
Abstract
Indoleamine 2,3-dioxygenase 1 (IDO1), a monomeric heme-containing enzyme, catalyzes the first and rate-limiting step in the kynurenine pathway of tryptophan metabolism, which plays an important role in immunity and neuronal function. Its implication in different pathophysiologic processes including cancer and neurodegenerative diseases has inspired the development of IDO1 inhibitors in the past decades. However, the negative results of the phase III clinical trial of the would-be first-in-class IDO1 inhibitor (epacadostat) in combination with an anti-PD1 antibody (pembrolizumab) in patients with advanced malignant melanoma call for a better understanding of the role of IDO1 inhibition. In this review, the current status of the clinical development of IDO1 inhibitors will be introduced and the key pre-clinical and clinical data of epacadostat will be summarized. Moreover, based on the cautionary notes obtained from the clinical readout of epacadostat, strategies for the identification of reliable predictive biomarkers and pharmacodynamic markers as well as for the selection of the tumor types to be treated with IDO1inhibitors will be discussed.
Similar content being viewed by others


Background
With increasing recognition of evading immunological destruction as a hallmark of cancer acquired during its development[1], immunotherapy has emerged as a novel and important pillar of cancer treatment in addition to surgery, radiation, chemotherapy, and targeted therapy. Instead of directly killing cancer cells or suppressing the abnormal signal transduction within cancers, cancer immunotherapy aims to leverage the patient’s immune system to eliminate tumor cells by enhancing tumor immunity mediated by blocking immune inhibitory pathways and/or inhibitory cells in the tumor microenvironment (TME) or enhancing the specificity of anti-tumor immunity by inducing the expansion of T cells and antibodies directed to well-defined tumor antigens[2]. The launch of immune checkpoint inhibitors (ICPIs) including the first cytotoxic T-lymphocyte antigen 4 (CTLA-4) blocking antibody ipilimumab in 2011[3] and programmed death receptor 1 or ligand 1 (PD-1/PD-L1) blocking antibodies pembrolizumab and nivolumab in 2014[4, 5] represents the substantive progress in the field and boosts immunotherapy as a new standard therapy for many cancer types. To date, the US Food and Drug Administration has approved the use of PD-1/PD-L1 and CTLA-4 targeted therapies for the treatment of more than 15 cancers including melanoma, non-small cell lung cancer (NSCLC), small cell lung cancer, squamous cell carcinoma of the head and neck, renal cell carcinoma (RCC), hepatocellular carcinoma (HCC), classical Hodgkin lymphoma, urothelial carcinoma, colorectal cancer (CRC) and other cancer types[3,4,5]. However, limited response due to innate and acquired resistance as well as serious adverse effects especially life-threatening immune-related toxicities of anti-CTLA-4 and anti-PD-1/PD-L1 antibodies call for a need to identify other immunotherapy options targeting different mechanisms of immunosuppression in tumors that could be used alone or in combination with existing treatments. Currently over three thousand drugs directing against more than four hundred active targets are being investigated under different stages from preclinical to post-approval[6]. In this context indoleamine 2,3-dioxygenase 1 (IDO1) is one of the most studied target of cancer immunotherapies by virtue of its effects on immune suppression in the TME[6].
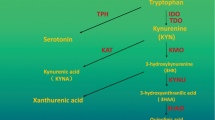
IDO1, a monomeric heme-containing enzyme, is one of the three enzymes that catalyze the first and rate-limiting step in the kynurenine pathway (KP), leading to the degradation of the essential amino acid tryptophan (TRP) and generation of kynurenine (KYN) and other downstream metabolites, in addition to tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase 2 (IDO2)[7, 8]. TRP is required for protein synthesis and TRP metabolites can act as neurotransmitters and signalling molecules. Thus, TRP and its metabolites play important roles in diverse physiological processes ranging from cell growth and survival to the coordination of responses to internal and external environmental changes[9]. IDO1 is expressed in various tissues, organs and cell types, catalyzing the conversion of a wide range of substrates including TRP and 5-hydroxytryptamine[10, 11]. TDO mainly exists in the liver and brain[12, 13], specifically catalyzing the conversion of TRP and some of its derivatives substituted in the 5- and 6-positions of the indole ring[14, 15]. TDO is thought to be the main modulator of TRP catabolism and is responsible for regulating systemic TRP levels[16], while IDO1 plays an important role in TRP catabolism under pathological conditions[17, 18]. The host-protective effect of IDO1 in parasitic infection has been first discovered in 1990s, which is attributable to a reduced availability of TRP in the inflammatory environment[19, 20]. In addition, as many KP metabolites are neuroactive, dysfunction of KP enzymes including IDO1 and TDO, often caused by inflammatory insults can trigger or facilitate diseases of the central nervous system, such as depression, Alzheimer’s disease, and Huntington’s disease[21,22,23]. IDO1 was first discovered to have a potent role in immune tolerance between a mother and fetus, since the inhibition of IDO1 in pregnant mice caused spontaneous immune rejection of allogeneic fetuses[24]. Later, the IDO1-mediated immune tolerance in tumors has been noted, experimental models suggest that IDO1 expression prevents rejection of tumor cells in immunogenic