduces tumor growth in a melanoma mouse model, proving that the TIGIT blockade effect is also mediated by Treg cells [31]. Recently, it was demonstrated that TIGIT blockade elicits NK-mediated antitumoral immunity in tumor-bearing mouse models [63]. Antibodies targeting TIGIT are currently being investigated in clinical trials to treat patients with different tumor types (Table 4).
PVRIG blockade
PVRIG, also known as CD112R, is another member of the immunoglobulin superfamily of paired receptors. It has recently been identified in human T and NK cells. PVRIG binds to nectin-2 but does not recognize PVR. In T cells, PVRIG signaling inhibits the TCR-mediated signal dampening T cell response by recruiting SHP-1 and SHP-2 phosphatases to its ITIM-like domains [64] (Fig. 2B). Human NK cells expressing PVRIG present reduced proliferation and cytokine release [56]. Recent results suggest that PVRIG expression is reduced during human NK-cell activation [65], while exhausted tumor-infiltrating NK cells express high levels of PVRIG [66]. Until now, expression of PVRIG has not been described in myeloid immune cell populations.
Targeting PVRIG with antibodies promoted T cell expansion in vitro, which was further increased by simultaneous blockade with TIGIT [64]. PVRIG blockade enhances in vitro NK cell antitumoral activity and increases IFN-γ release [67]. In AML patients, anti-PVRIG therapy promotes NK-cell cytotoxicity against AML blasts [65]. In mouse models of cancer, TILs show high PVRIG levels [68]. Ex vivo PVRIG blockade of patient-derived T cells upregulates T-cell function, an effect that is further enhanced when combined with anti-TIGIT or anti-PD-1 treatments [56]. Anti-PVRIG treatment delays tumor growth and prolongs survival of tumor-bearing mice by reversing exhaustion of NK and CD8+ T cells of solid tumors. Dual blockade of PVRIG and PD-L1 enhances the antitumoral effects in comparison to single-blockade therapies in solid tumors [66, 68]. Notably, PVRIG−/− mice exhibit reduced tumor growth in a CD8+ T cell-dependent manner, an effect that is synergistically enhanced by PD-L1 blockade. Finally, PVRIG is co-expressed with PD-1 and TIGIT in activated T cells and combinatory dual blockade of PVRIG with PD-1 or TIGIT additionally promoted T-cell activity [69]. An anti-PVRIG mAb is currently being tested in a clinical trial in patients with advanced solid tumors (Table 4).
KIR-L blockade
Killer immunoglobulin-like (KIRs) are a family of receptors that regulate self-tolerance and effector functions of NK cells by binding to classic HLA class I allotypes (HLA-A, HLA-B, and HLA-C). These receptors allow the identification and elimination of cells that fail to express a sufficient level of HLA, like many tumor cells, in a process called missing self-recognition. KIR receptors with a long cytoplasmic tail (KIR-L) mainly present NK coinhibitory capabilities, while short-tailed KIR receptors (KIR-S) enhance NK function [70]. The KIR-L cytoplasmic tail contains ITIM motifs that become phosphorylated upon binding with its ligand, and recruit tyrosine phosphatases such as SHP-1 and SHP-2, resulting in NK-cell inhibition [71] (Fig. 2A and B). KIRs were initially characterized as NK cell receptors, but they are also expressed in CD8+ T-cell subsets. It has been proposed that KIR expression can be sustained by TCR engagement, and may participate in T-cell tolerance to self-antigens [72].
Initial evidence that the blockade of KIR signaling could be beneficial in treating cancer came with the observation that acute myeloid leukemia (AML) patients did not experience recurrence within 5 years if they received a bone marrow transplantation from a donor who presented NK with KIRs that mismatched the interaction with the HLA of the patients [73]. Thus, the lack of recognition of HLA class I molecules by KIRs enhances NK cell cytotoxic activity, causing the elimination of malignant cells. Additionally, tumor-infiltrating NK cells have a high level of expression of inhibitory KIRs that are correlated with poor clinical outcome in non-small-cell lung cancer (NSCLC) [74]. A unique mAb targeting KIR2DL1, KIR2DL2, and KIR2DL3 receptors increases NK-cell cytotoxicity against autologous AML blasts and multiple myeloma cells [18]. Hence, KIR blockade appears to enhance NK cell function in the tumors. Different KIR inhibitors are being tested in clinical trials as single agents or combined with anti-PD-1 or anti-CTLA-4 antibodies (Table 4).
NKG2A blockade
The natural killer group 2A (NKG2A) is a member of the NKG2 receptor group, which dimerizes with CD94 to bind to non-classic HLA-E class I molecules, which are ubiquitously expressed. Upon binding to its ligand, CD94/NKG2A signaling downregulates NK cell-mediated cytotoxicity. Tumor-infiltrating NK cells from patients with liver cancer express high levels of NKG2A and are functionally exhausted [75]. NKG2A and CD94 expression is restricted to a subset of CD8+ T cells [76] and its expression in TILs generates negative regulatory signals. Mechanistically, NKG2A contains cytoplasmic ITIM motifs responsible for its inhibitory functions by recruiting the phosphatases SHP-1 and SHP-2 [77] (Fig. 2A and B).
Combined blockade of NKG2A and PD-1 or PD-L1 synergizes to reduce tumor growth in a mouse model of B-cell lymphoma by promoting NK and CD8+ T cell cytotoxic activity [78]. Anti-NKG2A blockade enhances NKG2A+ NK cells’ cytotoxic function, eliminating human leukemia cells engrafted in mice [79]. Monalizumab is a humanized mAb that specifically blocks NKG2A and enhances NK and T cell effector function and promotes anti-tumor immunity [78]. Monalizumab is under analysis in clinical trials in which it is used either alone or in combination with anti-PD-L1 antibodies to treat a variety of tumor types (Table 4).
CD200 blockade
CD200 is a cell-surface glycoprotein that binds to the coinhibitory receptor CD200R1 expressed in subsets of NK, T, B, and myeloid cells. CD200 expression is detected in various human tissues such as endothelium, central nervous system, retina, and in activated DCs, T, and B cells. It is of note that the interaction between CD200 and CD200R1 occurs between different cells expressing ligand and receptor (in trans), while the interaction in the same cell (in cis) is remote due to steric constraints [80]. CD200R1 signaling in NK cells inhibits their cytotoxic activity and cytokine release [18]. CD200R1 contains a NPXY motif in its cytoplasmic tail that when phosphorylated inhibits the Ras/MAPK signaling [81] (Fig. 2C). A direct effect of CD200R1 signaling on T cell activity is less clear. While one study in CD200R KO mice suggested greater T-cell cytotoxicity, another showed a normal T-cell response with a lack of CD200R1 signaling [82]. In DCs and macrophages, CD200R1 signaling reduces pro-inflammatory cytokine production, leading to a tolerogenic state [82]. CD200 may have indirect inhibitory effects on T-cell activity by modifying the cytokine landscape rather than a direct cell-intrinsic inhibitory signal. No direct effect of CD200R1 signaling in other CD200R-expressing immune populations has been reported.
CD200−/− mice displayed reduced rate of skin carcinogenesis [82]. CD200+ leukemic cells from AML patients reduced NK cytotoxic activity relative to CD200− leukemic cells and NK function was recovered with anti-CD200 mAb [83]. CD200 blockade increased antitumoral response in a mammary carcinoma mouse model [84]. Finally, CD200 blockade stimulated T cell antitumoral response in CLL patients [85]. Samalizumab, an antibody that targets CD200, is being tested in clinical trials (Table 4). No anti-CD200R1 antibodies have been evaluated in preclinical or clinical contexts as antitumoral therapy.
CD47 blockade
CD47, originally called integrin-associated protein (IAP), is a transmembrane glycoprotein of the immunoglobulin superfamily that is expressed ubiquitously by human cells. When CD47 binds to its ligands, the signal regulatory protein alpha (SIRPα) and thrombospondin-1 (TSP1) can induce cell activation or apoptosis, depending on the cellular context. CD47 activation in immune cells has been linked to tumor immune evasion, decreased antigen-presentation cell function, and impaired effector functions of NK and T cells [86]. In vivo experiments have shown that CD47 signaling inhibits NK and T cell cytotoxicity indirectly through impaired APC functions [87, 88]. Notably, the best characterized function of CD47 serves as an antiphagocytic signal for macrophages upon binding to SIRPα [86].
