
Abstract
Background
Hereditary Spastic Paraplegia (HSP) represents a large group of clinically and genetically heterogeneous disorders linked to over 70 different loci and more than 60 recognized disease-causing genes. A heightened vulnerability to disruption of various cellular processes inherent to the unique function and morphology of corticospinal neurons may account, at least in part, for the genetic heterogeneity.
Methods
Whole exome sequencing was utilized to identify candidate genetic variants in a four-generation Siberian kindred that includes nine individuals showing clinical features of HSP. Segregation of candidate variants within the family yielded a disease-associated mutation. Functional as well as in-silico structural analyses confirmed the selected candidate variant to be causative.
Results
Nine known patients had young-adult onset of bilateral slowly progressive lower-limb spasticity, weakness and hyperreflexia progressing over two-to-three decades to wheel-chair dependency. In the advanced stage of the disease, some patients also had distal wasting of lower leg muscles, pes cavus, mildly decreased vibratory sense in the ankles, and urinary urgency along with electrophysiological evidence of a mild distal motor/sensory axonopathy. Molecular analyses uncovered a missense c.2155C > T, p.R719W mutation in the highly conserved GTP-effector domain of dynamin 2. The mutant DNM2 co-segregated with HSP and affected endocytosis when expressed in HeLa cells. In-silico modeling indicated that this HSP-associated dynamin 2 mutation is located in a highly conserved bundle-signaling element of the protein while dynamin 2 mutations associated with other disorders are located in the stalk and PH domains; p.R719W potentially disrupts dynamin 2 assembly.
Conclusion
This is the first report linking a mutation in dynamin 2 to a HSP phenotype. Dynamin 2 mutations have previously been associated with other phenotypes including two forms of Charcot-Marie-Tooth neuropathy and centronuclear myopathy. These strikingly different pathogenic effects may depend on structural relationships the mutations disrupt. Awareness of this distinct association between HSP and c.2155C > T, p.R719W mutation will facilitate ascertainment of additional DNM2 HSP families and will direct future research toward better understanding of cell biological processes involved in these partly overlapping clinical syndromes.
Similar content being viewed by others
Background
Hereditary spastic paraplegia (HSP) comprises a group of clinically and genetically heterogeneous diseases that affect the upper motor neurons and their long axonal projections. The hallmark of hereditary spastic paraplegia (HSP) is progressive degeneration of the corticospinal tracts, often in a length-dependent manner, and progressive spasticity as the major clinical feature; neuropathological studies have identified axon degeneration of the corticospinal tracts and fasciculus gracilis fibers [1]. The key diagnostic findings are lower limb weakness, increased muscle tone, hyperreflexia, extensor plantar responses, and gait spasticity [2].
The burden inherent to corticospinal neurons with their long axonal processes spanning from motor cortex to distant segments of the spinal cord is likely to involve various cellular vulnerabilities that could account for the broad genetic heterogeneity of HSPs. Variant forms of HSP arise from mutations in genes directly implicated in membrane traffic, mitochondrial function, endoplasmic reticulum shaping/distribution, myelination, lipid/cholesterol metabolism, bone morphogenetic protein signaling, and endolysosomal function, as well as endosome and microtubule dynamics [3, 4]. Typically, the longest fibers — those connecting to the lower spinal cord segments — are the earliest to experience changes in their terminal regions that predate changes in cell bodies, leading to suggestions that impact on axonal traffic is a common mechanism of HSP. In this regard, HSP may be viewed as a central nervous system counterpart of the axonal form of Charcot-Marie-Tooth (CMT2) neuropathy, a clinically and genetically heterogeneous group of disorders of the peripheral nervous system marked by length-dependent distal axon degeneration of motor and sensory nerves [5].
We report on a novel association of an autosomal dominant HSP phenotype with a heterozygous DNM2 mutation. Dynamins are highly conserved large GTPases that play a critical role in clathrin-mediated endocytosis. They participate in converting the nascent invaginated clathrin-coated pit into a fully formed vesicle and in detaching the vesicle from the plasma membrane (membrane fission) [6]. In developing neurons, both endocytosis and exocytosis are critical for delivery of nutrients and building materials. Clathrin-mediated endocytosis plays a particularly important and specialized role at neuronal synapses [7]. Dynamin isoforms dynamin 1 and dynamin 3 are expressed in neurons, while dynamin 2 is found ubiquitously, including the CNS [8]. Dynamin 2 is involved in several other cellular processes, such as regulation of neuronal morphology, axonal growth and formation of growth cones, centrosome cohesion, actin- and microtubular dynamics [9].
