
Abstract
Dynamin 2 (DNM2) is an ubiquitously expressed large GTPase well known for its role in vesicle formation in endocytosis and intracellular membrane trafficking also acting as a regulator of cytoskeletons. During the last two decades, DNM2 involvement, through mutations or overexpression, emerged in an increasing number of cancers and often associated with poor prognosis. A wide panel of DNM2-dependent processes was described in cancer cells which explains DNM2 contribution to cancer pathomechanisms. First, DNM2 dysfunction may promote cell migration, invasion and metastasis. Second, DNM2 acts on intracellular signaling pathways fostering tumor cell proliferation and survival. Relative to these roles, DNM2 was demonstrated as a therapeutic target able to reduce cell proliferation, induce apoptosis, and reduce the invasive phenotype in a wide range of cancer cells in vitro. Moreover, proofs of concept of therapy by modulation of DNM2 expression was also achieved in vivo in several animal models. Consequently, DNM2 appears as a promising molecular target for the development of anti-invasive agents and the already provided proofs of concept in animal models represent an important step of preclinical development.
Similar content being viewed by others
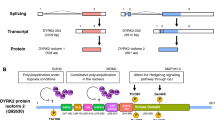


Background
Dynamin 2 (DNM2) belongs to a superfamily of large GTPases including three classical dynamins and several dynamin-like proteins sharing propensity to self-assemble into polymeric structures [1]. DNM2 is ubiquitously expressed and is encoded by the DNM2 gene producing four major splice isoforms using a combination of two alternative splice sites [2]. In addition, a muscle-specific isoform was identified [3]. DNM2 is a central actor of many membrane remodeling processes including clathrin-mediated endocytosis [4], clathrin-independent endocytosis [5, 6], coat-independent endocytosis [7, 8], intracellular vesicle trafficking [9,10,11,12,13], and exocytosis [14]. For its function of vesicle formation, DNM2 oligomerizes in a helical structure around the neck of nascent vesicles [4], and GTP hydrolysis leads to modifications in the helical structure associated with the release of the vesicles [15]. A function of regulator of actin [16,17,18] and microtubule [19, 20] cytoskeletons is now well recognized for DNM2 which sustains, in particular, its participation to mitosis and cell cycle progression [8, 21,22,23]. Heterozygous mutations in the DNM2 gene are responsible for autosomal dominant forms of three human diseases, i.e. centronuclear myopathy [24], Charcot-Marie-Tooth disease [25], and hereditary spastic paraplegia [26] and one homozygous mutation was shown to cause a lethal congenital syndrome [27]. DNM2 has also been described as a susceptibility gene for late-onset Alzheimer disease [28]. In addition, numerous studies are now available involving DNM2 in several cancers. The purpose here is to review involvement of DNM2 in pathophysiology of cancers and highlight the opened perspectives in medicine using DNM2 as a therapeutic target in these conditions.
DNM2 deregulation in cancers
Acute lymphoblastic leukaemia (ALL) is the most common malignancy in B cells, immature T lymphocytes or lymphoid progenitors. Somatic heterozygous mutations in the DNM2 gene were identified in children affected by early T-cell precursor acute lymphoblastic leukaemia (ETP-ALL), a form of ALL associated with a high risk of treatment failure [29]. In this study, DNM2 mutations have been identified in 17 patients (13 out of 64 ETP patients and 4 out of 42 non ETP ALL patients) including 2 cases with biallelic mutations. The 19 mutations include 3 frameshifts, 3 splice site mutations, 6 missense mutations, 2 nonsense mutations, and 5 in frame deletions or insertions not found in centronuclear myopathy, Charcot-Marie-Tooth disease and hereditary spastic paraplegia. Thereafter, 4 other DNM2 mutations (1 nonsense and 3 missense) have been described in adult T cell ALL patients extending the DNM2 involvement in ALL [30]. Clinical analysis of this small cohort of patients suggested that DNM2 mutations are associated with a poor prognosis.
In addition to the DNM2 mutations, an increased DNM2 expression was also identified in several cancer types (summarized in Table 1). DNM2 expression is increased in ALL, especially in B-ALL in which overexpression is associated with leukaemia cell proliferation and a poor prognosis [31]. DNM2 overexpression was also demonstrated in stem cells and progenitor cells of chronic myeloid leukemia (CML), i.e. a clonal myeloproliferative disorder originating from hematopoietic stem cells [32], in multiple ovarian cancer data sets [33], in bladder tumors in which the overexpression is correlated with the grade progression [34], in papillary thyroid cancer in which high DNM2 expression is associated with unfavourable prognosis (tumor recurrence, overall survival rate, distant metastases) [35], and in prostate cancer in which high DNM2 expression is associated with higher aggressiveness (Gleason score) and mortality [36, 37]. DNM2 overexpression was also reported in cervical cancer. In this cancer, the initial transformation of normal epithelium to preneoplastic cervical intraepithelial neoplasia (CIN) may be followed by a transformation to invasive cancer. DNM2 is not expressed in normal cervical epithelium but is overexpressed in preinvasive low grade lesions (CIN grade 1). With cancer progression, DNM2 expression decreases through an unidentified mechanism leading to low or no expression in high-grade lesions (CIN grades 2 and 3) especially, in case of deep tumor invasion and lymph node metastasis [38, 39]. These results establish DNM2 expression as a biomarker in grading of CIN, with a negative correlation between DNM2 expression and the severity of lesions, and suggest a crucial impact of DNM2 overexpression in the earliest steps of neoplasia of cervix. Both expression and localization of DNM2 were studied in tumor and adjacent normal tissue from 113 patients affected by several types of breast cancers [40]. All these breast tumor tissue samples exhibit a higher DNM2 expression compared to normal tissues. Among these cases, cytoplasmic overexpression was correlated with a specific cancer sub-type (invasive ductal carcinoma) whereas membranous DNM2 staining was associated with vascular invasion, an indicator of aggressiveness of breast cancer. Intriguingly, an increased expression of DNM2 was also noticed in the nucleus which is positively correlated with the histological grade and the tumor stage. This non-conventional nuclear localization also suggests a gain-of-function in the nucleus which needs to be deepen explored. In a particular sub-group of breast cancer, i.e. the triple negative breast cancers, defined by absence of estrogen receptor, progesterone receptor and without overexpression or amplification of EGF receptor 2, the level of DNM2 overexpression was negatively correlated with positive chemotherapy outcome [41]. The greater relapse rate in the patients exhibiting the higher DNM2 expression in tumor was attributed to the role of DNM2 in homology-directed repair of DNA, involving a DNM2 mediated trafficking of the RAD51 recombinase, which may mitigate over time the benefit of DNA-damaging chemotherapy in case of DNM2 overexpression. Finally, DNM2 expression was also shown to be markedly upregulated in tumors and metastases of patients affected by pancreatic ductal adenocarcinoma and this overexpression contributes to lamellipodia extension, cell migration and invasion [42]. In addition, patients with the highest levels of DNM2, associated with high level of α-actinin 4, had lower mean survival times compared to patients with lower expression [43]. Altogether, these data highlight the frequent up-regulation of DNM2 in cancers which can be used as a marker of poor prognosis.