CD47 blockade indirectly enhances anti-tumor cytotoxicity by stimulating macrophage phagocytosis and antigen presentation by APCs, which enhances CD8+ T cell cytotoxicity. In this sense, the antitumoral effects of anti-CD47 treatment are abrogated in T cell-deficient mouse models [88]. Head and neck squamous cell carcinoma (HNSCC) cells with a high level of expression of CD47 show lower NK cytotoxicity, which is reversed upon anti-CD47 treatment [87]. Another strategy that has been developed involves the direct blockage of the SIRPα ligand. An anti-SIRPα antibody reduced tumor growth in an NK- and T cell-dependent manner [86]. A recent study in a mouse model of breast cancer demonstrated that the simultaneous blockade of CD47 and PD-L1 inhibits tumor growth by enhancing T- and NK-cell activity [89]. The targeting of CD47 is being tested in clinical trials for the treatment of various tumor types (Table 4).
BTLA blockade
B and T cell lymphocyte attenuator (BTLA) is a coinhibitory receptor expressed in T cells, DCs, and B cells, but not in NK cells. BTLA binds to the herpesvirus entry mediator (HVEM) protein, which is expressed by B cells, DCs, and T cells. In naïve T cells, BTLA-HVEM interaction in cis inhibits T-cell activation, ensuring a quiescent state [90]. At the signaling level, BTLA cytoplasmic tail contains ITIM and ITSM motifs responsible for recruiting the tyrosine phosphatases SHP-1 and SHP-2 which reduces TCR downstream phosphorylation [91] (Fig. 2A and B). TILs exhibit upregulated expression of BTLA, less proliferation, and less extensive cytokine production when they interact with HVEM expressed by cancer cells. Indeed, BTLA and PD-1 co-expression is detected in exhausted TILs from patients with advanced melanoma [92]. Finally, DCs expressing BTLA, when interacting with HVEM, promote the differentiation of peripheral Treg cells, and induce antigen tolerance [93].
Anti-BTLA therapy combined with chemotherapy reduces tumor growth, and increases the survival of tumor-bearing mice [94]. Recent proteomic studies have revealed a rationale for simultaneously blocking PD-1 and BTLA in order to increase the T-cell antitumoral response [95]. In this regard, BTLA blockade upon anti-PD-1 and anti-TIM-3 treatment further increases CD8+ T cell proliferation [92]. Interestingly, in ovarian carcinomas BTLA expression was mainly identified in B lymphocytes rather than T or NK cells, and the BTLA blockade antitumoral effect was caused by inhibiting a specific subset of B lymphocytes rather than stimulating T or NK cell function [94]. Two mAbs targeting BTLA are currently being studied in early-phase clinical trials (Table 4).
VISTA blockade
V-domain Ig suppressor of T cell activation (VISTA), also known as programmed death-1 homolog (PD-1H), is an IC expressed by CTLs, Treg cells, DCs, macrophages, and neutrophils, but not NK cells. VISTA has been considered to be a ligand or a receptor in different studies. Here, we will consider it to be a receptor, since the interaction with its ligand leads to VISTA intracellular downstream signaling. VISTA is a functionally pH-selective receptor and interacts with the P-selectin glycoprotein ligand-1 (PSGL-1) under acid pH conditions [96]. PSGL-1 is expressed by different cell populations as T, B, endothelial cells, DCs, macrophages, monocytes, and neutrophils, and participates in leukocyte homing processes. VISTA signaling suppresses T-cell activation and proliferation in in vivo experiments [97]. The VISTA cytoplasmic tail contains conserved proline residues and an SH2 domain that may be responsible for its inhibitory functions [98] (Fig. 2C).
Anti-VISTA antibodies reduce tumor growth and increase T-cell tumor infiltration and effector functions in preclinical models. VISTA blockade decreases the suppressive functions of Treg cells, reduces the intratumoral presence of MDSCs, and increases tumor infiltration of activated DCs [99]. Dual blockade of PD-1 and VISTA synergistically enhance the antitumor T-cell response in various mouse models [100]. Finally, given that VISTA binds its ligand, PSGL-1, under low pH conditions, the possibility of using engineered pH-selective antibodies that bind its epitope in specific pH environments has arisen. The synergy between an acidic pH-selective VISTA-blocking antibody and an anti-PD-1 antibody has been shown to reduce tumor growth in a mouse model of colon adenocarcinoma [96]. VISTA blockade has been translated to the clinical milieu for testing as an anticancer therapy (Table 4).
B7-H3 blockade
The B7 homolog 3 protein (B7-H3) is a B7 family molecule that functions as a ligand, but its receptor remains to be discovered. T lymphocytes and NK cells respond to B7-H3, suggesting that the B7-H3 receptor is expressed in these immune cell types [18]. B7-H3 has been reported as playing a variety of roles in T cells. B7-H3 signaling increases T-cell proliferation, cytokine release, and enhances antitumor T cell activity in cancer mouse models [101]. Conversely, B7-H3 signaling in T cells is also able to inhibit them by blocking TCR signaling [102]. These contradictory effects might be due to specific TME features interfering with T-cell function. B7-H3 functions as an inhibitory ligand for NK cells, and anti-B7-H3 antibodies enhance NK-cell cytotoxicity in vitro. Various human cancer cells upregulate B7-H3 expression, and are related to impaired T-cell function, suppressed NK cytolytic activity, and tumor immune evasion [18].
Anti-B7-H3 treatment reduces tumor growth in cancer mouse models that express B7-H3. B7-H3 KO glioma-initiating cells show less invasiveness and higher susceptibility to NK cell-mediated cytolysis [103]. Other preclinical studies have indicated that higher levels of B7-H3 may be beneficial in the T-cell-mediated anti-tumor response against mastocytoma. A synergistic antitumoral response between anti-B7-H3 mAbs and chemotherapy has been observed in several preclinical models [104]. Thus, blockade of B7-H3 may be beneficial, boosting T and NK cell effector functions in specific tumor types and cellular contexts. Finally, the combination of anti-B7-H3 and anti-PD-L1 treatment promotes a synergistic antitumor response relative to single-agent blockades in a mouse model of NSCLC [105]. Antibodies blocking B7-H3 are being tested in clinical trials for the treatment of several tumor types (Table 4).
IC inhibitors under preclinical investigation
CD96 blockade
CD96, also known as T cell activation increased late expression protein (TACTILE), is a member of the immunoglobulin superfamily of paired receptors which contains intracellular inhibitory and activating motifs. The cytoplasmic tail of CD96 contains an inhibitory motif ITIM and a YXXM motif that is thought to mediate activating or inhibitory functions depending on the cell type [106] (Fig. 2C). CD96 binds to PVR and nectin-1 (CD111) and is expressed in T and NK cells. It has been reported that CD96 signaling in NK cells negatively controls cytokine release [56]. CD96’s role in CD8+ T cells is controversial. Cross-linking CD96 in CD8+ T cells induces proliferation and effector cytokine production [107]. However, recent findings indicate that CD96 blockade inhibits primary tumor growth in various tumor mouse models, an effect that is dependent on CD8+ T cell activity [108]. Further studies are needed to determine the role of CD96 in T cells under pathological conditions.
The presence of exhausted CD96+ NK cells is associated with poor clinical prognosis in HCC patients. CD96 blockade increases NK cell-mediated lysis and synergizes with an anti-TIGIT antibody to produce an enhanced antitumoral effect. CD96−/− mice injected with B16 melanoma cells develop fewer lung metastases, this reduction being dependent on NK cells. Another study reported decreased metastasis development after anti-CD96 treatment in several preclinical models [18]. Dual blockade of CD96 with PD-1, PD-L1, TIGIT, or CTLA-4 increases antitumor response, and triple blockade of CD96, PD-1 and TIGIT yields the highest level of antitumoral immunity in various mouse tumor models [108]. To date, antibodies blocking CD96 have not been evaluated in clinical trials.