mice[25,26,27,28,29,30]. The imbalances in the level of TRP and KYN as well as the overexpression of IDO1 have been reported in many cancers[25,26,27,28,29, 31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47]. High IDO1 expression was associated with a decreased survival[48] and the extent of IDO1 overexpression depends on the tumor type and risk factors[49]. Three downstream effector pathways, including the general control nonderepressible 2 (GCN2), mammalian target of rapamycin (mTOR), and aryl hydrocarbon receptor (AhR) pathways, have been implicated in the biological responses to IDO1-mediated TRP depletion or KYN accumulation. In addition, some studies have shown that these effector pathways mediate immunosuppression in the TME[50,51,52,53]. TRP depletion promotes the accumulation of uncharged tRNA, resulting in a GCN2-dependent inhibition of protein synthesis that is accompanied by cell cycle arrest and irresponsiveness to immunological challenges[54]. TRP depletion leads to inhibition of the immunoregulatory kinases mTOR and protein kinase C, along with the induction of autophagy[55]. KYN accumulation promotes the translocation of AhR from the cytosol to nucleus, where it binds target genes and activates their transcription, inducing tolerogenic immune responses with the consequent benefit to tumor progression and migration[53, 56]. The exact contribution of each effector function to tumor immunosuppression remains to be determined. And it is likely that each pathway counts for much or less in different tumors although all of them cooperate synergistically in the development of immunosuppression in the TME. Based on these data, the inhibition of IDO1 has become an exciting approach as cancer immunotherapy and IDO1 inhibitors have been intensively investigated during the recent years. Multiple IDO1 inhibitors with different structural skeleton have been developed[57,58,59,60,61,62,63,64,65,66,67] and some have entered clinical development (Table 1). However, the further development of IDO1 inhibitors has experienced a significant setback due to the recent failure of the phase III trial (ECHO-301/KEYNOTE-252) of epacadostat (INCB024360), the would-be first-in-class IDO1 inhibitor, which had been anticipated to receive regulatory approval on the basis of promising results from initial studies (Fig. 1) [24, 50, 68,69,70,71,72].

The roadmap of IDO1 inhibitor development. IDO1 indoleamine 2,3-dioxygenase 1. PD-1 programmed death receptor 1
Several reviews discussing the biochemical properties and physiological functions of IDO1 and the therapeutic applications of IDO1 inhibition in cancer and other diseases have been published [8, 16, 73,74,75]. The aims of this article are, (i) to introduce the current status of clinical testing of IDO1 inhibitors (ii) to review representative data of epacadostat and (iii) to discuss some open questions raised from the failure of epacadostat in advanced malignant melanoma.
IDO1 inhibitors under clinical development
Currently, as a focus of research and drug discovery efforts, there are at least eight small molecule IDO1 inhibitors under investigation in clinical trials indicated by the clinical trial registry website ClinicalTrials.gov. These include epacadostat (INCB024360; developer: Incyte), indoximod (D-1MT; developer: NewLink Genetics), navoximod (NLG919; developer: NewLink Genetics), BMS-986205 (Ono-7701/linrodostat; developer: Bristol-Myers Squibb/Ono), PF-06840003 (EOS-200,271; developer: Pfizer), KHK-2455 (developer: Kyowa Hakko Kirin), LY-3381916 (developer: Eli Lilly) and DN1406131 (developer: Jiangxi Qingfeng).
The first clinical trial of IDO1 inhibitor was started in 2007, which is the first-in-human (FIH) trial of indoxiomd with the purpose of assessing its safety and pharmacokinetics profile in patients with advanced solid tumors (NCT00567931). Since then to the cutoff date of 16 June 2020, a total of 98 clinical trials aiming to characterize the safety and efficacy profile of different IDO1 inhibitors have been initiated and the majority of them (88/98, 90 %) focuses on the exploration of treatment combinations (Table 1). Because one trial may have been tested in multiple cancer types, total 104 studies of 98 clinical trials have been carried out (Table 2). Seventy studies have been performed in 6 cancer types including lung cancer (16/70), head and neck cancer (12/70), gynecological tumors including ovarian and endometrial carcinoma (12/70), malignant melanoma (11/70), urothelial carcinoma (11/70) and brain tumors especially glioblastoma (8/70). Twenty studies did not specifically indicate the target cancer type or enrolled healthy volunteers only, and the rest studies have been performed in 15 cancer types including RCC, HCC and CRC (Table 2). In all studies, ICPIs, in particular anti-PD-1/PD-L1 antibodies were the most popular class selected as a combination agent, while others include chemotherapy, cancer vaccines, and other immunotherapies targeting CTLA-4, lymphocyte-activation gene 3, C-C motif chemokine receptor type 4 are also under investigation.
An analysis of the trial initiation (i.e. First posted date disclosed in ClinicalTrials.gov) over time (Fig. 2) shows that the clinical development of IDO1 inhibitors started from 2007, and the expansion significantly accelerated after 2014 with its peak in 2017. In 2017, the number of all initiated trials has grown to 27, representing a 125 % increase from 2016 to 2017 and the proportion of phase III trials has grown from 8 % (1/12) to 30 % (8/27) from 2016 to 2017. However, a rapid fade of interest in IDO1 inhibitors came up in 2018, the year that the failure of ECHO-301/KEYNOTE-252 was announced[72].