Mechanisms responsible for DNM2 overexpression in cancers
The mechanisms underlying DNM2 overexpression in cancers have not been extensively studied and remains an important open question. Of note, Ikaros was shown to bind to the DNM2 promoter in B cells [31] (Fig. 1). Ikaros is a DNA-binding zinc finger protein essential for normal haematopoiesis and immune development and acting as a tumor suppressor gene in acute B- and T-cell ALL. The correlation between low level of Ikaros mRNA and high level of DNM2 mRNA in ALL cells reinforces the hypothesis that Ikaros depletion may explain the high DNM2 expression in this pathological condition [31]. In addition, it was shown in ovarian cancer cell lines that DNM2 levels are transcriptionally down-regulated by the hypoxia-induced factor 1 (HIF-1) which directly binds to the DNM2 promoter [44]. Consequently, HIF-1 inhibition may participate to DNM2 overexpression. The finding that DNM2 overexpression conversely reduced HIF-1α expression [44] may create a vicious circle maintaining a high level of DNM2 transcription. Finally, a regulation of DNM2 expression by CD9, a member of the tetraspanin family playing important functions in signal transductions from the plasma membrane, was demonstrated in pancreatic cancer cell lines [45]. In these cells, overexpression of CD9 led to the upregulation of the DNM2 expression by an unsolved molecular pathway.

Regulation of DNM2 expression. CD9: CD9 tetraspanin. ER: estrogen receptors. HIF1: hypoxia-induced factor 1. P: phosphorylation of STAT3. PTEN: phosphatase and tensin homolog deleted on chromosome 10. STAT3: signal transducer and activator of transcription 3. Red light: inhibition of DNM2 transcription. Green light: Activation of DNM2 transcription. The figure was built using the Servier medical art database
Other potential regulators of DNM2 transcription in cancers can be inferred from studies relative to another disease in which deleterious DNM2 overexpression occurs. The myotubular myopathy is the severe X-linked form of centronuclear myopathy due to mutations in the MTM1 gene encoding Myotubularin 1. Patients and mouse model of myotubular myopathy exhibit an increased DNM2 expression and a forced DNM2 reduction restores phenotypes and lifespan in a mouse model of the disease [46, 47]. A recent study identified overexpression and activation by phosphorylation of the transcription factor STAT3 (signal transducer and activator of transcription 3) as responsible for the DNM2 upregulation in this pathological condition [48]. STAT3 is a key regulator of cell proliferation, survival and apoptosis and is constitutively activated in most human cancers including prostate, pancreas, breast cancers and leukaemia [49,50,51]. It is tempting to speculate that STAT3 activation is involved in DNM2 upregulation in cancers and that DNM2 overexpression may be an effector of features associated with abnormal STAT3 activation in cancers such as tumor growth and metastasis leading to poor prognosis [49,50,51]. In addition, the demonstration that tamoxifen, known to bind estrogen receptors, normalizes DNM2 expression and improves the phenotype of the Mtm1-deficient mouse model suggests that estrogen receptors may be involved in the regulation of the DNM2 expression [52].
Besides the above-mentioned transcriptional regulations of the DNM2 gene, post-transcriptional mechanisms may be also involved in DNM2 upregulation in cancers. DNM2 is a target of miR-133a-1 and miR-133a-2 (Fig. 1) and silencing of miR-133a in mouse induces an increase in DNM2 in skeletal muscle and a centronuclear myopathy phenotype [53]. It is tempting to speculate that downregulation of miR-133a, as reported in prostate, pancreas and bladder cancers [54], may participate to the DNM2 deregulation in cancers.
Involvement of DNM2 dysfunction in cancer pathophysiology
Several DNM2-dependent pathways have been identified in cancer cell lines and may be involved in cancer pathomechanisms. These mechanisms, summarized in Fig. 2, may be classified in 2 groups: mechanisms promoting cell migration and metastasis, and deregulation of intracellular signaling pathways linked to cell proliferation and survival.