ILT2 blockade
The immunoglobulin-like transcript-2 (ILT2), also known as leukocyte immunoglobulin-like receptor-1 (LIR1), is a coinhibitory receptor expressed by NK cells, subsets of T, B cells, and DCs. Non-classic HLA-G class I molecules are ILT2 ligands. HLA-G maintains fetal-maternal immune tolerance, and is expressed in adult tissues in cancer. ILT2 signaling inhibits NK-cell effector functions by decreasing cytotoxicity and IFN-γ release. ILT2 interaction with HLA-G inhibits T-cell proliferation and related cytolysis by recruiting SHP-1 phosphatase to its cytoplasmic ITIM domain [109] (Fig. 2A). In DCs, ILT2 signaling induces the development of tolerogenic DCs. Furthermore, exposure of HLA-G molecules to DCs induces anergy in CD4+ and CD8+ T cells, and impairs NK cytolytic functions [110].
Simultaneous blockade of ILT2 and NKG2A increases the cytotoxicity of NK cells to acute myelogenous leukemia and acute lymphoblastic leukemia human blasts. Moreover, ILT2 blockade increases NK-cell cytotoxicity, leading to the elimination of malignant cells from CLL patients [111]. At present, no ILT2 blockade antibodies have been reported in clinical trials.
Siglec-7 and Siglec-9 blockade
Sialic acid-binding immunoglobulin type lectins (Siglecs) are cell-surface receptors of the I-type lectin family that bind sialic acid-containing glycans present on glycoproteins and glycolipids. Siglec-7 and Siglec-9 are expressed by human NK cells, negatively regulating NK effector function [18]. CD8+ T cell subsets expressing Siglec-7 and Siglec-9 receptors present reduced activity. Specifically, Siglec-7 and Siglec-9 contain cytosolic inhibitory ITIM motifs responsible for recruiting the phosphatase SHP-1 [112] (Fig. 2A). In monocytes, Siglec-7 signaling induces the release of pro-inflammatory molecules upon pathogen recognition [113]. Siglec-9 signaling in macrophages reduces LPS-induced CCR7 expression, revealing a role in modulating innate immunity [114], while Siglec-9 ligand interaction with immature DCs reduces LPS-induced IL-12 release [115]. Finally, the presence of molecules containing sialic acid modifications, including hypersialylation and xenosialylation, has been linked to tumor progression [116].
Siglec-9 is co-expressed with PD-1 in TILs from patients with various cancer types [117]. Targeting Siglec signaling pathways enhances the antitumoral response, while genetically modified mice expressing Siglec-9 in T cells show accelerated growth of CRC tumors [117]. Isolated NK cells from cancer patients with upregulated Siglec-9 expression are less cytotoxic. Anti-Siglec-7 and anti-Siglec-9 antibodies strengthen the effector function of NK cells against cancer cell lines expressing Siglec-7 or Siglec-9 ligands [118]. At present, there are no reports of Siglec-7 or Siglec-9 antibodies being assessed in clinical trials.
KLRG1 blockade
KLRG1 is a coinhibitory receptor expressed exclusively in NK and T cells that binds to E-cadherin and N-cadherin. KLRG1 signaling inhibits NK-associated cytotoxicity and reduces IFN-γ production [119]. In T cells, KLRG1 signaling inhibits cell proliferation and cytotoxic activity [120]. Upon binding to its ligands, KLRG1 recruits SHIP-1 and SHP-2 phosphatases but not SHP-1 to its cytoplasmic ITIM domains resulting in interfered TCR signaling [121] (Fig. 2B and C).
Although KLRG1 KO mice do not display increased NK or T cell cytotoxic functions, pharmacological targeting of KLRG1 increases the effector functions of NK and T cells [122]. Antibody blockade of KLRG1 restores NK-cell cytotoxicity of genetically engineered KLRG1-expressing NK cells in vitro [123]. Administration of anti-KLRG1 antibody does not modify primary tumor growth but has been shown to reduce lung metastases in breast cancer mouse models [124]. Notably, dual blockade of KLRG1 and PD-1 has been shown to decrease primary tumor growth synergistically in melanoma and CRC mouse models [124]. No clinical trials involving anti-KLRG1 antibodies have so far been reported.
2B4 Blockade
2B4, also known as CD244, is a coinhibitory receptor expressed by myeloid cells, NK cells, and a subset of CD8+ T cells. CD48, which is 2B4 ligand, is ubiquitously expressed by hematopoietic cells and upregulated in hematological malignancies [125, 126]. 2B4 receptors contains inhibitory ITSM motifs responsible of recruiting the tyrosine phosphatases SHP-1, SHP-2 and SHIP-1 [127] (Fig. 2). In human NK cell precursors, 2B4 presents inhibitory functions, whereas in mature NK cells, 2B4 signaling enhances their cytotoxic activity [128]. NK cells and CD8+ T cells expressing 2B4 from human cancer patients present an exhausted phenotype that can be reversed by the blockade of the 2B4-CD48 interaction [129]. Furthermore, 2B4 expression levels in tumor-infiltrating DCs and MDSCs are correlated with tumor cell PD-L1 expression and MDSC production of immunosuppressive molecules [130].
In vitro blockade of 2B4 with mAbs increases NK-cell and T-cell functions in 2B4+-exhausted NK and T cells [129]. A 2B4 KO mouse model revealed increased rejection of engrafted melanoma cells [131]. In addition, 2B4 KO mice presented impaired HNSCC growth in a preclinical model. Anti-2B4 mAb treatment phenocopies 2B4 KO mice, inhibiting tumor growth and increasing the presence of TILs [130]. No 2B4 blockade therapies have so far been tested in clinical trials.
Current challenges: improving efficacy without increasing adverse events
Although immunotherapy based on IC blockade has produced promising results in a fraction of patients, a large number of patients do not benefit from the existing approved drugs (Table 3). Tumor-infiltrating immune cells present in the TME play a fundamental role in therapy response. Tumors that are not likely to generate a robust immune response have been classified as poorly immunogenic or cold tumors. These tumors have low quantities of immune infiltrate [1]. On the other hand, hot tumors have high levels of T cell infiltration and are highly immunogenic. Targeting the TME to transform cold tumors into hot tumors before IC blockade therapies is being investigated as a strategy to increase the responsiveness to these therapies. The approaches promoting immunogenicity of cold tumors include enhancing antigen presentation by DCs, reducing the presence and function of immunosuppressive cells, and delivering immunomodulatory factors to boost inflammation [132].
Combining IC blockade drugs with other immunotherapeutic agents also produces promising enhanced antitumoral responses. Chimeric antigen receptor (CAR) T cells are engineered autologous T cells with an artificial TCR that recognize a specific antigen in an MHC-independent manner. CAR T cells achieve long-lasting responses in hematological malignancies, whereas the clinical activity observed so far in solid tumors has been more modest. This may occur because the immunosuppressive TME that present solid tumors can lead to CAR T cell exhaustion [133]. Hence, combining CAR T cells with anti-IC drugs and/or drugs targeting the immunosuppressive TME may ideally produce a synergistic effect, unleashing CAR T cell activity against tumoral cells. Combining anti-PD-1 antibodies with adoptive transfer of CAR T cells might overcome PD-1 dependent T cell exhaustion, thereby improving single-treatment responses [134, 135]. Another interesting strategy to overcome the exhaustion of CAR T cells in solid tumors is to use PD-1 KO CAR T cells, which have enhanced cytotoxic activity [136]. Given the crucial role of APCs in priming antigen-specific T cell immunity, the modulation of APC function has also been used as an antitumoral strategy. In this regard, several DC-based immunotherapies are being clinically studied, such as treatment with immunostimulatory molecules that promote DC activity or vaccine administration of tumor-associated antigens (TAAs) that can be processed and presented by endogenous DCs [137]. The use of DC vaccines consisting of ex vivo-amplified autologous DCs, tumor antigen load and reinfusion to patients, has also proven clinical benefits [137]. Combining dendritic cell vaccines with anti-PD-1 therapies may boost efficacy by improving T cell antitumoral function in mouse models [138, 139]. In addition, DC vaccination can overcome IC blockade-resistant murine lung cancers by eliciting an antitumoral response [140]. Hence, the appropriate combination of immunotherapeutic agents is a promising strategy for treating single-agent-resistant tumors. In addition, chemotherapy and radiotherapy are standard cancer therapies used in the clinic setting that produce immunostimulatory effects and that can be combined with IC blockade therapies to increase antitumoral immunity [141, 142].