Dynamin 2 is a 100 kDa multidomain protein composed of a highly conserved catalytic N-terminal GTPase domain (GTPase), a middle domain (MD) driving dynamin oligomerization, a pleckstrin homology domain (PH) critical for the interaction with membrane phosphoinositides, a GTPase effector domain (GED) that activates GTPase upon assembly of dynamin oligomers into higher order structures, and a less conserved C-terminal proline/arginine rich domain (PRD) which directs dynamin to endocytic sites and is a major site for interacting with other proteins [10, 11] (Fig. 1).

Domain structure of dynamin 2 and location of known disease-causing mutations. Mutations associated with Central nuclear myopathy are shown in black in the upper set; mutations associated with subtypes of Charcot-Marie-Tooth neuropathy are shown in blue at the bottom of the diagram. A homozygous mutation associated with Lethal Congenital Contracture Syndrome appears in green in the upper set. The p.R719W mutation identified in the Siberian family with Hereditary Spastic Paraplegia is shown in red within the bottom set
Distinct mutations in dynamin 2 have previously been associated with other phenotypes including two forms of Charcot-Marie-Tooth disease: axonal CMT2M (MIM# 606482) originally reported by Züchner et al. [12] and intermediate form CMTDIB (MIM# 606482) reported by Kennerson et al. [13]; centronuclear myopathy ADCNM (MIM# 160150) originally reported by Bitoun et al. [14], and lethal congenital contractures syndrome type 5 LCCS5 (MIM#615368). The majority of CMT-associated DNM2 mutations is located in the N-terminal part of the pleckstrin homology domain (Fig. 1), while a distinct set of mutations, mainly those found in the middle, PH, and PH/GTPase-effector boundary, is linked to autosomal-dominant centronuclear myopathy. The mechanisms by which various DNM2 mutations affect discrete tissues and lead to distinct phenotypes remain unknown.
Methods
Family pedigree and patient evaluation
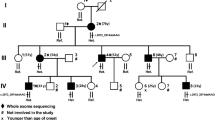
Chronic neurological disorders are highly prevalent in the Sakha (Yakut) Republic, Russian Federation. In the process of systematic ascertainment of patients with Viliuisk encephalomyelitis and related disorders [15], a multigenerational HSP family was identified. This 4-generational Siberian family included 9 patients. The oldest family member (I:2, Fig. 2) known to be affected by history had stiff gait and progressive muscle weakness of the lower extremities for at least 10 years before his death at 63. His 3 sons from two marriages (II:1, II:4, and II:6) developed the “family disease”. They were repeatedly examined over the following three decades and diagnosed with hereditary spastic paraplegia. In the third generation, three sons of patient II:4, also from separate marriages, and a daughter of patient II:6 developed the same disease (III:2, III:3, III:5, and III:7). Finally, a great-grandson (IV:1) was the youngest patient to be diagnosed. Pedigree was constructed based on cross interviews of patients and closest family members.

Pedigree of the Siberian family with Hereditary spastic paraplegia. Filled symbols indicate individuals affected with HSP; open symbols represent unaffected family members. The p.R719W mutation was identified in the affected family members II:6, III:2, III:3, III:7, IV:1, but the test for the mutation was negative in unaffected individuals III:1, IV:2, IV:3, IV:4
The study was approved by the Institutional Review Boards of the Institute of Health, North-Eastern Federal University, Yakutsk, and the Yakutsk Research Center, Siberian Branch of the Russian Academy of Medical Sciences. The statement confirmed in writing that the Clinical protocol complied with the Declaration of Helsinki. A written informed consent was obtained from each participant.
After obtaining informed consent, 5 affected and 5 unaffected family members underwent a neurological exam that included assessment of mental status, cranial nerves, muscle strength (MRC scale), coordination, tendon reflexes, muscle bulk, muscle tone, plantar responses, foot deformity, and gait. Evaluation of sensory impairment included clinical testing for pain and temperature sensation, vibration and position sense. Electrophysiological investigation performed in 3 patients (III:3, III:5, and IV:1) included motor and sensory nerve conduction velocities (NCV), compound muscle action potential (CMAP) amplitudes, distal motor latencies (DML), sensory NCV and sensory nerve action potential (SNAP) amplitudes recorded under standard conditions from the median, ulnar, peroneal, tibial, and sural nerves. Routine clinical MRI of the spinal cord was also obtained in three cases. Blood for DNA extraction was drawn from 9 family members.