DNM2-dependent processes in tumor cells. AHI-1: Abelson helper integration site-1. BCR-ABL: product of the BCR-ABL fusion gene. Cort: Cortactin. DNM2: Dynamin 2. FasL: Fas Ligand. FBP17: Formin Binding Protein 17. HIF1: hypoxia-induced factor 1. IL-7: interleukin 7. IL-24: interleukin 24. MAPK: mitogen-activated protein kinase. MT1-MMP: membrane type 1 matrix metalloproteinase. P: phosphorylated residue in DNM2. PRL: prolactin. ROS: reactive oxygen species. SFK: Src-family kinase. SHP2: tyrosine-protein phosphatase non-receptor type 11. STAT3: signal transducer and activator of transcription 3. The miR199a and miR638 are both produced from the DNM2 gene. The figure was built using the Servier medical art database
Migration, invasion and metastasis
Cancer cell migration requires formation of actin-based specialized structures such as lamellipodia and filopodia. By its role in the global organization of the actomyosin cytoskeleton through Rac- and cortactin-dependent processes [55,56,57,58,59], DNM2 appears as an key factor for formation and maintenance of these plasma membrane protrusions providing invasive behaviour of cancer cells. Several molecular pathways underlying the DNM2 involvement in cell migration and invasion have been identified in cancer cells. DNM2 mediates the Platelet Derived Growth Factor Receptor α (PDGFRα)-stimulated growth and invasion of glioblastoma cells through the formation of a Src-phosphorylated DNM2-PDGFRα-tyrosine-protein phosphatase non-receptor type 11 (SHP2) complex [60]. In pancreatic adenocarcinoma cell lines, the invasive phenotype was associated to the interaction between DNM2 and Vav1, a Rac1 guanine exchange factor abnormally expressed in pancreatic cancer [61]. In these cells, the physical DNM2-Vav1 interaction protect Vav1 from lysosomal degradation and then promotes activation of Rac1, a small GTPase known to regulate actin dynamics and branching, and the subsequent formation of lamellipodia and invasive cell migration [61]. Like for glioblastoma, importance of phosphorylation of DNM2, at known Src phosphorylation sites, was also demonstrated in migration of pancreatic cancer cell line as overexpression of a phospho-deficient DNM2 mutant is inefficient to promote lamellipodia formation and motile phenotype [42]. In addition to this DNM2 impact on actin cytoskeleton, its impact on microtubule cytoskeleton was demonstrated for migration of pancreatic cancer cells [62]. Indeed, through a direct interaction with Podocalyxin, a plasma membrane glycoprotein abnormally expressed in several cancers, DNM2 regulates the microtubule dynamics leading to reduction of focal adhesions promoting cell migration [62].
On the other hand, plasticity of cell environment, especially through modification of the extracellular matrix, plays an important role in tumor cell invasion and DNM2 was also implicated in this process. DNM2 activity is required for extracellular matrix degradation by invasive tumor cells at invadopodia, another specialized actin-based plasma membrane protrusion [63]. Formation of invadopodia and invasive phenotype of bladder tumor cells involve Formin Binding Protein 17 (FBP17) belonging to the family of formin-binding proteins which regulate the formin-dependent actin assembly [64]. FBP17 interacts with DNM2 in these cells and may then recruit DNM2 to the invadopodia. Extracellular matrix remodelling mediated by invadopodia depends on matrix-degrading proteases such as the secreted and membrane-anchored matrix metalloproteinases including the membrane type 1 matrix metalloproteinase (MT1-MMP). DNM2 provides the proper delivery from late recycling endosomes of MT1-MMP to the invadopodia of breast cancer cells and is required for their ability to degrade matrix [65]. Of note, MT1-MMP is overexpressed in 25% of triple-negative breast cancers [65], the type of aggressive and highly proliferative breast tumors in which DNM2 is also overexpressed [41]. Concomitant overexpression of both DNM2 and MT1-MMP may synergize to increase aggressiveness of breast tumors.
In summary, DNM2 directly impacts tumor cells invasiveness and consequent metastasis by stabilizing distinct actin-based structures involved in cell migration, by remodelling extracellular matrix through metalloproteinase delivery and by promoting focal adhesions disassembly. The regulation of the actin dynamics as the main DNM2 function involved in metastasis was confirmed by the role highlighted for the DNM2-α-actinin 4 complex in the lamellipodia-mediated migration and invadopodia-mediated matrix degradation in pancreatic ductal adenocarcinoma, one of the most aggressive cancer associated with high rates of metastasis [43].
Cell proliferation and survival
DNM2 is the main interactor of the ubiquitously expressed adapter Growth factor receptor-bound protein 2 (Grb2) in hepatocarcinoma cells, suggesting that various signal transduction pathways involving Grb2 may be impaired in case of DNM2 deregulation in cancer cells [66]. Over-activation of such signal transduction pathways, requiring a DNM2-dependent receptor internalization for their downstream signaling, may be deleterious by increasing tumor cell proliferation and survival as suggested in breast cancer and leukemia cells. In breast cancer cells, the association between prolactin exposure and development of invasive breast cancer is widely accepted. Interestingly, the ligand-induced prolactin receptor endocytosis, required for the activation of the downstream Src family kinase-mediated signaling cascade linked to cell proliferation, was shown to be DNM2-dependent [67]. In the same line, prolactin also enhances Insulin growth factor 1 (IGF1)-induced phosphorylation of IGF1-receptor increasing its AKT and ERK1/2 downstream signaling associated with proliferation, survival and invasion of breast cancer cells. It was demonstrated in breast cancer cells that DNM2-dependent endocytosis of the IGF1-receptor is required for the prolactin-induced increase of IGF1-receptor phosphorylation inducing IGF1-receptor signaling [68]. In chronic myeloid leukemia cells, a complex formed by DNM2, Abelson helper integration site-1 (AHI-1) and the fusion oncogene BCR-ABL leads to activation of DNM2 through its phosphorylation by the BCR-ABL tyrosine kinase [32]. This aberrant complex was involved in the increase in clathrin-mediated endocytosis, reactive oxygen species (ROS) production and autophagy in the chronic myeloid leukemia stem and progenitor cells [32] possibly at the origin of the cell survival, genomic instability and resistance to treatment. An increase in endocytosis following DNM2 overexpression may also help cancer cells to protect themselves to complement-dependent necrotic cell death as suggested by the DNM2-dependent internalization of complement complexes from the plasma membrane in leukemia cells [69].
In some other conditions, it is more difficult to anticipate the potential functional consequences of DNM2 overexpression. For example in melanoma cells, DNM2 regulates plasma membrane content of the Fas receptor which plays an important role in programmed cell death when bound to Fas ligand [70, 71]. Similarly, the DNM2-dependent endocytosis of the Interleukin 24 (IL-24) with its receptor is required in prostate cancer cells for the tumor suppressor action of the IL-24 (also called MDA-7) [72]. In these two examples, overexpression of DNM2 may reinforce Fas ligand-induced apoptosis or the cancer-specific cell killing activity of IL-24. alternatively, the DNM2 overexpression in these cells may reduce by endocytosis the basal content of Fas and IL-24 receptor in absence of ligands and consequently reduce the beneficial signaling pathways when Fas and IL-24 arrived.