Despite the positive results of IC blockade therapies as single agents or in combination, patients treated with IC inhibitors may suffer from secondary autoimmune events, also known as immune-related adverse events (irAEs) [143]. Immune cell exhaustion, promoted by the activation of IC pathways, among other mechanisms, prevents overactivation of effector immune cells and preserves normal homeostasis and self-tolerance. Genetic ablation of ICs leads to the development of autoimmune diseases in multiple mouse models. CTLA-4 KO leads to lymphoproliferative disorders and early lethality, and PD-1 gene abrogation promotes severe autoimmune diseases, while TIM-3 KO and TIGIT KO cause experimental autoimmune encephalomyelitis (EAE) [15]. Pharmacological blockade of IC receptors exacerbates autoimmune events in mouse models of autoimmune diseases [15]. Hence, IC blockade therapies in cancer patients can lead to the appearance of irAEs, which can be variable from person to person. Typically, these toxicities, which have recently been reviewed [144], affect barrier tissues such as the skin, gastrointestinal tract, liver, and respiratory epithelium.
The overall response rate (ORR) to IC blockade antibodies, as a measure of therapy response, varies between tumor types (Table 3). This variation may be due to tumor-specific biological differences. Tumor types with a higher mutation rate and that conserve the expression of HLA have more neo-antigens, and are more likely to be identified and attacked by T cells. Moreover, tumor types with an immunosuppressive TME, including the presence of M2 macrophages, MDSCs, Treg cells, and immunosuppressive soluble factors, are less responsive to anti-IC therapies [145]. In addition, differences in ORR within same tumor type between drugs targeting the same pathway also arise (Table 3), possibly as a consequence of the intertumoral heterogeneity observed in patients with the same tumor type [146]. As previously described, IC blockade therapy response is affected by the TME profile, which also varies between patients [1].
To improve efficacy, the immune infiltrate and IC ligand expression in tumor and tumor-infiltrating immune cells could be characterized to determine the IC pathway most likely to be responsible for the attenuation of cytotoxic immune cell activity. Despite this, the overall response rates to single IC blockade therapies are generally low. Combining the blockade of multiple ICs is a strategy to increase the response of these therapies against certain tumor types but potentially increases the risk of irAE development. In order to minimize the risk of irAEs and maximize the response, the combinatory blockade of ICs with non-redundant downstream signaling could be a good strategy for enhancing antitumor immunity. Ideally, the blockade of an IC that recruits the phosphatases SHP-1 and/or SHP-2, such as PD-1, KLRG1, PVRIG, 2B4, Siglec-7/− 9, ILT2, TIGIT, NKG2A, KIR-L, or BTLA, should be combined with the blockade of another IC that exhibits alternative downstream signaling, such as CTLA-4, TIM3, LAG-3, CD47, CD200R1, or VISTA. Non-redundant IC combinatory blockade therapies may have synergistic effects that boost antitumoral immunity. However, further studies should be carried out to address this hypothesis. In addition, it needs to be considered that combining the blockade of any IC with CTLA-4 might present stronger secondary events given the role of CTLA-4 in regulating central tolerance [147]. Moreover, combinatory treatments that promote T and NK cell function simultaneously to reduce the presence of immunosuppressive cells might also be of value. The use of novel small-molecule inhibitors of PD-1/PD-L1 currently under clinical development might be beneficial because of their reduced immunogenicity. Indeed, immune checkpoint antibodies have a longer pharmacokinetic half-life than small-molecule inhibitors, which manifests as sustained immune system activation and a greater quantity of derived irAEs [148]. Combining small-molecule inhibitors of ICs concomitantly with the blockade of antibodies for different ICs may result in increased effector cell function and reduced tumor growth without any more frequent occurrence of irAEs.
Finding the correct IC blockade combination for each tumor setting to ensure that efficacy is increased without raising the risk of irAEs occurring is a major challenge. Understanding the mechanisms leading to irAEs will allow biomarkers to be developed that can classify patients according to the administration of the most effective and safe IC blockade therapy. Some of the plausible biomarkers studied for this purpose include peripheral blood cell counts, circulating cytokines and chemokines, the presence of autoantibodies, and the composition of gut microbiota [149]. However, the predictive information of these biomarkers has been studied in very few tumor types. The study of genetic polymorphisms associated with autoimmune diseases may also be of importance in identifying patients who are more likely to develop irAEs. Omics studies are of particular interest when reliable biomarkers across multiple tumor types need to be established. The expression of the lymphocyte cytosolic protein 1 (LCP1) and the adenosine diphosphate- dependent glucokinase (ADPGK) have been proposed as biomarkers of irAEs [150]. Further prospective trials are needed to identify the probable combinations of biomarkers that will allow us to categorize patients with respect to the determining the therapy that is safest for them to receive.
Conclusions
The stimulation of antitumoral immunity through immunotherapy has revolutionized cancer treatment in recent years. Antibodies against CTLA-4, PD-1, and PD-L1 have been approved for the treatment of several types of tumors but have been of limited clinical benefit in some patients. This could be related to the existence of the many mechanisms that tumor cells use to evade the immune response, such as the expression of a long repertoire of IC ligands and the infiltration of several immunosuppressive immune cell populations. Blockade of novel ICs is being evaluated in clinical trials. Antibodies against LAG-3, TIM-3, TIGIT, CD47, and B7-H3 are the most advanced IC blockade drugs and may be approved for the treatment of specific tumor types in the near future, depending on the results of the trials. However, other challenges need to be overcome to fully exploit the therapeutic potential of blockade ICs, and thereby boost antitumoral immunity.
A full understanding of the crosstalk between cancer cells and the TME of every tumor type is needed to identify the specific immune-evasion mechanisms exploited by cancer cells and, subsequently, to apply proper therapy. Tumor cells can upregulate the expression of various IC ligands and promote the activation of multiple IC receptors of tumor-infiltrating immune cells. Hence, activation of alternative IC signals in the tumors may diminish the effect of single-blockade antibodies. Combinatory IC blockade treatments might present synergistic antitumoral responses in specific tumor types, and potentially increase the risk of secondary events such as irAEs. Hence, finding the best IC blockade combination to achieve increased effectiveness while minimizing the risk of irAEs should be a priority. Given that some IC receptors share downstream mechanisms to interfere with T and NK cell activation, the co-blockade of IC with non-redundant signaling to improve antitumoral immunity and prevent overlapping signals could be a good strategy. Furthermore, the use of small-molecule IC inhibitors might be advantageous compared with blockade antibodies, given their reduced immunogenicity. Importantly, development and analysis of biomarkers that allow patients to be classified according to their specific pathology settings and IC activation status should improve response rates to IC blockade therapies. The rational blockade of ICs, based on the specific tumor characteristics of each patient, may represent a breakthrough in our pursuit of a more personalized medicine.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the Drug Approvals and Databases FDA repository, www.fda.gov and in the Data clinical trials repository, www.clinicaltrials.gov.