Exome sequencing
WES was performed using genomic DNA extracted from peripheral white blood cells of patients II:6 and III:3. Exome capture utilized TruSeq Kit v1 (Illumina, Sand Diego, California) in accordance with manufacturer’s instructions. Library construction, sequence generation, sequence alignment to the reference genome (UCSC GRCh37/hg19), variant calling, and identification of potentially pathogenic variants were performed as described [16]. Variant analysis was performed using an autosomal dominant genetic model. Only variants satisfying stringent coverage and genotype quality criteria were included in further downstream variant analysis. WES generated variants were further validated by segregation analysis to 3 additional affected and 4 unaffected members of the Siberian family using standard Sanger sequencing of amplified DNA fragments.
Endocytosis assay
The wild-type human DNM2 in pmCherry-N1 vector (Addgene plasmid 27689) was a kind gift from Dr. C. Merrifield [17]. The c.2155C > T, p.R719W mutation was introduced into the pmCherry-N1 DNM2 plasmid using the Quick-Change Site Directed Mutagenesis kit according to manufacturer’s protocol (Agilent Technologies, Santa Clara, CA). The integrity of all plasmids was confirmed by DNA sequencing.
HeLa cells were grown in Dulbecco's modified Eagle’s medium (Life Technologies, Grand Island, NY) supplemented with 10 % fetal bovine serum. 50-60 % confluent cells were transfected with plasmids containing wild type or mutant DNM2 by using transfection reagent HilyMax (Dojindo Molecular Technologies, Rockville, MD) according to the manufacturer’s protocol. Cultures were then incubated in growth medium. At 20 hrs post-transfection, the cells were serum starved for 1 h and treated with 25ug/ml Alexa-Flour 488 conjugated Transferrin (Life Technologies, Frederick, MD) for 15 min at 37 °C. Cells labeled with fluorescent transferrin were then washed and fixed with 4 % paraformaldehyde. Immunofluorescene analysis was performed as previously described [18]. Cells were visualized with a Zeiss LSM 510 confocal microscope. Transferrin immunofluorescent signal was measured by utilizing ImageJ (Image Processing and Analysis in Java, National Institutes of Health) software. Mean fluorescence was first measured in the background; the corrected total transferrin fluorescence signal was then compared between cells expressing WT and mutant DNM2 constructs. Statistical significance was evaluated using the Student’s t-test. P-value <0.05 was considered significant.
Structural studies
Molecular models of dynamin 2 wild type and p.R719W were generated by I-TASSER [19–21], using dynamin 1 as a template [22]. The sequence homology between dynamin 1 and 2 is 78 % identical overall and 87 % in the GTPase domain. The position of R719 in dynamin 1 is R725. The molecular models were visualized and H-bonds calculated by Chimera [23]. The dynamin 1 tetramer image was generated from docked crystal structures in the 3D density map of K44A-dynamin 1 [24].
Results
Phenotypic features and disease outcomes
The pattern of disease inheritance in this family was autosomal dominant (Fig. 2). Clinical information obtained at evaluation of 5 affected family members (II:6, III:3, III:5, III:7, and IV:1) is summarized in Table 1. The illness had an insidious onset between the ages of 10 and 37 years (mean, 26 years) with impaired gait, muscle stiffness and weakness in the lower limbs. Further progression of illness in patients II:6 and III:5 led to severe bilateral lower limb muscle weakness requiring the use of cane and eventual wheel-chair dependency by the 28th and 23rd years of illness. Three patients (I:2, II:1, and III:2) died after an illness lasting for 23 to 32 years, and one (II:4) died in an accident.
At examination (Table 1), a single patient had mild developmental cognitive delay. No optic nerve atrophy, nystagmus, or signs of ataxia were found. Cranial nerves were intact, bulbar functions preserved until late in the illness, none showed dysphagia, and only a single patient (III:5) had moderate spastic dysarthria. Upper extremities muscle bulk, strength, muscle tone, reflexes, sensation and coordination were not affected. In the lower limbs, typical features of upper motor neuron involvement were present in all patients, including spasticity, reduced muscle strength, tightness of Achilles heels. Deep tendon reflexes were increased in all patients with clonus in 3 and bilateral Babinski sign in 4. Moderate muscle atrophy of lower leg muscles was noticed in advanced illness in patients II:6, III:3, III:5, and one patient had flexion contracture of leg muscles and chronic decubitus ulceration (II:6). All patients had profoundly spastic gait. Sphincter control abnormalities manifesting as urinary urgency were observed late in the illness in two patients (II:6 and III:5). Clinical testing for pain, temperature, and position sense did not reveal any deficits. Mild distal vibratory sense impairment was detected in patients II:6 (inconsistently), III:3, and III:5 late in the course of illness. Scoliosis was present in one patient and bilateral pes cavus in all examined patients. MRI of the spinal cord performed in patients III:3, III:5, and IV:1 showed no evidence for cord compression, cord atrophy or intrinsic spinal cord abnormalities. There was no evidence of multisystem involvement as determined by routine blood count and routine blood chemistry. In summary, the clinical course in five affected individuals over a multi-decade observation period was overwhelmingly consistent with upper motor neuron dysfunction leading to progressive spasticity. Only late in the course of illness symptoms suggestive of a mild peripheral involvement in the form of mild sensory changes and distal muscle atrophy became apparent. No chronic neurological diseases were found among other members of the extended family or other inhabitants of this village.