DNM2 overexpression may have also indirect deleterious impacts through concomitant upregulation of miRNA located at the DNM2 locus as already demonstrated for the miR199a-1 in the pathophysiology of the myotubular myopathy [48]. This hypothesis may be of particular interest for the miR-638 located in the intron 1 of DNM2 and known to target two of the main tumor suppressor genes, i.e. PTEN (phosphatase and tensin homolog deleted on chromosome 10) and p53 considered as the ‘‘guardian of the genome’’ [33]. A miR-638 upregulation in DNM2 overexpressing cells may reduce expression of PTEN and p53 (Fig. 1) and then deregulate their specific signaling pathways controlling cell growth and survival, DNA repair, cell-cycle arrest, and apoptosis.
Finally, only one study addressed the functional effects of the DNM2 mutations and their contribution to the T-ALL pathogenesis [73]. This study suggested that DNM2 mutations affect the clathrin-mediated endocytosis through a dominant-negative effect. By this pathomechanism, DNM2 mutations increase the plasma membrane content of Interleukin 7 (IL-7) receptor in pre-leukemic thymocytes leading to enhancing IL-7 signaling and development of more immature T-ALL.
Therapeutic developments
Benefit of DNM2 inhibition was largely documented for almost all the DNM2-related phenotypes in tumor cells in vitro. The function of the DNM2 in cytokinesis was targeted in order to influence the cell cycle progression and reduce proliferation of tumor cells. With this objective, pharmacological inhibitors of DNM2 [74, 75] were used to induce cytokinesis failure at the membrane abscission step leading to growth arrest and death of cervical, lung, and leukaemia cancer cell lines [76,77,78]. This induced cancer cell death following cytokinesis failure results from the intrinsic caspases 3 and 9-mediated apoptotic pathway and is more efficient in cancer cell lines harbouring low level of the anti-apoptotic Bcl-2 and Mcl-1 proteins [79]. Interestingly, nontumorigenic fibroblasts treated with DNM2 inhibitors appear less sensitive to cell death than cancer cells [76, 77]. A benefit a DNM2 inhibition to reduce cell proliferation and/or induce apoptosis was confirmed in a wide range of cancer cells including cervical epithelial cancer cells [80], prostate cancer cells [36, 42], non-small-cell lung cancer cells [81], glioblastoma cells [82], chronic myeloid leukemia cells [32], B- and T-ALL cells [31] and hepatocellular carcinoma cells [83]. The similar benefit using pharmacological DNM2 inhibitors [74, 75, 82, 84,85,86,87,88] or DNM2 gene silencing demonstrates the requirement of the DNM2 GTPase activity in the phenotypes occurring in these cancer cells. In hepatocellular carcinoma cells, dynamin inhibition accelerates degradation of c-Met, a tyrosine kinase receptor involved in hepatocellular carcinoma development and progression, and decreases the c-Met downstream signaling [83]. DNM2 inhibition can also reduce the invasive phenotype of tumor cells in vitro. The benefit of pharmacological inhibitors [75, 84, 85, 89] or DNM2 mRNA silencing on cell invasion in prostate, lung, bladder, and pancreatic cancer cells is provided through an impact on actin dynamics and formation of the actin-based protrusions (lamellipodia, filopodia, and invadopodia) and/or stabilizing focal adhesion which impede invasive behaviour [36, 42, 43, 57, 58, 61, 62, 89]. In melanoma and breast cancer cell lines, DNM2 inhibition impacts cell invasion by reducing extracellular matrix remodelling and the number of degrading cells [63, 65]. DNM2 inhibition was also achieved by targeting the DNM2 gene transcription. Indeed, due to the role of Ikaros in DNM2 expression in ALL cells, effect of 4,5,6,7-Tetrabromobenzotriazole (TBB), an enhancer of Ikaros tumor suppressor activity, was investigated. Interestingly, TBB increased Ikaros binding to the DNM2 promoter and inhibited expression of DNM2 mRNA in ALL cells [31]. A better understanding of the mechanisms underlying the DNM2 overexpression in the different types of cancers will be helpful for developing similar therapeutic approaches.
Importantly, the proof of concept of therapy by modulating DNM2 expression was also achieved in vivo in several animal models (Table 2). In order to assess whether lowering DNM2 levels would increase sensitivity to chemotherapy, breast cancer cells expressing an inducible shRNA to silence DNM2 by RNA interference were implanted into mammary fat pads of mice. Reduced expression of DNM2 significantly improved tumor response to cyclophosphamide, a drug widely used in chemotherapy of breast cancers, leading to reduced tumor volume [41]. A proof of concept was also done using pancreatic cancer cells in vivo [42]. Orthotopic injection of cells overexpressing DNM2 promotes tumor cells dissemination distal from the injection area compared to mice injected with pancreatic cancer cells with basal DNM2 expression. Inhibition of DNM2 by overexpressing phospho-deficient mutant in the injected pancreatic cancer cells drastically reduces the number of large tumors outside the injection site. A third study in which prostate cancer cells were injected in prostate of mice showed that tumor weight and lymph node metastases are reduced when DNM2 is inhibited by RNA interference in the injected cells [36]. A similar result was reported for the PDGFRα-stimulated glioma cell growth and invasion in brain of mice [60]. In this case, injection of DNM2-depleted cells is associated with decrease in tumor cell proliferation and increase in apoptosis. Proofs of concept were also achieved using pharmacological compounds. In a prostate cancer mouse model in which formation of subcutaneous tumors are induced after injection of human prostatic adenocarcinoma cells, intratumoral injection of a DNM2 inhibitor (N’-[4-(dipropylamino)benzylidene]-2-hydroxybenzohydrazide, DBHA) rapidly reduces tumor size without apparent adverse effect [89]. In the same line, continuous delivery of the CyDyn4-36 DNM2 inhibitor by subcutaneous osmotic pumps reduces the size of established tumors formed by prior intracranial injection of glioma stem cells [82] and intraperitoneal injection of the Dynole 34.2 DNM2 inhibitor leads to progressive exhaustion of pre-leukemia stem cells in a mouse model of acute leukemia [90]. Dynole 34.2 also reduces the number of leukemic cells in the bone marrow and spleen of mice after T-ALL and acute myeloid leukemia cells xenografts [90]. DBHA, Dynole 34.2 and CyDyn4-36 represent a new generation of DNM2 inhibitors opening the way for future clinical use. DBHA, is an analogue of the non-competitive inhibitor dynasore, identified through a screening for inhibitors of dynamin GTPase activity, which suppress actin dynamics and cancer cells migration more efficiently with less cytotoxicity compared to dynasore [85, 89]. Dynoles are also non-competitive inhibitors of dynamin GTPase activity [75] and the Dynole 34–2 was shown as the most effective at causing cancer cell death [77]. CyDyn4-36 is a next-generation Dynole designed to be more brain penetrant through a reduction in polar surface area and the number of hydrogen bond donors and acceptors [82]. Altogether, these data highlight Dynamin 2 as a promising molecular target for the development of anti-invasive agents and the proof of concept of therapy already achieved by reducing DNM2 expression in animal models is an important step of preclinical development.