Abbreviations
- ICs:
-
Immune checkpoints
- CTLs:
-
Cytotoxic T lymphocytes
- NK:
-
Natural killer
- CTLA-4:
-
Cytotoxic T lymphocyte-associated molecule-4
- PD-1:
-
Programmed cell death receptor-1
- PD-L1:
-
Programmed death ligand-1
- PD-L2:
-
Programmed cell death 1 ligand 2
- MHC:
-
Major histocompatibility complex
- MDSCs:
-
Myeloid-derived suppressor cells
- Treg:
-
T regulatory
- TME:
-
Tumor microenvironment
- CAR:
-
Chimeric antigen receptor
- IFN:
-
Interferon
- TNF:
-
Tumor necrosis factor
- TCR:
-
T-cell receptor
- HLA:
-
Human leukocyte antigen
- APCs:
-
Antigen-presenting cells
- DCs:
-
Dendritic cells
- Th:
-
T-helper
- Th1:
-
T-helper 1
- Th2:
-
T-helper 2
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- TILs:
-
Tumor-infiltrating lymphocytes
- FDA:
-
US Food and Drug Administration
- EMA:
-
European Medicines Agency
- KO:
-
Knockout
- mAb:
-
Monoclonal antibody
- CRC:
-
Colorectal cancer
- RCC:
-
Renal cell cancer
- NSCLC:
-
Non-small cell lung cancer
- LAG-3:
-
Lymphocyte activation gene-3
- FGL-1:
-
Fibrinogen-like protein 1
- TIM-3:
-
T cell immunoglobulin and mucin domain-containing protein 3
- PS:
-
Phosphatidylserine
- CEACAM1:
-
Carcinoembryonic antigen-related cell adhesion molecule 1
- HMGB1:
-
High-mobility group protein 1
- TIM-3:
-
T-cell immunoglobulin and mucin-domain containing-3
- TIGIT:
-
T cell immunoglobulin and ITIM domain
- PVRIG:
-
Poliovirus receptor-related immunoglobulin
- PVR:
-
Poliovirus receptor
- TACTILE:
-
T cell activation increased late expression protein
- KIR:
-
Killer immunoglobulin-like
- NKG2A:
-
Natural killer group 2A
- ILT-2:
-
Immunoglobulin-like transcript-2
- KIR-L:
-
Long-tailed KIR
- KIR-S:
-
Short-tailed KIR
- AML:
-
Acute myeloid leukemia
- IAP:
-
Integrin-associated protein
- SIRPα:
-
Signal regulatory protein alpha
- TSP1:
-
Thrombospodin-1
- HNSCC:
-
Head and neck squamous cell carcinoma
- Siglecs:
-
Sialic acid-binding immunoglobulin type lectins
- LPS:
-
Lipopolysaccharide
- KLRG1:
-
Killer cell lectin-like receptor subfamily G member 1
- BTLA:
-
B and T cell lymphocyte attenuator
- HVEM:
-
Herpesvirus entry mediator
- VISTA:
-
V-domain Ig suppressor of T cell activation
- PD-1H:
-
Programmed death.1 homolog
- PSGL-1:
-
P-selectin glycoprotein ligand-1
- B7-H3:
-
B7 homolog 3 protein
- EAE:
-
Experimental autoimmune encephalomyelitis
- ORR:
-
Overall response rate
- mPFS:
-
Median progression-free survival
References
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–50. https://doi.org/10.1038/s41591-018-0014-x Nature Publishing Group.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (80- ). 2015:69–74. https://doi.org/10.1126/science.aaa4971.
Kim ES, Kim JE, Patel MA, Mangraviti A, Ruzevick J, Lim M. Immune checkpoint modulators: an emerging antiglioma armamentarium. J Immunol Res. 2016;2016:4683607.
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017:761–73. https://doi.org/10.7150/jca.17648.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015;15:388–400.
Moretta A, Marcenaro E, Parolini S, Ferlazzo G, Moretta L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ. 2008;15:226–33.
Kim R, Emi M, Tanabe K. Cancer immunosuppression and autoimmune disease: beyond immunosuppressive networks for tumour immunity. Immunology. 2006;119:254–64.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99.
Bi J, Tian Z. NK cell exhaustion. Front Immunol. 2017;8. https://doi.org/10.3389/fimmu.2017.00760.
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (80- ). 1996;271:1734–6.
Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–95.
Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118:9–16.
Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (80- ). Science. 2011;332:600–3. https://doi.org/10.1126/science.1202947.
Schnell A, Bod L, Madi A, Kuchroo VK. The yin and yang of co-inhibitory receptors: toward anti-tumor immunity without autoimmunity. Cell Res. 2020:285–99. https://doi.org/10.1038/s41422-020-0277-x Springer Nature.
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (80- ). Science. 2008;322:271–5. https://doi.org/10.1126/science.1160062.
Chan DV, Gibson HM, Aufiero BM, Wilson AJ, Hafner MS, Mi QS, et al. Differential CTLA-4 expression in human CD4+ versus CD8 + T cells is associated with increased NFAT1 and inhibition of CD4+ proliferation. Genes Immun. 2014;15:25–32. https://doi.org/10.1038/gene.2013.57 Nature Publishing Group.
Khan M, Arooj S, Wang H. NK cell-based immune checkpoint inhibition. Front Immunol. 2020;11:167.
Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–80.
Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (80- ). 2017;355:1428–33. https://doi.org/10.1126/science.aaf1292 American Association for the Advancement of Science.
Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–44. https://doi.org/10.1182/blood-2008-12-195792.
MacFarlane AW, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, et al. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol Res. 2014;2:320–31. https://doi.org/10.1158/2326-6066.CIR-13-0133 American Association for Cancer Research Inc.
Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J Allergy Clin Immunol. Mosby Inc. 2017;139:335–346.e3. https://doi.org/10.1016/j.jaci.2016.04.025.
Purdy AK, Campbell KS. SHP-2 expression negatively regulates NK cell function. J Immunol. The American Association of Immunologists. 2009;183:7234–43. https://doi.org/10.4049/jimmunol.0900088.
Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed Death-1 ligand 1 interacts specifically with the B7-1 Costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–22. https://doi.org/10.1016/j.immuni.2007.05.016.
Ni X, Song Q, Cassady K, Deng R, Jin H, Zhang M, et al. PD-L1 interacts with CD80 to regulate graft-versus-leukemia activity of donor CD8+ T cells. J Clin Invest. 2017;127:1960–77. https://doi.org/10.1172/JCI91138.
Cai J, Wang D, Zhang G, Guo X. The role of PD-1/PD-L1 axis in treg development and function: implications for cancer immunotherapy. Onco Targets Ther Dove Medical Press Ltd. 2019:8437–45. https://doi.org/10.2147/OTT.S221340.
Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, et al. Successful anti-PD-1 Cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. 2018;49:1148–61.
Wan C, Keany MP, Dong H, Al-Alem LF, Pandya UM, Lazo S, et al. Enhanced efficacy of simultaneous PD-1 and PD-L1 immune checkpoint blockade in high-grade serous ovarian Cancer. Cancer Res. American Association for Cancer Research Inc. 2021;81:158–73. https://doi.org/10.1158/0008-5472.CAN-20-1674.
Stecher C, Battin C, Leitner J, Zettl M, Grabmeier-Pfistershammer K, Höller C, et al. PD-1 blockade promotes emerging checkpoint inhibitors in enhancing T cell responses to allogeneic dendritic cells. Front Immunol. Frontiers Media S.A. 2017;8. https://doi.org/10.3389/fimmu.2017.00572.
Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44:989–1004.
Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 expression defines a subset of CD4 + CD25 high Foxp3 + regulatory T cells that are expanded at tumor sites. J Immunol. 2010;184:6545–51.
Narayanan S, Ahl PJ, Bijin VA, Kaliaperumal N, Lim G, Wang C, et al. LAG3 is a Central Regulator of NK Cell Cytokine Production. bioRxiv. 2020;01.31.9282.
Graydon CG, Mohideen S, Fowke KR. LAG3’s enigmatic mechanism of action. Front Immunol Frontiers Media SA. 2021;11:615317. https://doi.org/10.3389/fimmu.2020.615317.
Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. 2015;6:27359–77.
Burova E, Hermann A, Dai J, Ullman E, Halasz G, Potocky T, et al. Preclinical development of the anti-LAG-3 antibody REGN3767: characterization and activity in combination with the anti-PD-1 antibody cemiplimab in human PD-1xLAG-3–knockin mice. Mol Cancer Ther. American Association for Cancer Research Inc. 2019;18:2051–62. https://doi.org/10.1158/1535-7163.MCT-18-1376.
Wierz M, Pierson S, Guyonnet L, Viry E, Lequeux A, Oudin A, et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood American Society of Hematology. 2018:1617–21. https://doi.org/10.1182/blood-2017-06-792267.
Marcq E, Van Audenaerde JRM, De Waele J, Merlin C, Pauwels P, Van Meerbeeck JP, et al. The search for an interesting partner to combine with PD-L1 blockade in mesothelioma: focus on TIM-3 and LAG-3. Cancers (Basel). MDPI AG. 2021;13:1–14. https://doi.org/10.3390/cancers13020282.
Jie HB, Gildener-Leapman N, Li J, Srivastava RM, Gibson SP, Whiteside TL, et al. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer. 2013;109:2629–35.