Electrodiagnostic studies
Motor and sensory nerve conduction studies were performed in patients III:3, III:5, and IV:1 (Table 2). Patients III:3 and III:5 were in advanced phases of illness at the 17th and 32nd year from disease onset, patient IV:1 was the youngest in this family at the age of 17 years, in the 7th year after the disease onset. In the n.medianus, conduction studies revealed mostly preserved distal motor latency (DML), motor nerve conduction velocities (NCV) and compound muscle action potentials (CMAP). In the lower limbs, stimulation of n.peroneus profundus and n.tibialis posterior showed reduced NCV and CMAP, most significantly in patients with advanced disease. Sensory nerve action potential (SNAP) amplitudes in n.suralis were also severely affected in patients with advanced disease. The electrodiagnostic study revealed an axonal impairment in motor and sensory peripheral nerves confined to the distal lower extremities .
Genetic analysis
Prior to exome sequencing, coding regions of common dominant HSP genes had been analyzed by Sanger sequencing, including SPAST (spastin) linked to autosomal dominant SPG4 (MIM# 18601) and ATL1 (atlastin) causing SPG3A (MIM# 182600), genes that account for 37-46 % and 6-11 % of dominant HSP cases [25]. No pathogenic changes were identified.
Exome sequencing was performed in two affected individuals, II:6 and III:3 (Fig. 2). A number of filtering steps were used to prioritize sequence variants (Table 3), beginning with the requirement that the variant had to be shared by two studied affected members of the Siberian family in heterozygous state, assuming a dominant model. Variants in non-coding regions and synonymous SNPs were excluded. Candidates were further restricted by excluding variants reported to have occurred in adult cohort of over 900 subjects participating in the ClinSeq project (ClinSeq www.genome.gov/20519355 ) or the 1000 genome project. Variant population frequency was additionally examined using the ExAC data set. Missense variants were further prioritized by the degree of functional disruption predicted by PolyPhen-II ( www.genetics.bwh.harvard.edu/pph2/ ), SIFT [26], MutationTaster ( www.mutationtaster.org/ ), and CADD (Combined Annotation–Dependent Depletion) - a single-annotation method used to prioritize functional, deleterious and pathogenic variants across many functional categories, effect sizes, and genetic architectures [27]. Exome sequencing did not identify any predicted or known pathogenic changes in previously identified HSP-associated genes. Selected variants were then assessed based on encoded protein expression and function. Considering that our patients had an adult onset neurodegenerative disorder with high penetrance, we excluded genes with no expression in the CNS or genes involved in early embryonic development. Based on analysis of whole exome sequencing data, 4 heterozygous variants were identified under autosomal dominant model. Table 4 provides information regarding the encoded proteins function, expression, and the known disease associations. These four strongest candidates were tested for segregation within the Siberian family (Table 5). The results of this analysis indicated that DNM2 p.R719W variant is the only one that segregated perfectly.
DNM2 variant
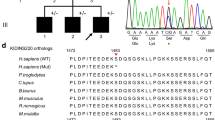
The identified DNM2 missense substitution is located at NM_001005360:c.2155C > T; Chr19(GRCh37):g.10939808C > T in exon 19 of the DNM2 gene, which replaces Arginine (R) with Tryptophan (W) (p.R719W) in the encoded dynamin 2 (Fig. 3a). The region is highly conserved at the amino acid level, up to the worm (Fig. 3b). The p.R719W mutation is in the GTPase effector (GED) domain (Fig. 1). The variant is predicted to be damaging or deleterious by all methods employed in the study. The variant was seen only once in an individual of South Asian descent in the entire Exome Aggregation Consortium data set that comprises exomes of 60,706 unrelated individuals sequenced as part of various disease-specific and population genetic studies (www.exac.broadinstitute.org).

Identification of the disease-causing genetic variant. a Sequence chromatograph of a fragment of the DNM2 gene showing the position of nucleotide substitution (arrow) responsible for the p.R719W mutation. b Protein alignment of the GTPase effector domain in various dynamins. Mutated residue is colored red
Validation by Sanger sequencing and further segregation to other family members shows that only the affected members II:6, III:2, III:3, III:7, and IV:1 are heterozygous for the c.2155C > T, p.R719W change whereas unaffected individuals III:1, IV:2, IV:3, and IV:4 are homozygous reference (Table 5). We conclude that the DNM2 c.2155C > T, p.R719W variant is the underlying molecular basis for the HSP phenotype in the Siberian family.