Conclusions
Numerous studies are now available demonstrating the interest of DNM2 as a biomarker for prognosis of tumor invasion and metastasis, as exemplified for cervical cancers, or to predict therapy efficacy as shown in a sub-group of breast cancer. The available data strongly suggest that DNM2 participates to the maintenance of the necessarily favourable environment for tumor to grow, invade and migrate. Interestingly, modulation of DNM2 expression may prove a valuable therapeutic target with proof of concept achieved in vitro and in vivo. Molecules targeting DNM2 expression and/or activity may expand the panoply of anti-mitotic and anti-metastatic agents in cancer treatments. Therapeutic potential of DNM2-based approaches may be of particular interest to counteract metastasis which mark the transition from a benign tumor to a lethal, malignant cancer with dissemination of tumor cells. However, future therapeutic intervention should avoid to excessively reduce DNM2 expression for maintaining the beneficial aspects of DNM2 function in cancer cells as DNA repair already mentioned [41] or the metastasis suppressor function of NME proteins [91]. Indeed, when re-expressed or overexpressed in metastatic tumor cells, NME proteins suppress cell motility and migration in vitro and metastatic colonization in vivo through a mechanism requiring DNM2-dependent endocytosis [91]. The importance to preserve DNM2 expression is also highlighted by the reported analysis of 131 hepatocellular carcinoma showing that patients with low DNM2 expression displayed a significantly worse overall survival [92]. This study concludes that drastic DNM2 downregulation by siRNA or drug in hepatocellular carcinoma cell lines provides colony formation, migration and invasion of tumor cells by reducing the role of DNM2 as a negative regulator of epidermal growth factor (EGF) signaling through the endocytosis of the EGF receptor [92]. Overall, the challenge of future DNM2-based therapies will be to simultaneously maintain DNM2 positive roles and counteract its deleterious functions in tumor cells.
Availability of data and materials
Not applicable.
Abbreviations
- AHI-1:
-
Abelson helper integration site-1
- ALL:
-
Acute lymphoblastic leukaemia
- CML:
-
Chronic myeloid leukemia
- DNM2:
-
Dynamin 2
- ETP-ALL:
-
Early T-cell precursor acute lymphoblastic leukaemia
- EGF:
-
Epidermal growth factor
- FBP17:
-
Formin Binding Protein 17
- Grb2:
-
Growth factor receptor-bound protein 2
- HIF-1:
-
Hypoxia-induced factor 1
- IGF1:
-
Insulin growth factor 1
- IL-7:
-
Interleukin 7
- IL-24:
-
Interleukin 24
- MT1-MMP:
-
Membrane type 1 matrix metalloproteinase
- PDGFRα:
-
Platelet Derived Growth Factor Receptor α
- PTEN:
-
Phosphatase and tensin homolog deleted on chromosome 10
- ROS:
-
Reactive oxygen species
- SHP2:
-
Tyrosine-protein phosphatase non-receptor type 11
- STAT3:
-
Signal transducer and activator of transcription 3
- TBB:
-
4,5,6,7-Tetrabromobenzotriazole
References
Ramachandran R, Schmid SL. The dynamin superfamily. Curr Biol. 2018;28:R411–6.
Cao H, Garcia F, McNiven MA. Differential distribution of dynamin isoforms in mammalian cells. Mol Biol Cell. 1998;9:2595–609.
Cowling BS, Prokic I, Tasfaout H, Rabai A, Humbert F, Rinaldi B, et al. Amphiphysin (BIN1) negatively regulates dynamin 2 for normal muscle maturation. J Clin Invest. 2017;127(12):4477–87.
Warnock DE, Baba T, Schmid SL. Ubiquitously expressed dynamin-II has a higher intrinsic GTPase activity and a greater propensity for self-assembly than neuronal dynamin-I. Mol Biol Cell. 1997;8:2553–62.
Gold ES, Underhill DM, Morrissette NS, Guo J, McNiven MA, Aderem A. Dynamin 2 is required for phagocytosis in macrophages. J Exp Med. 1999;190:1849–56.
Henley JR, Krueger EW, Oswald BJ, McNiven MA. Dynamin-mediated internalization of caveolae. J Cell Biol. 1998;141:85–99.
Cao H, Chen J, Awoniyi M, Henley JR, McNiven MA. Dynamin 2 mediates fluid-phase micropinocytosis in epithelial cells. J Cell Sci. 2007;120:4167–77.
Liu YW, Surka MC, Schroeter T, Lukiyanchuk V, Schmid SL. Isoform and splice-variant specific functions of dynamin-2 revealed by analysis of conditional knock-out cells. Mol Biol Cell. 2008;19:5347–59.
van Dam EM, Stoorvogel W. Dynamin-dependent transferrin receptor recycling by endosome-derived clathrin-coated vesicles. Mol Biol Cell. 2002;13:169–82.
Nicoziani P, Vilhardt F, Llorente A, Hilout L, Courtoy PJ, Sandvig K, et al. Role for dynamin in late endosome dynamics and trafficking of the cation-independent mannose 6-phosphate receptor. Mol Biol Cell. 2000;11:481–95.
Jones SM, Howell KE, Henley JR, Cao H, McNiven MA. Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science. 1998;279:573–7.
Mesaki K, Tanabe K, Obayashi M, Oe N, Takei K. Fission of tubular endosomes triggers endosomal acidification and movement. PLoS One. 2011;6:e19764.