Comparison of all initiated clinical trials by start date† and trial phase. †The calculation is according to the time of first posted date on clinicaltrials.gov
Epacadostat is an IDO1 selective inhibitor, which is the most advanced in the clinical development and would be supposed to be the first IDO1 inhibitor to get registration approval. Epacadostat showed preliminary promising anti-cancer activity in its early phase I/II trials (ECHO-202/KEYNOTE-037 and ECHO-204) when combined with anti-PD-1 drugs including pembrolizumab and nivolumab in patients with advanced malignant melanoma[76, 77]. However, its clinical value was not further validated in subsequently pivotal phase III trial (ECHO-301/KEYNOTE-252)[78] when the number of enrolled patients increased – the addition of epacadostat to pembrolizumab failed to improve progression-free survival (PFS) in patients with advanced malignant melanoma compared to pembrolizumab monotherapy.
Consequently, the unexpected setback of epacadostat has had a major impact on the entire field and tremendously slowed the pace of clinical development of all other IDO1-targeting compounds. In turn, total 27 trials involving 5 compounds, including epacadostat, BMS-986205, indoximod, PF-06840003 and LY-3381916, have been terminated, suspended or withdrawn and none of the other phase III programs of epacadostat is enrolling patients as originally planned. Just as every coin has its two sides, this failure also contributes to the understanding of the role of IDO1 inhibition and changes the clinical development program of inhibitors. Since the second half of 2018, seventeen new trials have been initiated and a large proportion of the clinical development is taking place in phase I/II, with only one agent-BMS-986205 proceeded to Phase III trial in bladder cancer (Fig. 2). Of note, the target patient populations in these new trials were narrowed down to certain tumor types, such as head and neck cancer, glioblastoma and bladder cancer. This may reflect that IDO1 is still regarded as a potential target to break the immune suppressive TME and restore the immune surveillance system. The critical challenges would be what we can learn from the failure and how to address the outstanding questions.
The development path of epacadostat
In preclinical studies, epacadostat selectively and competitively inhibited the catalytic activity of IDO1 in multiple cell-based assays with little activity against IDO2 and TDO2[65, 79](Table 3). In co-cultures of human allogeneic lymphocytes with dendritic cells (DCs) or tumor cells, epacadostat promoted the growth of effector T cells and NK cells, reduced the conversion of naïve T cells to regulatory T cells (Tregs) and increased the number of CD86high DC[79]. Along with these, administration of epacadostat to CT26 tumor-bearing mice reduced tumor growth with up to 57 % tumor growth control (TGC) in a dose-dependent and lymphocyte-dependent fashion and similarly decreased kynurenine levels in tumor tissues, plasma and draining lymph nodes ranging from 78–87 %[65]. As the most advanced IDO1 inhibitor, epacadostat was selected as a positive control in several preclinical studies of other IDO1 inhibitors in some animal models and showed similar IDO1 inhibitory activities and variable anti-tumor activities (Tables 3 and 4). Meanwhile, a synergy of INCB23843, an analogue of epacadostat, with anti-PD-L1 or anti-CTLA-4 antibodies, respectively, was investigated in B16 melanoma models. The combination with either anti-PD-L1 or anti-CTLA-4 antibody could suppress tumor growth more effectively than either agent alone, primarily through reactivation of anti-tumor immunity[80]. The combination of epacadostat with a MerTK inhibitor or an agonistic CD40 antibody also exhibited positive anti-tumor activities but epacadostat alone not[81, 82]. Furthermore, anti-PD-1 antibody, anti-CTLA-4 antibody and agonistic CD40 antibody therapies have been found to induce IDO1 expression[82, 83], which might be one reason for the limited response rate of these therapies. Therefore, the combination of IDO1 inhibitor with these therapies to defeat cancer has been paid high attention.
With the support of the results generated from the above summarized preclinical data, the activity of epacadostat as single agent was evaluated in a FIH phase I trial (NCT01195311) and subsequently in a phase II trial (NCT01685255). In the FIH phase I trial, the safety, pharmacokinetics (PK), pharmacodynamics (PD) and preliminary anti-tumor efficacy profile of epacadostat were characterized in 52 patients with advanced solid malignancies. The results showed that the safety profile of epacadostat was manageable and it can be generally well tolerated at doses of up to 700 mg twice daily (BID). The PK of epacadostat was characterized by a time of maximum concentration at around 2 hours, a dose-independently biphasic disposition with an apparent terminal-phase disposition half-life of 2.9 hours and a relatively longer effective half-life of 4 to 6 hours in the context of systemic accumulation following BID dosing[91]. Regarding PD assessments, in addition to the direct evaluation of the plasma concentration of TRP and KYN in patients at different time points, the levels of TRP and KYN in patients blood pretreated with interferon gamma (IFN-γ) or lipopolysaccharide (LPS) were also determined. IFN-γ or LPS was used to induce IDO1 expression and minimize the confounding effect of TDO. Tracking KYN levels in plasma has been considered as a sufficient method for assessing IDO1 activity and selected as a valuable pharmacodynamic marker in the clinical studies of epacadostat[65]. The percentage of IDO1 inhibition was finally defined as the reduction in plasma KYN levels from the predose to the postdose value. It was found that epacadostat could reduce KYN levels at doses ≥ 100 mg BID and a 90 % inhibition was achieved when the plasma concentration of epacadostat was greater than 500 nmol/L[91]. Epacadostat’s on-target potency, i.e., its concentration exerting 50 % of the maximal inhition effect (IC50) against IDO1, is important for dose selection but complicated by the bioconversion of TRP to KYN catalyzed by both IDO1 and TDO. In vitro and ex vivo, the IC50 was estimated following the selective induction of IDO1, rendering the TDO activity relatively insignificant; however, it was desirable to determine the in vivo IC50 without inducing IDO1. Based on the data from the FIH trial, a mechanism-based population PD model was developed, and epacadostat in vivo IC50 was estimated to be ~ 70 nM, consistent with the ex vivo value independently determined. And this model also suggests that approximately 60 % of TRP to KYN bioconversion was attributed to IDO1 in the cancer patients at baseline, the mean concentration of plasma KYN was 2–3 % of that of TRP, and no significant changes in TRP concentrations were observed before or during epacadostat treatment[92]. Based on PK and PD data, 300 mg BID was selected as the recommended phase II monotherapy dose to provide sufficient drug exposure that would inhibit > 90 % of IDO1 activity[91]. The therapeutic efficacy profile from this FIH trial suggested a limited anti-tumor activity of epacadostat in human, only stable disease (SD), which is one of response category[93], was observed as the best overall response in 18 of 52 patients, and SD lasting ≥ 16 weeks was seen in 7 patients [91].