Functional study
To investigate the effects of the disease-related dynamin 2 p.R719W mutant, we evaluated clathrin-mediated endocytosis in HeLa cells transiently transfected with the wild type and mutant dynamin 2 following incubation with fluorescently labeled transferrin. We observed a prominent punctate staining in cells transfected with p.R719W DNM2 (Fig. 4), a phenomenon morphologically similar to that reported for other DNM2 mutations causing CMT and CNM [28]. There was a significant decrease in transferrin uptake in cells expressing mutant dynamin 2. This decrease was evident in all transfected cells and was especially significant (more than 50 %) in cells marked by arrows. The granules are localized most prominently in the perinuclear area, likely in the endosomal compartment. The results suggest that inhibited endocytosis is part of the pathophysiological mechanisms leading to HSP in this family.

Results of functional analysis of the c.2155C > T, p.R719W mutation. a HeLa cells transiently transfected with vectors containing the wild-type (upper panel) and mutant (bottom panel) DNM2. The p.R719W mutant exhibits punctate pattern of DNM2 expression (arrowheads), whereas cells transfected with wild type DNM2 show diffuse staining of the cytoplasm. Uptake of transferrin is reduced in cells expressing mutant DNM2 (arrows). Transferrin is labeled by Alexa-Flour 488 (green); nuclei are labeled with blue stain. b Quantification of transferrin uptake. The histogram represents the mean +/− standard error of the corrected total transferrin fluorescence of WT (n = 30 cells) and p.R719W (n = 25 cells); P < 0.001
Structural models
The HSP mutation p.R719W is located in the three-helix bundle of dynamin called the bundle-signaling element (BSE) (Fig. 5a). It is intriguing that p.R719W is the only disease-associated mutation found in this region. Both CMT and CNM mutations are located in the stalk (Middle/GED) and PH domains of the protein (Fig. 5a). The BSE undergoes a large conformational change upon GTP hydrolysis that has been described as the power stroke of dynamin [29, 30]. A mutation in this region suggests a defect in propagating the BSE conformational change to the rest of the molecule and assembled helical polymer. In addition, the mutation is located near a hinge between the BSE and the stalk and as indicated by fewer H-bonds (1 H-bond in W719 compared to 3 H-bonds in R719), p.R719W may disrupt the connection between these domains (Fig. 5b). The potential disruption of R719W in the assembled protein is further illustrated by examining the dynamin tetramer (Fig. 5c, asterisks).

Structural modeling of dynamin 2 c.2155C > T, p.R719W mutation. a Molecular model of dynamin 2 based on the crystal structure of dynamin 1 [22] indicating the HSP p.R719W mutation (cyan), CNM mutations (magenta) and CMT mutations (green). Dynamin domains are colored as follows: GTPase domain (red), Middle (blue), PH (yellow) and GED (orange). p.R719W is located at the hinge region between the BSE (a three helix bundle consisting of N and C termini of the G domain and the C termini of GED) and the stalk (Middle and GED) of dynamin. Generated by I-TASSER. b Overlay of wild-type and p.R719W dynamin 2 molecular models with R719 (purple) and W719 (cyan) side chains shown. On the right panel: there are three putative H bonds connecting R719 to the rest of the molecule (labeled H1–3-R719) compared to only one for W719 (H1-W719). c The assembled tetramer of dynamin 1 was generated from docking crystal structures into a 3D density map of K44A-dynamin 1 [24]. Dynamin monomers are colored red, yellow, blue and purple. Asterisks indicate the location of R725W (equivalent to R719W in dynamin 2) in the assembled dynamin 1
Discussion
The Siberian family under study had a history compatible with autosomal dominant transmission of a young-adult onset slowly progressive bilateral leg spasticity corresponding to typical clinical manifestations of HSP: each studied patient had lower extremity hyperreflexia, muscle weakness, and spastic gait. Some patients also showed wasting of lower leg muscles, pes cavus, decreased vibratory sense in the ankles, and urinary urgency as late features of illness. There was no optic neuropathy, retinopathy, extrapyramidal symptoms, dementia, ataxia, ichthyosis, or deafness described as part of the “complicated” subtypes of HSP. Alternative diagnoses were excluded based on the results of clinical studies and routine laboratory investigations. Nerve conduction studies in patients with advanced disease revealed motor and sensory axonal neuropathy in distal lower extremities, a feature reported in other subtypes of HSP [31].