Cao H, Weller S, Orth JD, Chen J, Huang B, Chen JL, et al. Actin and Arf1-dependent recruitment of a cortactin-dynamin complex to the Golgi regulates post-Golgi transport. Nat Cell Biol. 2005;7:483–92.
Gonzalez-Jamett AM, Momboisse F, Guerra MJ, Ory S, Baez-Matus X, Barraza N, et al. Dynamin-2 regulates fusion pore expansion and quantal release through a mechanism that involves actin dynamics in neuroendocrine chromaffin cells. PLoS One. 2013;8:e70638.
Morlot S, Galli V, Klein M, Chiaruttini N, Manzi J, Humbert F, et al. Membrane shape at the edge of the dynamin helix sets location and duration of the fission reaction. Cell. 2012;151:619–29.
Krueger EW, Orth JD, Cao H, McNiven MA. A dynamin-cortactin-Arp2/3 complex mediates actin reorganization in growth factor-stimulated cells. Mol Biol Cell. 2003;14:1085–96.
Sever S, Chang J, Gu C. Dynamin rings: not just for fission. Traffic. 2013;14(12):1194–9.
Gu C, Yaddanapudi S, Weins A, Osborn T, Reiser J, Pollak M, et al. Direct dynamin-actin interactions regulate the actin cytoskeleton. EMBO J. 2010;29:3593–606.
Tanabe K, Takei K. Dynamic instability of microtubules requires dynamin 2 and is impaired in a Charcot-Marie-Tooth mutant. J Cell Biol. 2009;185:939–48.
Henmi Y, Tanabe K, Takei K. Disruption of microtubule network rescues aberrant actin comets in dynamin2-depleted cells. PLoS One. 2011;6:e28603.
Thompson HM, Skop AR, Euteneuer U, Meyer BJ, McNiven MA. The large GTPase dynamin associates with the spindle midzone and is required for cytokinesis. Curr Biol. 2002;12:2111–7.
Chircop M, Sarcevic B, Larsen MR, Malladi CS, Chau N, Zavortink M, et al. Phosphorylation of dynamin II at serine-764 is associated with cytokinesis. Biochim Biophys Acta. 2011;1813:1689–99.
Morita M, Hamao K, Izumi S, Okumura E, Tanaka K, Kishimoto T, et al. Proline-rich domain in dynamin-2 has a low microtubule-binding activity: how is this activity controlled during mitosis in HeLa cells? J Biochem. 2010;148:533–8.
Bitoun M, Maugenre S, Jeannet PY, Lacène E, Ferrer X, Laforêt P, et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. 2005;37:1207–9.
Zuchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, De Jonghe P, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 2005;37:289–94.
Sambuughin N, Goldfarb LG, Sivtseva TM, Davydova TK, Vladimirtsev VA, Osakovskiy VL, et al. Adult-onset autosomal dominant spastic paraplegia linked to a GTPase-effector domain mutation of dynamin 2. BMC Neurol. 2015;15:223.
Koutsopoulos OS, Kretz C, Weller CM, Roux A, Mojzisova H, Bohm J, et al. Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur J Hum Genet. 2013;21:637–42.
Aidaralieva NJ, Kamino K, Kimura R, Yamamoto M, Morihara T, Kazui H, et al. Dynamin 2 gene is a novel susceptibility gene for late-onset Alzheimer disease in non-APOE-epsilon4 carriers. J Hum Genet. 2008;53:296–302.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63.
Ge Z, Li M, Zhao G, Xiao L, Gu Y, Zhou X, et al. Novel dynamin 2 mutations in adult T-cell acute lymphoblastic leukemia. Oncol Lett. 2016;12:2746–51.
Ge Z, Gu Y, Han Q, Zhao G, Li M, Li J, et al. Targeting high Dynamin-2 (DNM2) expression by restoring Ikaros function in acute lymphoblastic leukemia. Sci Rep. 2016;6:38004.
Liu X, Rothe K, Yen R, Fruhstorfer C, Maetzig T, Chen M, et al. A novel AHI-1–BCR-ABL–DNM2 complex regulates leukemic properties of primitive CML cells through enhanced cellular endocytosis and ROS-mediated autophagy. Leukemia. 2017;31:2376–87.
Tay Y, Tan SM, Karreth FA, Lieberman J, Pandolfi PP. Characterization of dual PTEN and p53-targeting microRNAs identifies microRNA-638/Dnm2 as a two-hit oncogenic locus. Cell Rep. 2014;8:714–22.
Raja S, Shah S, Tariq A, Bibi N, Sughra K, Yousuf A, et al. Caveolin‑1 and dynamin‑2 overexpression is associated with the progression of bladder cancer. Oncol Lett. 2019;18:219–26.
Ren N, Tian Z, Sun H, Lu X. Dynamin 2 is correlated with recurrence and poor prognosis of papillary thyroid cancer. Med Sci Monit. 2020;26:e924590.
Xu B, Teng LH, Silva SD, Bijian K, Al Bashir S, Jie S, et al. The significance of dynamin 2 expression for prostate cancer progression, prognostication, and therapeutic targeting. Cancer Med. 2014;3:14–24.
Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8:393–406.
Lee YY, Do IG, Park YA, Choi JJ, Song SY, Kim CJ, et al. Low dynamin 2 expression is associated with tumor invasion and metastasis in invasive squamous cell carcinoma of cervix. Cancer Biol Ther. 2010;10:329–35.
Lee YY, Song SY, Do IG, Kim TJ, Kim BG, Lee JW, et al. Dynamin 2 expression as a biomarker in grading of cervical intraepithelial neoplasia. Eur J Obstet Gynecol Reprod Biol. 2012;164:180–4.
Sajed R, Saeednejad Zanjani L, Rahimi M, Mansoori M, Zarnani A-H, Madjd Z, et al. Overexpression and translocation of dynamin 2 promotes tumor aggressiveness in breast carcinomas. EXCLI J. 2020;19:1423–35.
Chernikova SB, Nguyen RB, Truong JT, Mello SS, Stafford JH, Hay MP, et al. Dynamin impacts homology-directed repair and breast cancer response to chemotherapy. J Clin Invest. 2018;128:5307–21.