Ohs I, Ducimetière L, Marinho J, Kulig P, Becher B, Tugues S. Restoration of natural killer cell antimetastatic activity by IL12 and checkpoint blockade. Cancer Res. American Association for Cancer Research Inc. 2017;77:7059–71. https://doi.org/10.1158/0008-5472.CAN-17-1032.
Sakuishi K, Ngiow SF, Sullivan JM, Teng MWL, Kuchroo VK, Smyth MJ, et al. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology. 2013;2:e23849.
Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–52. https://doi.org/10.1038/ni1271.
Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of TIM-3+FoxP3+ regulatory T cells. Eur J Immunol. 2014;44:2703–11.
van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun. 2006;351:571–6. https://doi.org/10.1016/j.bbrc.2006.10.079.
Davidson D, Schraven B, Veillette A. PAG-associated FynT regulates calcium signaling and promotes Anergy in T lymphocytes. Mol Cell Biol. American Society for Microbiology. 2007;27:1960–73. https://doi.org/10.1128/mcb.01983-06.
Sehrawat S, Reddy PBJ, Rajasagi N, Suryawanshi A, Hirashima M, Rouse BT. Galectin-9/TIM-3 interaction regulates virus-specific primary and memory CD8 T cell response. PLoS Pathog. 2010;6:1–16. https://doi.org/10.1371/JOURNAL.PPAT.1000882.
Yang R, Sun L, Li CF, Wang YH, Yao J, Li H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun Nature Research. 2021;12. https://doi.org/10.1038/s41467-021-21099-2.
Liu JF, Ma SR, Mao L, Bu LL, Yu GT, Li YC, et al. T-cell immunoglobulin mucin 3 blockade drives an antitumor immune response in head and neck cancer. Mol Oncol. 2017;11:235–47.
Yang M, Du W, Yi L, Wu S, He C, Zhai W, et al. Checkpoint molecules coordinately restrain hyperactivated effector T cells in the tumor microenvironment. Oncoimmunology. 2020;9:1708064.
Oweida A, Hararah MK, Phan A, Binder D, Bhatia S, Lennon S, et al. Resistance to radiotherapy and PD-L1 blockade is mediated by TIM-3 upregulation and regulatory T-cell infiltration. Clin Cancer Res. American Association for Cancer Research Inc. 2018;24:5368–80. https://doi.org/10.1158/1078-0432.CCR-18-1038.
Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. American Association for Cancer Research Inc. 2017;23:124–36. https://doi.org/10.1158/1078-0432.CCR-15-1535.
Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol. 2015;29:635–41.
Seo H, Kim BS, Bae EA, Min BS, Han YD, Shin SJ, et al. IL21 therapy combined with PD-1 and Tim-3 blockade provides enhanced NK cell antitumor activity against MHC class I-deficient tumors. Cancer Immunol Res. American Association for Cancer Research Inc. 2018;6:685–95. https://doi.org/10.1158/2326-6066.CIR-17-0708.
Maurya N, Gujar R, Gupta M, Yadav V, Verma S, Sen P. Immunoregulation of dendritic cells by the receptor T cell Ig and Mucin Protein-3 via Bruton’s tyrosine kinase and c-Src. J Immunol. 2014;193:3417–25.
Wang T, Zhang J, Li N, Li M, Ma S, Tan S, et al. Spatial distribution and functional analysis define the action pathway of Tim-3/Tim-3 ligands in tumor development. Mol Ther. 2021. https://doi.org/10.1016/J.YMTHE.2021.11.015.
Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM, et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers (Basel). 2019;11:877.
Alteber Z, Kotturi MF, Whelan S, Ganguly S, Weyl E, Pardoll DM, et al. Therapeutic targeting of checkpoint receptors within the DNAM1 Axis. Cancer Discov. 2021;11:1040–51. https://doi.org/10.1158/2159-8290.CD-20-1248.
Li M, Xia P, Du Y, Liu S, Huang G, Chen J, et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. J Biol Chem. American Society for Biochemistry and Molecular Biology Inc. 2014;289:17647–57. https://doi.org/10.1074/jbc.M114.572420.
Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol. The American Association of Immunologists. 2011;186:1338–42. https://doi.org/10.4049/jimmunol.1003081.
Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10:48–57. https://doi.org/10.1038/ni.1674.
Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The Immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell. 2014;26:923–37.
Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. Taylor and Francis Inc. 2018;7. https://doi.org/10.1080/2162402X.2018.1466769.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19:723–32.
Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, et al. Identification of CD112R as a novel checkpoint for human T cells. J Exp Med Rockefeller University Press. 2016;213:167–76. https://doi.org/10.1084/jem.20150785.
Li J, Whelan S, Kotturi MF, Meyran D, D’Souza C, Hansen K, et al. PVRIG is a novel natural killer cell immune checkpoint receptor in acute myeloid leukemia. Haematologica. 2021;106. https://doi.org/10.3324/HAEMATOL.2020.258574.
Li Y, Zhang Y, Cao G, Zheng X, Sun C, Wei H, et al. Blockade of checkpoint receptor PVRIG unleashes anti-tumor immunity of NK cells in murine and human solid tumors. J Hematol Oncol. 2021;14:100. https://doi.org/10.1186/S13045-021-01112-3.
Xu F, Sunderland A, Zhou Y, Schulick RD, Edil BH, Zhu Y. Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol Immunother. Springer Science and Business Media Deutschland GmbH. 2017;66:1367–75. https://doi.org/10.1007/s00262-017-2031-x.
Murter B, Pan X, Ophir E, Alteber Z, Azulay M, Sen R, et al. Mouse PVRIg has CD8 + T cell-specific coinhibitory functions and dampens antitumor immunity. Cancer Immunol Res. 2019;7:244–56.
Whelan S, Ophir E, Kotturi MF, Levy O, Ganguly S, Leung L, et al. PVRIG and PVRL2 are induced in Cancer and inhibit CD8 + T-cell function. Cancer Immunol Res. 2019;7:257–68. https://doi.org/10.1158/2326-6066.CIR-18-0442.
Long EO, Barber DF, Burshtyn DN, Faure M, Peterson M, Rajagopalan S, et al. Inhibition of natural killer cell activation signals by killer cell immunoglobulin-like receptors (CD158). Immunol Rev. 2001;181:223–33.
Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev NIH Public Access. 2008:70–84. https://doi.org/10.1111/j.1600-065X.2008.00660.x.
Huard B, Karlsson L. KIR expression on self-reactive CD8+ T cells is controlled by T-cell receptor engagement. Nature. 2000;403:325–8.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell aloreactivity in mismatched hematopoietic transplants. Science (80- ). 2002;295:2097–100.
He Y, Bunn PA, Zhou C, Chan D. KIR 2D (L1, L3, L4, S4) and KIR 3DL1 protein expression in non-small cell lung cancer. Oncotarget. 2016;7:82104–11.
Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology. 2016;6:e1264562.
Braud VM, Aldemir H, Breart B, Ferlin WG. Expression of CD94-NKG2A inhibitory receptor is restricted to a subset of CD8+ T cells. Trends Immunol. 2003;24:162–4.
Le Dréan E, Vély F, Olcese L, Cambiaggi A, Guia S, Krystal G, et al. Inhibition of antigen-induced T cell response and antibody-induced NK cell cytotoxicity by NKG2A: association of NKG2A with SHP-1 and SHP-2 protein-tyrosine phosphatases. Eur J Immunol; 1998;28:264–276. doi: 10.1002/(SICI)1521-4141(199801)28:01<264::AID-IMMU264>3.0.CO;2-O.
André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175:1731–43.
Ruggeri L, Urbani E, André P, Mancusi A, Tosti A, Topini F, et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica Ferrata Storti Foundation. 2016;101:626–33. https://doi.org/10.3324/haematol.2015.135301.
Hatherley D, Lea SM, Johnson S, Barclay AN. Structures of CD200/CD200 receptor family and implications for topology, regulation, and evolution. Structure Elsevier. 2013;21:820–32. https://doi.org/10.1016/j.str.2013.03.008.