Exome sequencing methodology was used for genetic analysis of this HSP family. There are well known limitations to exome sequencing as a method of detection of the causative mutation. Structural genomic alterations, changes in regions not captured by this technology such as translocations, changes in regulatory regions or introns, may be responsible for the clinical phenotypes. But subsequent analysis of tissue expression, structural and functional characteristics of the candidate genetic variants, and segregation within the pedigree as applied in this study are helpful in the process of detection of the disease-associated variant.
The identification of the DNM2 p.R719W mutation as the cause of HSP in this family brought attention to the fact that mutations in this same gene are responsible for other phenotypes, including two forms of CMT peripheral neuropathy. DNM2-associated autosomal dominant CMT2 neuropathy is characterized by progressive distal muscle weakness and atrophy, predominantly in the lower extremities, decreased or absent tendon reflexes, steppage gait, foot drop, pes cavus [32, 33]. These clinical features are in contrast with the presentation observed in patients with DNM2-associated HSP described here, although some overlap with CMT2 neuropathy exists, especially late in the disease. There has been active discussion on the subject of similarity between some forms of CMT neuropathy and HSP based on the identification of shared mechanisms involving central and peripheral motor neurons (for review, see [5]). Interestingly, mutations in KIF5A, a gene encoding the neuronal kinesin heavy chain implicated in anterograde axonal transport, have been associated with “pure” HSP, although a fraction of family members shows electrophysiological evidence of sensory-motor peripheral axonal neuropathy [34, 35]. Our findings give some weight to the view that HSP and CMT caused by DNM2 mutations, although clinically and anatomically separate syndromes, may have some overlap due to shared pathological vulnerabilities in their extended axonal processes.
Another DNM2-associated disease, autosomal-dominant Centronuclear myopathy, is a slowly progressive congenital myopathy characterized by delayed motor milestones, generalized muscle weakness, ptosis, and ophthalmoplegia [14]. It includes a wide spectrum of phenotypes ranging from severe neonatal to mild late-onset familial forms [36, 37]. CNM is characterized pathologically by myofiber atrophy, abnormal nuclear centralization and internalization, and type 1 muscle fiber predominance [38]. Some patients with CNM show clinical overlap of myopathy with mild axonal involvement in peripheral nerves [39].
It remains to be determined why specific DNM2 mutations result in involvement of skeletal muscle, peripheral nerves, peripheral or corticospinal neurons. Existing hypotheses concentrate on different roles of dynamin 2 domains involved in CMT or CNM. CMT mutations are located within the pleckstrin homology [12] and proline-rich domains [33], whereas the CNM mutations are found within the PH domain. The significance of involvement of specific dynamin domains is further underlined by the evidence we obtained in this study that the HSP dynamin 2 mutation, p.R719W, is the only known mutation in the bundle-signaling element of dynamin, which is structurally and most likely functionally different from the stalk and PH domains in which CMT and CNM mutations are located. A mutation in this region may cause a conformational change to the helical element and affect dynamin assembly. Moreover, the mutation results in fewer hydrogen bonds between the bundle-signaling element and the stalk. A weaker connection potentially uncouples the power stroke of dynamin from the rest of the molecule, a step that is predicted to be essential for endocytosis [30].
Modeling in cell cultures of all known DNM2 mutations causing either disease show their destructive effect on clathrin-mediated endocytosis considered a major mechanism for uptaking molecules into eukaryotic cells regulated by dynamin [40, 41]. Clathrin-coated pits capture cargo molecules via adaptors, the pits invaginate and pinch off to form clathrin-coated vesicles that carry the cargo into the cell. Data presented here show that the HSP-causing dynamin 2 p.R719W mutation induces a marked decrease of clathrin-mediated endocytosis similarly to other disease-causing dynamin 2 mutations, suggesting that impairment of the membrane trafficking process contributes to the pathogenesis of all DNM2-associated diseases. Prominent punctate staining in mutant cells also suggests that the HSP DNM2 mutation affects cellular morphology by mechanisms similar to previously characterized CMT and CNM mutations [12, 28].
The superfamily of dynamin GTPases comprises a group of proteins that are involved in diverse cellular functions and are often closely associated with biological membranes; they function in vesicle scission, as well as in fusion and fission of organelles [9]. The large GTPase atlastin belongs to the dynamin superfamily and shares the domain architecture with dynamin 2. Atlastin is involved in ER fusion, vesicular trafficking, axon formation and elongation. Mutations in atlastin isoform 1 (atlastin 1) have been identified in patients with early-onset HSP (SPG3). Curiously, mutations in atlastin 1 have also been associated with hereditary sensory neuropathy type 1D (HSN1D; MIM# 613708), a condition characterized by distal axonal sensory deficits leading to late distal skin ulceration and amputation, with some patients showing upper motor involvement [42]. The majority of atlastin 1 mutations are located in the GTPase domain, although some have been identified in the stalk, the BSE, as well as in the transmembrane domains [43] suggesting that membrane association that is common to both representatives of the dynamin family, dynamin 2 and atlastin 1, may be critical in the pathophysiology of HSP.