The following phase II trial aiming to compare the efficacy of epacadostat versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)–only epithelial ovarian cancer reported similar results. Epacadostat was generally well tolerated, but its efficacy was not satisfactory enough, i.e. no significant difference in efficacy was found between epacadostat and tamoxifen, and thus the trial was terminated accordingly[94]. These results are not that unexpected, since multiple mechanisms are involved in the evasion of tumors from immune surveillance and IDO1 inhibition strategy alone may be insufficient. Given that the synergy of IDO1 inhibitor and ICPIs was confirmed in B16 melanoma models[80], several phase I/II clinical trials were then initiated to investigate epacadostat in combination with ipilimumab[95] (NCT01604889), pembrolizumab[76] (NCT02178722) and nivolumab[77] (NCT02327078) for the treatment of unresectable or metastatic malignant melanoma.
In the phase I/II trial investigating epacadostat in combination with ipilimumab (NCT01604889), epacadostat ≤ 50 mg BID was demonstrated to bear clinical and pharmacologic activity and was generally well tolerated in patients with advanced melanoma[95]. Objective responses were observed in 9 of 39 immunotherapy-naive patients (23 %), while no objective response was seen in the rest 11 patients who previously received immunotherapy although 3 of them (27 %) had SD. Using the same method and definition of IDO1 inhibition as in the previous FIH trial, PD data from this trial showed that a dose-dependent inhibition of IDO1 by epacadostat and the average IDO1 inhibiton exceeded 50 % at doses of 25 mg BID and 50 mg BID[95]. These results were generally consistent with previously reported data in patients with advanced solid malignancies[91]. The emerging success of anti-PD-1 antibodies for the treatment melanoma led to an early termination of this trial (NCT01604889, only phase I part was conducted) and shift more attention to exploring epacadostat in combination with anti-PD-1 antibodies.
ECHO-202/ KEYNOTE-037 (NCT 02178722) was a phase I/II trial aiming to evaluate epacadostat plus pembrolizumab in patients with advanced solid tumors. The results from its phase I part showed that the combination regimen of epacadostat and pembrolizumab was well tolerated and a maximum tolerated dose (MTD) of epacadostat did not reach within pre-defined dose levels up to 300 mg BID; ≥50 % time-averaged IDO1 inhibition (i.e. the level of pharmacodynamic activity associated with inhibition of tumor growth seen in nonclinical models) determined with the above mentioned PD model was yielded in all patients treated with epacadostat 100 mg BID or 300 mg BID; objective responses were observed in 25 of total 62 patients (42 %) and 12 of 22 patients with advanced malignant melanoma (55 %)[76]. The phase II trial of epacadostat in combination with nivolumab, i.e. ECHO-204 (NCT02327078) having similar study design and patient population demonstrated consistently promising anti-tumor activity with objective response at 62 % and tolerable safety profile in 50 patients with advanced malignant melanoma[77]. On the basis of these results, epacadostat 100 mg BID was selected to combine with anti-PD-1 antibodies for additional investigation in phase III trials including ECHO-301/KEYNOTE-252 (NCT02752074)[78].
A non-randomized trial using a small number of patients is prone to biased selection that may confound the result and be misleading. Thus, a further confirmation with a randomized phase II trial as gatekeeper before entering into pivotal phase III trial might be necessary. However, regardless of the lack of evidence from at least one randomized, active controlled (anti-PD-1 antibody monotherapy) phase II trial as a validation step, the phase III trial ECHO-301/KEYNOTE-252 was initiated to further clarify the clinical benefit of epacadostat plus pembrolizumab compared to pembrolizumab alone in patients with advanced malignant melanoma previously untreated with a ICPI using PFS as primary endpoint. In this trial, the testing of IDO1 expression status by use of in situ hybridization RNA scope technology was retrospectively completed after patients were enrolled, but was not taken into account in the treatment assignment or primary analysis of the endpoints. Among all enrolled 706 patients, IDO1 expression was positive in 451 (90 %) of 502 patients with evaluable tumor specimens, and the IDO1 expression positivity was determined as higher than 1 % of tumor or immune cells expressed IDO1. The positive expression of IDO1 in tumor cells, intratumoral immune cells and both kinds of cells was found in 393 of 502 samples (78 %), 422 of 472 samples (89 %) and 364 (77 %) of 472 samples, respectively. As of data cutoff, surprisingly, the trial failed to prove the clinical value of epacadostat in terms of PFS, overall survival (OS), and objective response with a median PFS of 4.7 months (95 % CI 2.9–6.8) in the epacadostat plus pembrolizumab group and 4.9 months (2.9–6.8) in the placebo plus pembrolizumab group (HR 1.00, one-sided P = 0.52). Furthermore, the absence of a significant PFS benefit for epacadostat was evident in all prespecified and post-hoc subgroups examined including IDO1 status (Table 5).