Eppinga RD, Krueger EW, Weller SG, Zhang L, Cao H, McNiven MA. Increased expression of the large GTPase dynamin 2 potentiates metastatic migration and invasion of pancreatic ductal carcinoma. Oncogene. 2012;31:1228–41.
Burton KM, Cao H, Chen J, Qiang L, Krueger EW, Johnson KM, et al. Dynamin 2 interacts with α-actinin 4 to drive tumor cell invasion. Parent C, editor. MBoC. 2020;31:439–51.
Joshi HP, Subramanian IV, Schnettler EK, Ghosh G, Rupaimoole R, Evans C, et al. Dynamin 2 along with microRNA-199a reciprocally regulate hypoxia-inducible factors and ovarian cancer metastasis. Proc Natl Acad Sci U S A. 2014;111:5331–6.
Tang M, Yin G, Wang F, Liu H, Zhou S, Ni J, et al. Downregulation of CD9 promotes pancreatic cancer growth and metastasis through upregulation of epidermal growth factor on the cell surface. Oncol Rep. 2015;34:350–8.
Cowling BS, Chevremont T, Prokic I, Kretz C, Ferry A, Coirault C, et al. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J Clin Invest. 2014;124:1350–63.
Tasfaout H, Buono S, Guo S, Kretz C, Messaddeq N, Booten S, et al. Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat Commun. 2017;8:15661.
Chen X, Gao Y-Q, Zheng Y-Y, Wang W, Wang P, Liang J, et al. The intragenic microRNA miR199A1 in the dynamin 2 gene contributes to the pathology of X-linked centronuclear myopathy. J Biol Chem. 2020;295:8656–67.
Gharibi T, Babaloo Z, Hosseini A, Abdollahpour-alitappeh M, Hashemi V, Marofi F, et al. Targeting STAT3 in cancer and autoimmune diseases. Eur J Pharmacol. 2020;878:173107.
Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234–48.
Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in cancer immunotherapy. Mol Cancer. 2020;19:145.
Gayi E, Neff LA, Massana Muñoz X, Ismail HM, Sierra M, Mercier T, et al. Tamoxifen prolongs survival and alleviates symptoms in mice with fatal X-linked myotubular myopathy. Nat Commun. 2018;9:4848.
Liu N, Bezprozvannaya S, Shelton JM, Frisard MI, Hulver MW, McMillan RP, et al. Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J Clin Invest. 2011;121:3258–68.
Mitchelson KR. Roles of the canonical myomiRs miR-1, -133 and -206 in cell development and disease. WJBC. 2015;6:162.
Schlunck G, Damke H, Kiosses WB, Rusk N, Symons MH, Waterman-Storer CM, et al. Modulation of Rac localization and function by dynamin. Mol Biol Cell. 2004;15:256–67.
Yamada H, Abe T, Li SA, Masuoka Y, Isoda M, Watanabe M, et al. Dynasore, a dynamin inhibitor, suppresses lamellipodia formation and cancer cell invasion by destabilizing actin filaments. Biochem Biophys Res Commun. 2009;390:1142–8.
Yamada H, Takeda T, Michiue H, Abe T, Takei K. Actin bundling by dynamin 2 and cortactin is implicated in cell migration by stabilizing filopodia in human non-small cell lung carcinoma cells. Int J Oncol. 2016;49:877–86.
Zhang Y, Nolan M, Yamada H, Watanabe M, Nasu Y, Takei K, et al. Dynamin2 GTPase contributes to invadopodia formation in invasive bladder cancer cells. Biochem Biophys Res Commun. 2016;480:409–14.
Mooren OL, Kotova TI, Moore AJ, Schafer DA. Dynamin2 GTPase and cortactin remodel actin filaments. J Biol Chem. 2009;284:23995–4005.
Feng H, Liu KW, Guo P, Zhang P, Cheng T, McNiven MA, et al. Dynamin 2 mediates PDGFRalpha-SHP-2-promoted glioblastoma growth and invasion. Oncogene. 2012;31:2691–702.
Razidlo GL, Wang Y, Chen J, Krueger EW, Billadeau DD, McNiven MA. Dynamin 2 potentiates invasive migration of pancreatic tumor cells through stabilization of the Rac1 GEF Vav1. Dev Cell. 2013;24:573–85.
Wong BS, Shea DJ, Mistriotis P, Tuntithavornwat S, Law RA, Bieber JM, et al. A direct podocalyxin–dynamin-2 interaction regulates cytoskeletal dynamics to promote migration and metastasis in pancreatic cancer cells. Cancer Res. 2019;79:2878–91.
Baldassarre M, Pompeo A, Beznoussenko G, Castaldi C, Cortellino S, McNiven MA, et al. Dynamin participates in focal extracellular matrix degradation by invasive cells. Mol Biol Cell. 2003;14:1074–84.
Yamamoto H, Sutoh M, Hatakeyama S, Hashimoto Y, Yoneyama T, Koie T, et al. Requirement for FBP17 in invadopodia formation by invasive bladder tumor cells. J Urol. 2011;185:1930–8.
Rosse C, Lodillinsky C, Fuhrmann L, Nourieh M, Monteiro P, Irondelle M, et al. Control of MT1-MMP transport by atypical PKC during breast-cancer progression. Proc Natl Acad Sci. 2014;111:E1872–9.
Yoon SY, Jeong MJ, Yoo J, Lee KI, Kwon BM, Lim DS, et al. Grb2 dominantly associates with dynamin II in human hepatocellular carcinoma HepG2 cells. J Cell Biochem. 2001;84:150–5.
Piazza TM, Lu JC, Carver KC, Schuler LA. SRC family kinases accelerate prolactin receptor internalization, modulating trafficking and signaling in breast cancer cells. Mol Endocrinol. 2009;23:202–12.
Carver KC, Piazza TM, Schuler LA. Prolactin enhances insulin-like growth factor I receptor phosphorylation by decreasing its association with the tyrosine phosphatase SHP-2 in MCF-7 breast cancer cells. J Biol Chem. 2010;285:8003–12.
Moskovich O, Herzog L-O, Ehrlich M, Fishelson Z. Caveolin-1 and dynamin-2 are essential for removal of the complement C5b–9 complex via endocytosis. J Biol Chem. 2012;287:19904–15.