Lack of DNM2 mutations in other Siberian HSP families suggests that more than a single gene is responsible for this phenotype; HSP is an extremely heterogeneous disorder with mutations in more than 60 genes associated with this syndrome.
Conclusions
Patients in the 4-generation Siberian family suffered from hereditary spastic paraplegia and mild peripheral axonopathy. Our molecular, structural and functional analyses and segregation studies allowed to identify DNM2 c.2155C > T, p.R719W mutation as a cause of this disease. The mutation is located in a unique position of the multidomain dynamin 2 protein and its pathogenic effects may depend on structural relationships this mutation disrupts. Awareness of the distinct, but partly overlapping clinical phenotypes associated with mutations in DNM2, will facilitate identification of these disorders in additional families and direct future research toward better understanding of the role dynamin 2 and related networks have in health and disease.
References
Fink JK. Hereditary spastic paraplegia: clinical principles and genetic advances. Semin Neurol. 2014;34:293–305.
Durr A, Brice A, Serdaru M, et al. The phenotype of “pure” autosomal dominant spastic paraplegia. Neurology. 1994;44:1274–7.
Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci. 2012;35:25–47.
Zhu PP, Denton KR, Pierson TM, Li XJ, Blackstone C. Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum Mol Genet. 2014;23:5638–48.
Timmerman V, Clowes VE, Reid E. Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp Neurol. 2013;246:14–25.
Neumann S, Schmid SL. Dual role of BAR domain-containing proteins in regulating vesicle release catalyzed by the GTPase, dynamin-2. J Biol Chem. 2013;288:25119–28.
Raimondi A, Ferguson SM, Lou X, Armbruster M, Paradise S, Giovedi S, et al. Overlapping role of dynamin isoforms in synaptic vesicle endocytosis. Neuron. 2011;70:1100–14.
Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol. 2012;13:75–88.
Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane abulation and fission molecules? Nature Rev Mol Cell Biol. 2004;5:133–47.
Muhlberg AB, Warnock DE, Schmid SL. Domain structure and intramolecular regulation of dynamin GTPase. EMBO J. 1997;16:6676–83.
Kenniston JA, Lemmon MA. Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. 2010;29:3054–67.
Züchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 2005;37:289–94.
Kennerson ML, Zhu D, Gardner RJ, Storey E, Merory J, Robertson SP, et al. Dominant intermediate Charcot-Marie-Tooth neuropathy maps to chromosome 19p12-p13.2. Am J Hum Genet. 2001;69:883–8.
Bitoun M, Maugenre S, Jeannet PY, Lacene E, Ferrer X, et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. 2005;37:1207–9.
Goldfarb LG, Gajdusek DC. Viliuisk Encephalomyelitis in the Yakut people of Siberia. Brain. 1992;115:961–78.
Adams DR, Sincan M, Fuentes Fajardo K, Mullikin JC, Pierson TM, Toro C, et al. Analysis of DNA sequence variants detected by high-throughput sequencing. Hum Mutat. 2012;33:599–608.
Taylor MJ, Perrais D, Merrifield CJ. A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PloS Biol. 2011;9(3), e1000604.
Sambuughin N, Swietnicki W, Techtmann S, Matrosova V, Wallace T, Goldfarb L, et al. KBTBD13 interacts with Cullin 3 to form functional ubiquitin ligase. Biochem Biophys Res Comm. 2012;421:743–9.
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: Protein structure and function prediction. Nature Methods. 2015;12:7–8.
Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature Protocols. 2010;5:725–38.
Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40.
Faelber K, Posor Y, Gao S, Held M, Roske Y, Schulze D, et al. Crystal structure of nucleotide-free dynamin. Nature. 2011;477:556–60.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
Sundborger AC, Fang S, Heymann JA, Ray P, Chappie JS, Hinshaw JE. A dynamin mutant defines a superconstricted prefission state. Cell Rep. 2014;8:734–42.
Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. 2014;42:174–83.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Koutspoulos OS, Koch C, Tosch V, Bohm J, North KN, Laporte J. Mild functional differences of dynamin 2 mutations associated to Centronuclaer myopathy and Charcot-Marie-Tooth peripheral neuropathy. PloS One. 2011;6, e27498.