The failure of ECHO-301 has been discussed in terms of the sufficiency of the dose of epacadostat and drug combination selection[96, 97]. Furthermore, the signaling activity of IDO1 besides its catalytic activity may be attributed to another reason for the failure[97]. It has been described that the immunoregulatory activity of IDO1 is related to its signaling activity in addition to its catalytic activity[98, 99]. It could be the case and would offer a hypothesis that some IDO1 inhibitors, which have been designed for blocking the catalytic activity of IDO1 only, may not in reality block its signaling activity and thus could not be sufficient to “neutralize” the effects of IDO1. Although the failure of ECHO-301 trial upset the whole field as the benchmarker of IDO1 inhibitors, all preclinical and clinical data from epacadostat discussed above is like a treasure for the successors to excavate and get potential inspiration for future direction.
Future directions
Despite the failure of epacadostat to demonstrate clinical benefits in the pivotal phase III trial, there is much to be learned from the available data, which is likely to ultimately benefit the field. It is too arbitrary and premature to deny the future of the entire field as the result of a single trial failure in the context of some reasonable doubt raised accordingly.
Which biomarker should be used for IDO1 selective inhibitor therapy?
The term biomarker is commonly understood as referring to a characteristic that is measured as an indicator of normal biologic processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions[100]. At present, no biomarker has been used for patients’ selection in IDO1 inhibitors clinical trials although the IDO1 expression status has being explored in some trials to assess its correlation with response.
In a way, IDO1 expression is a kind of “straightforward choice” for the selection of IDO inhibitor predictive biomarker. A series of evidence could support this choice. As a part of a tumor immune escape mechanism, IDO1 overexpression has been described in numerous human cancer types not only in tumor lesions by tumor cells and other components of the TME like endothelial cells, myeloid derived suppressor cells (MDSCs), DCs, but also in tumor draining lymph nodes[30, 101, 102]. Several lines of studies have demonstrated that the overexpression of IDO1 in either tumor cells or in other cells within TME is associated with a more aggressive cancer phenotype, advanced disease stage and worse clinical outcome of various cancers including ovarian carcinoma, CRC, endometrial cancer, melanoma, cervical cancer, glioblastoma, lung adenocarcinoma, diffuse large B-cell lymphoma (DLBCL)[25,26,27,28,29, 31,32,33,34,35,36, 38]. Furthermore, a recent meta-analysis of 2706 patients from 24 articles found that high IDO1 expression is correlated with poor clinical outcomes in all cancers[37]. However, the results from the phase III trial (ECHO-301/KEYNOTE-252) did not show significant PFS benefit with epacadostat plus pembrolizumab therapy in overall population as well as the post-hoc IDO1-positive subgroup, despite 90 % patients were IDO1-positive[78]. This finding suggests that the expression of IDO1 in both tumor cells or immune cells is an important - but not a definitive - predictive biomarker of response to IDO1 inhibition.
Some reports give clues to explain the indefiniteness of IDO1 expression being a biomarker. First, there is an uncertainty of an exact correlation between IDO1 expression and tumor progression or patients’ outcome. Despite the positive correlation between high IDO1 expression and advanced cancer stage or poor patient prognosis[25,26,27,28,29, 31,32,33,34,35,36, 38], the negative correlation is found in patients with RCC[103] and contradictory correlations are documented in patients with HCC[104, 105]. Using Kaplan-Meier survival analyses, it has been found that no statistically significant relationship between OS, post-progression survival and distant-metastasis-free survival rates and tumour cell-derived IDO1 expression exists in patients with various types of cancers, such as lung, ovarian, breast or gastric cancer[88].
In addition, the method used to assess IDO1 expression was different from one study to another. Furthermore, the definition and grade of IDO1-positive tumors, as well as the nature of the IDO1-expressing cells were not always consistent. In the phase II trial (ECHO-202) and the phase III trial (ECHO-301/KEYNOTE-252), the methods used to assess IDO1 expression were the same (in situ hybridization using RNAscope technology), while the nature of expressing cells and threshold of positivity were different[76, 78]. In the phase II trial, only tumor-infiltrated immune cells were tested for IDO1 expression, and a histoscore ≥ 5 was used as an arbitrary cutoff for the IDO1-positive status. In the phase III trial, IDO1 positivity was defined as expression in more than 1 % of tumor or intra-tumoral immune cells.