Zhang S, Cherwinski H, Sedgwick JD, Phillips JH. Molecular mechanisms of CD200 inhibition of mast cell activation. J Immunol The American Association of Immunologists. 2004;173:6786–93. https://doi.org/10.4049/jimmunol.173.11.6786.
Liu JQ, Hu A, Zhu J, Yu J, Talebian F, Bai XF. CD200-CD200R pathway in the regulation of tumor immune microenvironment and immunotherapy. Adv Exp Med Biol Springer. 2020:155–65. https://doi.org/10.1007/978-3-030-35582-1_8.
Coles SJ, Wang ECY, Man S, Hills RK, Burnett AK, Tonks A, et al. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia. 2011;25:792–9.
Gorczynski RM, Chen Z, Erin N, Khatri I, Podnos A. Comparison of immunity in mice cured of primary/metastatic growth of EMT6 or 4THM breast cancer by chemotherapy or immunotherapy. PLoS One. 2014;9:e113597.
Pallasch CP, Ulbrich S, Brinker R, Hallek M, Uger RA, Wendtner CM. Disruption of T cell suppression in chronic lymphocytic leukemia by CD200 blockade. Leuk Res. 2009;33:460–4.
Veillette A, Chen J. SIRPα–CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol Elsevier Ltd. 2018:173–84. https://doi.org/10.1016/j.it.2017.12.005.
Kim MJ, Lee JC, Lee JJ, Kim S, Lee SG, Park SW, et al. Association of CD47 with natural killer cell-mediated cytotoxicity of head-and-neck squamous cell carcinoma lines. Tumor Biol. 2008;29:28–34.
Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21:1209–15.
Lian S, Xie R, Ye Y, Xie X, Li S, Lu Y, et al. Simultaneous blocking of CD47 and PD-L1 increases innate and adaptive cancer immune responses and cytokine release. EBioMedicine. 2019;42:281–95.
Cheung TC, Oborne LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, et al. T cell intrinsic Heterodimeric complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. J Immunol. The American Association of Immunologists. 2009;183:7286–96. https://doi.org/10.4049/jimmunol.0902490.
Gavrieli M, Watanabe N, Loftin SK, Murphy TL, Murphy KM. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem Biophys Res Commun. Academic Press Inc. 2003;312:1236–43. https://doi.org/10.1016/j.bbrc.2003.11.070.
Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8 + T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012;72:887–96.
Jones A, Bourque J, Kuehm L, Opejin A, Teague RM, Gross C, et al. Immunomodulatory functions of BTLA and HVEM govern induction of Extrathymic regulatory T cells and tolerance by dendritic cells. Immunity. 2016;45:1066–77.
Chen YL, Lin HW, Chien CL, Lai YL, Sun WZ, Chen CA, et al. BTLA blockade enhances Cancer therapy by inhibiting IL-6/IL-10-induced CD19high B lymphocytes. J Immunother Cancer. 2019;7:313.
Celis-Gutierrez J, Blattmann P, Zhai Y, Jarmuzynski N, Ruminski K, Grégoire C, et al. Quantitative Interactomics in primary T cells provides a rationale for concomitant PD-1 and BTLA Coinhibitor blockade in Cancer immunotherapy. Cell Rep. 2019;27:3315–30.
Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature. 2019;574:565–70.
Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O’Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014;74:1924–32.
Huang X, Zhang X, Li E, Zhang G, Wang X, Tang T, et al. VISTA: an immune regulatory protein checking tumor and immune cells in cancer immunotherapy. J Hematol Oncol BioMed Central. 2020:83. https://doi.org/10.1186/s13045-020-00917-y.
Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. 2014;74:1933–44.
Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A. 2015;112:6682–7.
Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-γ production. Nat Immunol. 2001;2:269–74.
Prasad DVR, Nguyen T, Li Z, Yang Y, Duong J, Wang Y, et al. Murine B7-H3 is a negative regulator of T cells. J Immunol. 2004;173:2500–6.
Lemke D, Pfenning PN, Sahm F, Klein AC, Kempf T, Warnken U, et al. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape and invasiveness. Clin Cancer Res. 2012;18:105–17.
Yamato I, Sho M, Nomi T, Akahori T, Shimada K, Hotta K, et al. Clinical importance of B7-H3 expression in human pancreatic cancer. Br J Cancer. 2009;101:1709–16.
Yonesaka K, Haratani K, Takamura S, Sakai H, Kato R, Takegawa N, et al. B7-h3 negatively modulates ctl-mediated cancer immunity. Clin Cancer Res. 2018;24:2653–64.
Georgiev H, Ravens I, Papadogianni G, Bernhardt G. Coming of age: CD96 emerges as modulator of immune responses. Front Immunol. Frontiers Media S.A. 2018. https://doi.org/10.3389/fimmu.2018.01072.
Chiang EY, Almeida PE, Almeida Nagata DE, Bowles KH, Du X, Chitre AS, et al. CD96 functions as a co-stimulatory receptor to enhance CD8 + T cell activation and effector responses. Eur J Immunol. 2020;50:891–902.
Mittal D, Lepletier A, Madore J, Aguilera AR, Stannard K, Blake SJ, et al. CD96 is an immune checkpoint that regulates CD8+ T-cell antitumor function. Cancer Immunol Res. 2019;7:559–71.
Sayós J, Martínez-Barriocanal A, Kitzig F, Bellón T, López-Botet M. Recruitment of C-terminal Src kinase by the leukocyte inhibitory receptor CD85j. Biochem Biophys Res Commun. 2004;324:640–7. https://doi.org/10.1016/j.bbrc.2004.09.097.
Rouas-Freiss N, Moreau P, Lemaoult J, Carosella ED. The dual role of HLA-G in cancer. J Immunol Res. 2014;2014:359748.
Villa-Álvarez M, Sordo-Bahamonde C, Lorenzo-Herrero S, Gonzalez-Rodriguez AP, Payer AR, Gonzalez-Garcia E, et al. Ig-like transcript 2 (ILT2) blockade and Lenalidomide restore NK cell function in chronic lymphocytic leukemia. Front Immunol. 2018;9:2917.
Ikehara Y, Ikehara SK, Paulson JC. Negative regulation of T cell receptor signaling by Siglec-7 (p70/AIRM) and Siglec-9. J Biol Chem. 2004;279:43117–25.
Varchetta S, Brunetta E, Roberto A, Mikulak J, Hudspeth KL, Mondelli MU, et al. Engagement of Siglec-7 receptor induces a pro-inflammatory response selectively in monocytes. PLoS One. 2012;7:e45821.
Higuchi H, Shoji T, Iijima S, Nishijima KI. Constitutively expressed Siglec-9 inhibits LPS-induced CCR7, but enhances IL-4-induced CD200R expression in human macrophages. Biosci Biotechnol Biochem. 2016;80:1141–8.
Ohta M, Ishida A, Toda M, Akita K, Inoue M, Yamashita K, et al. Immunomodulation of monocyte-derived dendritic cells through ligation of tumor-produced mucins to Siglec-9. Biochem Biophys Res Commun. 2010;402:663–9.
Adams OJ, Stanczak MA, Von Gunten S, Läubli H. Targeting sialic acid-Siglec interactions to reverse immune suppression in cancer. Glycobiology. 2018;28:640–7.
Stanczak MA, Siddiqui SS, Trefny MP, Thommen DS, Boligan KF, Von Gunten S, et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest. 2018;128:4912–23.
Jandus C, Boligan KF, Chijioke O, Liu H, Dahlhaus M, Démoulins T, et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J Clin Invest. 2014;124:1810–20.
Robbins SH, Nguyen KB, Takahashi N, Mikayama T, Biron CA, Brossay L. Cutting edge: inhibitory functions of the killer cell Lectin-like receptor G1 molecule during the activation of mouse NK cells. J Immunol. 2002;168:2585–9.
Gründemann C, Bauer M, Schweier O, von Oppen N, Lässing U, Saudan P, et al. Cutting edge: identification of E-cadherin as a ligand for the murine killer cell Lectin-like receptor G1. J Immunol. 2006;176:1311–5.
Tessmer MS, Fugere C, Stevenaert F, Naidenko OV, Chong HJ, Leclercq G, et al. KLRG1 binds cadherins and preferentially associates with SHIP-1. Int Immunol. 2007;19:391–400. https://doi.org/10.1093/intimm/dxm004.