Chappie JS, Acharya S, Leonard M, Schmid SL, Dyda F. G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature. 2010;465:435–40.
Chappie JS, Mears JA, Fang S, Leonard M, Schmid SL, Milligan RA, et al. A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Cell. 2011;147:209–22.
Kumar KR, Sue CM, Burke D, Ng K. Peripheral neuropathy in hereditary spastic paraplegia due to spastin (SPG4) mutation–a neurophysiological study using excitability techniques. Clin Neurophysiol. 2012;123:1454–9.
Fabrizi GM, Ferrarini M, Cavallaro T, Cabrini I, Cerini R, Bertolasi L, et al. Two novel mutations in dynamin-2 cause axonal Charcot-Marie-Tooth disease. Neurology. 2007;69:291–5.
Claeys KG, Züchner S, Kennerson M, Berciano J, Garcia A, Verhoeven K, et al. Phenotypic spectrum of dynamin 2 mutations in Charcot-Marie-Tooth neuropathy. Brain. 2009;132:1741–52.
Goizet C, Boukhris A, Mundwiller E, et al. Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum Mutat. 2009;30(2):E376–385.
Crimella C, Baschirotto C, Arnoldi A, Tonelli A, Tenderini E, Airoldi G, et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot–Marie–Tooth type 2. Clinical Genetics. 2012;82:157–64.
Bitoun M, Durieux AC, Prudhon B, Bevilacqua JA, Herledan A, et al. Dynamin 2 mutations associated with human diseases impair clathrin-mediated receptor endocytosis. Hum Mutat. 2009;30:1419–27.
Schessl J, Medne L, Hu Y, Zou Y, Brown MJ, Huse JT, et al. MRI in DNM2-related centronuclear myopathy: evidence for highly selective muscle involvement. Neuromuscul Disord. 2007;17:28–32.
Romero NB, Bitoun M. Centronuclear myopathies. Semin Pediatr Neurol. 2011;18:250–6.
Fischer D, Herasse M, Bitoun M, Barragán-Campos HM, Chiras J, Laforêt P, et al. Characterization of the muscle involvement in dynamin 2-related centronuclear myopathy. Brain. 2006;129:1463–9.
Schmid SL, Frolov VA. Dynamin: functional design of a membrane fission catalyst. Annu Rev Cell Dev Biol. 2011;27:79–105.
Srinivasan S, Mattila JP, Schmid SL. Intrapolypeptide interactions between the GTPase effector domain (GED) and the GTPase domain form the bundle signaling element in dynamin dimers. Biochemistry. 2014;53:5724–6.
Guelly C, Zhu P-P, Leonardis L, Papic L, Zidar J, Schabhuttl M, et al. Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I. Am J Hum Genet. 2011;88:99–105.
Alvarez V, Sánchez-Ferrero E, Beetz C, Díaz M, Alonso B, Corao AI, et al. Mutational spectrum of the SPG4 (SPAST) and SPG3A (ATL1) genes in Spanish patients with hereditary spastic paraplegia. BMC Neurol. 2010;10:89.
Acknowledgements
We wish to thank Dr. C. Merrifield for providing the cloned wild-type human DNM2 and Dr. W. Gall for helpful comments and suggestions. The study was funded in part by the Ministry of Education and Science of the Russian Federation (project No. 3095). Research was supported by the Uniformed Services University (USU) R080148613 grant to N.S., Intramural Research Programs of the National Institute of Neurological Disorders and Stroke, the National Institute of Diabetes, Digestive and Kidney Diseases, and the National Human Genome Research Institute, NIH. Technical support was provided by the NIH Intramural Sequencing Center (NISC), Bioinformatics core of the NIH Undiagnosed Diseases Program, and the Biomedical Instrumentation Center of USU.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NS, LGG, FAP, and CT contributed to the study concept and design, carried out molecular and experimental studies, were involved in analysis and interpretation of data, and writing the manuscript. TMS, TKD, VAV, APD, RSN, ANY, and MPD contributed to the clinical evaluations and electro-diagnostic studies, generating data for the manuscript and participating in the analysis and interpretation of the results. VLO and NMR contributed to the study concept and design, molecular analysis and interpretation of data, and drafting the manuscript. ACS and JEH contributed to the structural analysis, generating and interpretation of data, and writing the manuscript. Each author read and approved the final version of the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sambuughin, N., Goldfarb, L.G., Sivtseva, T.M. et al. Adult-onset autosomal dominant spastic paraplegia linked to a GTPase-effector domain mutation of dynamin 2. BMC Neurol 15, 223 (2015). https://doi.org/10.1186/s12883-015-0481-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-015-0481-3