Furthermore, the heterogeneity of IDO1 expression in tumor tissues also affect the detection results of IDO1 expression. Theate et al. assessed IDO1 expression by immunohistochemistry on tissue microarrays in 15 common solid tumor types with about 17–60 samples of each and found that the IDO1 expression could be identified in total 383 of 624 samples (61 %), but was limited to a small fraction of cells, and the proportion of IDO1-expressing cells widely varies according to the tumor type[101]. Among these 15 tumor types, cervical carcinoma (17/17, 100 %), endometrial carcinoma (45/48, 94 %) and bladder urothelial carcinoma (15/16, 94 %) most frequently expressed IDO1, followed by RCC (43/53, 81 %), NSCLC (41/51, 80 %), CRC (46/59, 78 %), head and neck cancer (14/24, 58 %) and melanoma (32/60, 53 %) while most glioblastoma (5/60, 8 %) were negative. Of note, the staining patterns of IDO1 varied widely according to the tumor type. For example, vascular staining was predominated in RCC (36/43, 84 %), which was consistent with previous report[103], whereas DC-like staining and tumor cell staining were predominated in CRC (45/46, 98 %) and endometrial carcinomas (39/45, 87 %), respectively[101]. It can be found that IDO1 is mainly expressed in the vasculature in RCC lesions rather than in tumor cells as described for other cancers. This variation may contribute to the contradictory phenomenon that the expression of IDO1 correlates with long survival of patients with RCC. Furthermore, in the phase III trial (ECHO-301/KEYNOTE-252), the high proportion of IDO1-positive samples was mainly attributable to the staining of immune cells rather than tumor cells[78]. Establishing a standard and uniform procedure of IDO1 expression assessment, for instance, a deep semi supervised generative learning for automated tumor proportion scoring which has been applied to standardization of PD-L1 expression and status[106], is required so that the extrapolation from non-clinical to clinical and from small scale to large scale may be more reliable.
Based on the clinical trial information, PD-L1 expression, tumor mutation burden (TMB), microsatellite instability-high (MSI-H) and mismatch-repair deficiency (MMR) have been identified as predictive biomarkers for anti-PD-1/PD-L1 antibody therapies[107,108,109,110,111]. Although biomarkers of IDO1 inhibitors gleaned from clinical trials is awaited, we here suggest that leveraging existing databases, such as The Cancer Genome Atlas, to figure out the molecular characteristics associated with the effect of IDO1 on prognosis might be a further approach. In fact, by this way, we have identified that the expression of 43 key genes is a better biomarker than the patient category regarding to the efficacy of IDO1 inhibitors (unpublished data).
Whether plasma KYN concentration is an efficient pharmacodynamic marker?
Due to the lack of assays, which allow to directly determine the activity of the IDO1 enzyme, measuring the changes of the TRP, KYN, or KYN/TRP ratio has been selected in different studies as a surrogate indicator for activity evaluation. Ideally, these measurements should be tested in tumor tissues, which can provide intuitive evidence of IDO1 inhibition. Nevertheless, the majority of current available preclinical data on IDO1 activity are derived from serum. In clinical trials of epacadostat, IDO1 activity was determined by only measuring the inhibition of the plasma KYN concentration. However, the depletion of TRP is not always synchronized with accumulation of KYN in human cancers (Table 6). In fact, TRP depletion is more sensitive to disease condition than Kyn accumulation, not only in cancers, but also in other diseases such as Alzheimer’s disease and atherosclerosis[112,113,114]. TRP deprivation is significantly correlated with clinical outcomes like the quality of life in CRC, survival in malignant melanoma and advanced disease stage in lung cancer[39,40,41] while the KYN generation has a prognostic significance in adult T-cell leukemia/lymphoma (ATL)[42]. Thus, it will be more informative to use of TRP, KYN concentrations and the KYN/TRP ratio for the IDO1 activity assessment and the dose selection from PD perspective.
Badawy et al. suggested that IDO1 induction is not the only determinant of the increase in the plasma KYN/TRP ratio in vivo[115]. Other determinants are liver TDO activity, increased flux of Trp through TDO and KP enzymes influencing KYN, especially kynurenine 3-monooxygenase, kynureninase and to a lesser extent kynurenine aminotransferase. Measurements of these additional determinants has been suggested to be able to contribute to a better understanding of the TRP status and IDO1 activity in IDO1 involved cancer and other diseases such as schizophrenia[115]. Furthermore, KYN/TRP ratio has also been known to be positively correlated with the concentrations of neopterin[40, 43], IL-6 as well as sIL-2Rα[44]. In summary, we suggest to determine the parameters TRP, KYN and the ratio of both in tumor and peripheral blood as primary PD marker in small studies for dose selection, before moving forward to large scale studies, and try to collect more information on other determinants of the KYN/TRP ratio if possible.
What is the most possible candidate tumor type?
Until now, lung cancer is the most-studied tumor type in clinical trials of IDO1 inhibitors (Table 2). However, the reason, why so much attention has been paid to lung cancer, is not sufficient. Most clinical trials of IDO1 inhibitors have been carried out to test the anti-PD-1 antibody combination in a tumor type where anti-PD-1 antibody alone has activity.