Gründemann C, Schwartzkopff S, Koschella M, Schweier O, Peters C, Voehringer D, et al. The NK receptor KLRG1 is dispensable for virus-induced NK and CD8 + T-cell differentiation and function in vivo. Eur J Immunol. 2010;40:1303–14.
Ito M, Maruyama T, Saito N, Koganei S, Yamamoto K, Matsumoto N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med. 2006;203:289–95.
Greenberg SA, Kong SW, Thompson E, Gulla SV. Co-inhibitory T cell receptor KLRG1: human cancer expression and efficacy of neutralization in murine cancer models. Oncotarget. 2019;10:1399–406.
Latchman Y, McKay PF, Reiser H. Cutting edge: identification of the 2B4 molecule as a counter-receptor for CD48. J Immunol. 1998;161:5809–12.
Hosen N, Ichihara H, Mugitani A, Aoyama Y, Fukuda Y, Kishida S, et al. CD48 as a novel molecular target for antibody therapy in multiple myeloma. Br J Haematol. 2012;156:213–24. https://doi.org/10.1111/j.1365-2141.2011.08941.x.
Pahima H, Puzzovio PG, Levi-Schaffer F. 2B4 and CD48: a powerful couple of the immune system. Clin Immunol Academic Press Inc. 2019:64–8. https://doi.org/10.1016/j.clim.2018.10.014.
Lanuza PM, Pesini C, Arias MA, Calvo C, Ramirez-Labrada A, Pardo J. Recalling the biological significance of immune checkpoints on NK cells: a chance to overcome LAG3, PD1, and CTLA4 inhibitory pathways by adoptive NK cell transfer? Front Immunol. Frontiers Media SA. 2020. https://doi.org/10.3389/fimmu.2019.03010.
Wu Y, Kuang DM, Pan WD, Le WY, Lao XM, Wang D, et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology. 2013;57:1107–16.
Agresta L, Lehn M, Lampe K, Cantrell R, Hennies C, Szabo S, et al. CD244 represents a new therapeutic target in head and neck squamous cell carcinoma. J Immunother Cancer. 2020;8:1–12.
Vaidya SV, Stepp SE, McNerney ME, Lee J-K, Bennett M, Lee K-M, et al. Targeted disruption of the 2B4 gene in mice reveals an in vivo role of 2B4 (CD244) in the rejection of B16 melanoma cells. J Immunol. 2005;174:800–7.
Duan Q, Zhang H, Zheng J, Zhang L. Turning cold into hot: firing up the tumor microenvironment. Trends Cancer. 2020;6:605–18. https://doi.org/10.1016/J.TRECAN.2020.02.022.
Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 2019;36:471–82. https://doi.org/10.1016/J.CCELL.2019.09.006.
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–44. https://doi.org/10.1172/JCI83092.
John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636–46. https://doi.org/10.1158/1078-0432.CCR-13-0458.
Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. 2019;68:365–77. https://doi.org/10.1007/S00262-018-2281-2.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol Nature Research. 2020:7–24. https://doi.org/10.1038/s41577-019-0210-z.
Kodumudi KN, Ramamoorthi G, Snyder C, Basu A, Jia Y, Awshah S, et al. Sequential anti-PD1 therapy following dendritic cell vaccination improves survival in a HER2 mammary carcinoma model and identifies a critical role for CD4 T cells in mediating the response. Front Immunol. 2019;10. https://doi.org/10.3389/FIMMU.2019.01939.
Yazdani M, Gholizadeh Z, Nikpoor AR, Mohamadian Roshan N, Jaafari MR, Badiee A. Ex vivo dendritic cell-based (DC) vaccine pulsed with a low dose of liposomal antigen and CpG-ODN improved PD-1 blockade immunotherapy. Sci Rep. 2021;11. https://doi.org/10.1038/S41598-021-94250-0.
Sun C, Nagaoka K, Kobayashi Y, Nakagawa H, Kakimi K, Nakajima J. Neoantigen dendritic cell vaccination combined with anti-CD38 and CpG elicits anti-tumor immunity against The immune checkpoint therapy-resistant murine lung Cancer cell line LLC1. Cancers (Basel). 2021;13:5508. https://doi.org/10.3390/CANCERS13215508.
Patel SA, Minn AJ. Combination Cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48:417–33. https://doi.org/10.1016/J.IMMUNI.2018.03.007.
Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020;17:725–41. https://doi.org/10.1038/S41571-020-0413-Z.
Pauken KE, Dougan M, Rose NR, Lichtman AH, Sharpe AH. Adverse events following Cancer immunotherapy: obstacles and opportunities. Trends Immunol. Elsevier Ltd. 2019:511–23. https://doi.org/10.1016/j.it.2019.04.002.
Dougan M, Luoma AM, Dougan SK, Wucherpfennig KW. Understanding and treating the inflammatory adverse events of cancer immunotherapy. Cell. 2021;S0092-8674:00161–6. https://doi.org/10.1016/j.cell.2021.02.011.
Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for Cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. 2016;22:1865–74. https://doi.org/10.1158/1078-0432.CCR-15-1507.
Sutherland KD, Visvader JE. Cellular mechanisms underlying Intertumoral heterogeneity. Trends Cancer. 2015;1:15–23. https://doi.org/10.1016/J.TRECAN.2015.07.003.
Verhagen J, Genolet R, Britton GJ, Stevenson BJ, Sabatos-Peyton CA, Dyson J, et al. CTLA-4 controls the thymic development of both conventional and regulatory T cells through modulation of the TCR repertoire. Proc Natl Acad Sci U S A. 2013:110. https://doi.org/10.1073/pnas.1208573110.
Sasikumar PG, Ramachandra M. Small-molecule immune checkpoint inhibitors targeting PD-1/PD-L1 and other emerging checkpoint pathways. BioDrugs Springer International Publishing. 2018:481–97. https://doi.org/10.1007/s40259-018-0303-4.
Hommes JW, Verheijden RJ, Suijkerbuijk KPM, Hamann D. Biomarkers of checkpoint inhibitor induced immune-related adverse events—a comprehensive review. Front Oncol Frontiers Media SA. 2021;10:585311. https://doi.org/10.3389/fonc.2020.585311.
Jing Y, Liu J, Ye Y, Pan L, Deng H, Wang Y, et al. Multi-omics prediction of immune-related adverse events during checkpoint immunotherapy. Nat Commun. Nature Research. 2020;11. https://doi.org/10.1038/s41467-020-18742-9.
Acknowledgements
We thank G. Jorba (SOM Innovation Biotech, S.A.) for exhaustively analyzing ongoing clinical trials.
Funding
AAO acknowledges the Government of Andorra for a predoctoral grant, ATC0XX - AND-2019/2020. P. Muñoz’s laboratory is supported by the Spanish Ministry of Science and Innovation (PID2020-113495RB-I00; co-funded by FEDER funds/European Regional Development Fund (ERDF) – A way to make Europe) and by the Catalan Department of Health (CERCA; Generalitat de Catalunya, 2017/SGR565). JML acknowledges funding by Instituto de Salud Carlos III through the project “PI19/01210” (Co-funded by European Regional Development Fund, A way to make Europe).
Author information
Authors and Affiliations
Contributions
AAO and PMM designed the study. AAO and CD reviewed the information. AAO wrote the manuscript. CD, JML, and PMM critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
JML has received advisory fees from Bristol-Myers Squibb, MSD, Novartis, Pierre Fabre, Roche, Sanofi; lecture fees from Astellas, Bristol-Myers Squibb, MSD, Novartis, Pierre Fabre, Pfizer, Roche; and travel grants from Bristol-Myers Squibb, MSD, Novartis, Pierre Fabre, Pfizer, Roche, Ipsen. All other authors declare no potential conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Phase III clinical trials of single-blockade immune checkpoint inhibitors including CTLA-4, PD-1, and PD-L1 in 2021, according to www.clinicaltrials.gov.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Archilla-Ortega, A., Domuro, C., Martin-Liberal, J. et al. Blockade of novel immune checkpoints and new therapeutic combinations to boost antitumor immunity. J Exp Clin Cancer Res 41, 62 (2022). https://doi.org/10.1186/s13046-022-02264-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02264-x