Ivanov VN, Ronai Z, Hei TK. Opposite roles of FAP-1 and dynamin in the regulation of Fas (CD95) translocation to the cell surface and susceptibility to Fas ligand-mediated apoptosis. J Biol Chem. 2006;281:1840–52.
Ivanov VN, Zhou H, Hei TK. Sequential treatment by ionizing radiation and sodium arsenite dramatically accelerates TRAIL-mediated apoptosis of human melanoma cells. Cancer Res. 2007;67:5397–407.
Pradhan AK, Bhoopathi P, Talukdar S, Das SK, Emdad L, Sarkar D, et al. Mechanism of internalization of MDA-7/IL-24 protein and its cognate receptors following ligand-receptor docking. Oncotarget. 2019;10:5103–17.
Tremblay CS, Brown FC, Collett M, Saw J, Chiu SK, Sonderegger SE, et al. Loss-of-function mutations of Dynamin 2 promote T-ALL by enhancing IL-7 signalling. Leukemia. 2016;30:1993–2001.
Hill TA, Odell LR, Quan A, Abagyan R, Ferguson G, Robinson PJ, et al. Long chain amines and long chain ammonium salts as novel inhibitors of dynamin GTPase activity. Bioorg Med Chem Lett. 2004;14:3275–8.
Hill TA, Gordon CP, McGeachie AB, Venn-Brown B, Odell LR, Chau N, et al. Inhibition of dynamin mediated endocytosis by the dynoles–synthesis and functional activity of a family of indoles. J Med Chem. 2009;52:3762–73.
Joshi S, Perera S, Gilbert J, Smith CM, Mariana A, Gordon CP, et al. The dynamin inhibitors MiTMAB and OcTMAB induce cytokinesis failure and inhibit cell proliferation in human cancer cells. Mol Cancer Ther. 2010;9:1995–2006.
Chircop M, Perera S, Mariana A, Lau H, Ma MP, Gilbert J, et al. Inhibition of dynamin by dynole 34–2 induces cell death following cytokinesis failure in cancer cells. Mol Cancer Ther. 2011;10:1553–62.
Hee Kim Y, Kim KY, Jun DY, Kim J-S, Kim YH. Inhibition of autophagy enhances dynamin inhibitor-induced apoptosis via promoting Bak activation and mitochondrial damage in human Jurkat T cells. Biochem Biophys Res Commun. 2016;478:1609–16.
Joshi S, Braithwaite AW, Robinson PJ, Chircop M. Dynamin inhibitors induce caspase-mediated apoptosis following cytokinesis failure in human cancer cells and this is blocked by Bcl-2 overexpression. Mol Cancer. 2011;10:78.
Lee Y-Y, Jeon H-K, Lee J, Hong JE, Do I-G, Choi CH, et al. Dynamin 2 inhibitors as novel therapeutic agents against cervical cancer cells. Anticancer Res. 2016;36:6381–8.
Shen F, Gai J, Xing J, Guan J, Fu L, Li Q. Dynasore suppresses proliferation and induces apoptosis of the non-small-cell lung cancer cell line A549. Biochem Biophys Res Commun. 2018;495:1158–66.
Luwor R, Morokoff AP, Amiridis S, D’Abaco G, Paradiso L, Stylli SS, et al. Targeting glioma stem cells by functional inhibition of dynamin 2: a novel treatment strategy for glioblastoma. Cancer Invest. 2019;37:144–55.
Zaky MY, Liu X, Wang T, Wang S, Liu F, Wang D, et al. Dynasore potentiates c-Met inhibitors against hepatocellular carcinoma through destabilizing c-Met. Arch Biochem Biophys. 2020;680:108239.
Quan A, McGeachie AB, Keating DJ, van Dam EM, Rusak J, Chau N, et al. Myristyl trimethyl ammonium bromide and octadecyl trimethyl ammonium bromide are surface-active small molecule dynamin inhibitors that block endocytosis mediated by dynamin I or dynamin II. Mol Pharmacol. 2007;72:1425–39.
Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–50.
Newton AJ, Kirchhausen T, Murthy VN. Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc Natl Acad Sci U S A. 2006;103:17955–60.
Lee S, Jung K-Y, Park J, Cho J-H, Kim Y-C, Chang S. Synthesis of potent chemical inhibitors of dynamin GTPase. Bioorg Med Chem Lett. 2010;20:4858–64.
McCluskey A, Daniel JA, Hadzic G, Chau N, Clayton EL, Mariana A, et al. Building a better dynasore: the dyngo compounds potently inhibit dynamin and endocytosis. Traffic. 2013;14(12):1272–89.
Yamada H, Abe T, Li S-A, Tago S, Huang P, Watanabe M, et al. N′-[4-(dipropylamino)benzylidene]-2-hydroxybenzohydrazide is a dynamin GTPase inhibitor that suppresses cancer cell migration and invasion by inhibiting actin polymerization. Biochem Biophys Res Commun. 2014;443:511–7.
Tremblay CS, Chiu SK, Saw J, McCalmont H, Litalien V, Boyle J, et al. Small molecule inhibition of Dynamin-dependent endocytosis targets multiple niche signals and impairs leukemia stem cells. Nat Commun. 2020;11:6211.
Khan I, Gril B, Steeg PS. Metastasis suppressors NME1 and NME2 promote dynamin 2 oligomerization and regulate tumor cell endocytosis, motility, and metastasis. Cancer Res. 2019;79:4689–702.
Gong C, Zhang J, Zhang L, Wang Y, Ma H, Wu W, et al. Dynamin2 downregulation delays EGFR endocytic trafficking and promotes EGFR signaling and invasion in hepatocellular carcinoma. Am J Cancer Res. 2015;5:702–13.
Acknowledgements
Not applicable.
Funding
This work was supported by the Institut National de la Santé et de la Recherche Médicale (Inserm), Sorbonne Université, and the Association Institut de Myologie.
Author information
Authors and Affiliations
Contributions
DT and MB wrote the article and prepared Tables and Figures. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
There is no conflict of interest to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Trochet, D., Bitoun, M. A review of Dynamin 2 involvement in cancers highlights a promising therapeutic target. J Exp Clin Cancer Res 40, 238 (2021). https://doi.org/10.1186/s13046-021-02045-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-02045-y