There are 17 new trials initiated after the failure of ECHO-301. The newest ones are a phase II trial (NCT04231864) to evaluate epacadosat plus durvalumab, a anti-PD-L1 antibody, for treatment of advanced Epstein-Barr virus (EBV) positive nasopharyngeal carcinoma (NPC) and a phase III trial (NCT03661320) to assess IDO1 inhibitor (BMS-986205) in combination with nivolumab and chemotherapy (gemcitabine and cisplatin) as neoadjuvant treatment in patients with muscle-invasive bladder cancer. Despite clinical trials in bladder cancer have been initiated before, the newly initiated phase II trial (NCT04231864) is the first trial of IDO1 inhibitor in NPC. In vitro, exposure to the milieu created by IDO1-positive NPC cell line CNE2 cell impaired the lymphocyte cytotoxicity[118]. IDO1 activity defined as KYN/TRP ratio is significantly higher in patients with NPC and could be a relevant marker for advanced NPC progression[119]. Meanwhile, IDO1 is expressed by either tumor cells or tumor-associated immune cells in 99 % or 94 % patients with NPC using 1 % cutoff value of immunohistochemistry and negatively associated with OS[120]. Furthermore, an immune checkpoint-based signature classifier, in which IDO1 is one out of five features was significantly correlated with the survival in patients’ with high EBV-DNA load[120]. Furthermore, the upregulation of PD-L1 expression induced by the latent membrane protein 1 and IFN-γ was detected in EBV-positive NPC cell lines[121]. Together, these findings provide solid evidence that IDO1 is involved in immune evasion of NPC, which forms the rationale for pursuing clinical trials.
Recently, we have reported that the expression of both IDO1 and TDO was associated with advanced stage of disease and poor prognosis in patients with glioblastoma, which attributed to Kyn-AhR-AQP signaling pathway and an IDO1/TDO (IDO1 and TDO) dual inhibitor could exert anti-glioma effects in GL261 orthotopic glioma mice[122]. TDO has also been regarded as a logical candidate to mediate resistance to IDO1-selective inhibition besides its direct roles in cancer, the expression of TDO was detected in lots of cancer types such as HCC, glioblastoma, bladder carcinoma, pancreatic carcinoma, colon carcinoma, melanoma and breast cancer[56, 123,124,125]. About one-third of cancer cell lines harboring high levels of KYN have simultaneous IDO1 and TDO expression, while the others are driven by either IDO1 or TDO[126].
In conclusion, aberrant activation of KP as well as a solid correlation between high expression of IDO1 or IDO1/TDO and tumor progression or poor patients’ outcome should be kept in mind when selecting the candidate tumor type for clinical trials of IDO1 inhibitors.
Conclusions
Extensive evidence supports the notion that an elevated TRP catabolism contributes to immune regulation that promotes tumor growth and therapy resistance. Targeting this biochemical pathway in patients with cancer offers promise. Preclinical and preliminary clinical data demonstrate evidence to anti-tumor activity of IDO1 inhibitors. However, setbacks in a recent clinical trial suggested an urgent need for more research to better understand what is the exact mechanism responsible for the immunosuppresion of IDO1 in cancer, how the TRP catabolism contributes to immune regulation mechanisms in the TME, what is the role of KYN and TRP in different cancer types, and to identify biomarkers to select patients that ultimately benefit from this treatment.
Availability of data and materials
All the data and materials are available upon reasonable request from the corresponding author.
Abbreviations
- AhR:
-
Aryl hydrocarbon receptor
- AML:
-
Acute myeloid leukemia
- ATL:
-
Adult T-cell leukemia/lymphoma
- BID:
-
Twice daily
- CRC:
-
Colorectal cancer
- CTLA-4:
-
Cytotoxic T-lymphocyte antigen 4
- DC:
-
Dendritic cell
- DLBCL:
-
Diffuse large B-cell lymphoma
- EBV:
-
Epstein-Barr virus
- FIH:
-
First-in-human
- GCN2:
-
Nonderepressible 2
- HCC:
-
Hepatocellular carcinoma
- ICPI:
-
Immune checkpoint inhibitor
- IDO1:
-
Indoleamine 2,3-dioxygenase 1
- IFN-γ:
-
Interferon gamma
- IDO2:
-
Indoleamine 2,3- dioxygenase 2
- KP:
-
Kynurenine pathway
- KYN:
-
Kynurenine
- LPS:
-
Lipopolysaccharide
- mTOR:
-
Mammalian target of rapamycin
- NK:
-
Natural killer
- NPC:
-
Nasopharyngeal carcinoma
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PD:
-
Pharmacodynamics
- PD-1:
-
Programmed death receptor 1
- PD-L1:
-
Programmed death ligand 1
- PFS:
-
Progression-free survival
- PK:
-
Pharmacokinetics
- RCC:
-
Renal cell carcinoma
- SD:
-
Stable disease
- TDO:
-
Tryptophan 2,3 dioxygenase
- TME:
-
Tumor microenvironment
- TGC:
-
Tumor growth control
- Treg:
-
Regulatory T cell
- TRP:
-
Tryptophan
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol. 2012;23(Suppl 8):viii6–9.
Ipilimumab Highlights of Prescribing Information US. https://packageinserts.bms.com/pi/pi_yervoy.pdf. Published 2020. Accessed.
Pembrolizumab Highlights of Prescribing Information US. https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf. Published 2020. Accessed.
Nivolumab Highlights of Prescribing Information US. https://packageinserts.bms.com/pi/pi_opdivo.pdf. Published 2020. Accessed.
Xin Yu J, Hubbard-Lucey VM, Tang J. Immuno-oncology drug development goes global. Nat Rev Drug Discov. 2019;18(12):899–900.
Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4(10):762–74.
Platten M, Nollen EAA, Rohrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. 2019;18(5):379–401.
Cervenka I, Agudelo LZ, Ruas JL. Kynurenines. Tryptophan’s metabolites in exercise, inflammation, and mental health. Science. 2017; 357:6349.
Mellor AL, Lemos H, Huang L. Indoleamine 2,3-Dioxygenase and Tolerance: Where Are We Now? Front Immunol. 2017;8:1360.
King NJ, Thomas SR. Molecules in focus: indoleamine 2,3-dioxygenase. Int J Biochem Cell Biol. 2007;39(12):2167–72.
Haber R, Bessette D, Hulihan-Giblin B, Durcan MJ, Goldman D. Identification of tryptophan 2,3-dioxygenase RNA in rodent brain. J Neurochem. 1993;60(3):1159–62.
Knox WE, Mehler AH. The conversion of tryptophan to kynurenine in liver. I. The coupled tryptophan peroxidase-oxidase system forming formylkynurenine. J Biol Chem. 1950;187(1):419–30.
Capece L, Lewis-Ballester A, Marti MA, Estrin DA, Yeh SR. Molecular basis for the substrate stereoselectivity in tryptophan dioxygenase. Biochemistry. 2011;50(50):10910–8.
Leeds JM, Brown PJ, McGeehan GM, Brown FK, Wiseman JS. Isotope effects and alternative substrate reactivities for tryptophan 2,3-dioxygenase. J Biol Chem. 1993;268(24):17781–6.
Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72(21):5435–40.
Larkin PB, Sathyasaikumar KV, Notarangelo FM, Funakoshi H, Nakamura T, Schwarcz R, et al. Tryptophan 2,3-dioxygenase and indoleamine 2,3-dioxygenase 1 make separate, tissue-specific contributions to basal and inflammation-induced kynurenine pathway metabolism in mice. Biochim Biophys Acta. 2016;1860(11 Pt A):2345–54.
Song P, Ramprasath T, Wang H, Zou MH. Abnormal kynurenine pathway of tryptophan catabolism in cardiovascular diseases. Cell Mol Life Sci. 2017;74(16):2899–916.
Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5(11):2516–22.
Pfefferkorn ER. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc Natl Acad Sci U S A. 1984;81(3):908–12.
Bonda DJ, Mailankot M, Stone JG, Garrett MR, Staniszewska M, Castellani RJ, et al. Indoleamine 2,3-dioxygenase and 3-hydroxykynurenine modifications are found in the neuropathology of Alzheimer’s disease. Redox Rep. 2010;15(4):161–8.
Mazarei G, Leavitt BR. Indoleamine 2,3 Dioxygenase as a Potential Therapeutic Target in Huntington’s Disease. J Huntingtons Dis. 2015;4(2):109–18.
Schwarcz R, Stone TW. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology. 2017;112(Pt B):237–47.
Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–3.
Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res. 2006;12(4):1144–51.
Brody JR, Costantino CL, Berger AC, Sato T, Lisanti MP, Yeo CJ, et al. Expression of indoleamine 2,3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle. 2009;8(12):1930–4.
Speeckaert R, Vermaelen K, van Geel N, Autier P, Lambert J, Haspeslagh M, et al. Indoleamine 2,3-dioxygenase, a new prognostic marker in sentinel lymph nodes of melanoma patients. Eur J Cancer. 2012;48(13):2004–11.
Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18(22):6110–21.
Chevolet I, Speeckaert R, Haspeslagh M, Neyns B, Kruse V, Schreuer M, et al. Peritumoral indoleamine 2,3-dioxygenase expression in melanoma: an early marker of resistance to immune control? Br J Dermatol. 2014;171(5):987–95.
Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9(10):1269–74.
Okamoto A, Nikaido T, Ochiai K, Takakura S, Saito M, Aoki Y, et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin Cancer Res. 2005;11(16):6030–9.
Ino K, Yoshida N, Kajiyama H, Shibata K, Yamamoto E, Kidokoro K, et al. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br J Cancer. 2006;95(11):1555–61.
Inaba T, Ino K, Kajiyama H, Yamamoto E, Shibata K, Nawa A, et al. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol Oncol. 2009;115(2):185–92.
Inaba T, Ino K, Kajiyama H, Shibata K, Yamamoto E, Kondo S, et al. Indoleamine 2,3-dioxygenase expression predicts impaired survival of invasive cervical cancer patients treated with radical hysterectomy. Gynecol Oncol. 2010;117(3):423–8.
Rubel F, Kern JS, Technau-Hafsi K, Uhrich S, Thoma K, Hacker G, et al. Indoleamine 2,3-Dioxygenase Expression in Primary Cutaneous Melanoma Correlates with Breslow Thickness and Is of Significant Prognostic Value for Progression-Free Survival. J Invest Dermatol. 2018;138(3):679–87.
Kozuma Y, Takada K, Toyokawa G, Kohashi K, Shimokawa M, Hirai F, et al. Indoleamine 2,3-dioxygenase 1 and programmed cell death-ligand 1 co-expression correlates with aggressive features in lung adenocarcinoma. Eur J Cancer. 2018;101:20–9.
Yu CP, Fu SF, Chen X, Ye J, Ye Y, Kong LD, et al. The Clinicopathological and Prognostic Significance of IDO1 Expression in Human Solid Tumors: Evidence from a Systematic Review and Meta-Analysis. Cell Physiol Biochem. 2018;49(1):134–43.
Ninomiya S, Hara T, Tsurumi H, Hoshi M, Kanemura N, Goto N, et al. Indoleamine 2,3-dioxygenase in tumor tissue indicates prognosis in patients with diffuse large B-cell lymphoma treated with R-CHOP. Ann Hematol. 2011;90(4):409–16.
Huang A, Fuchs D, Widner B, Glover C, Henderson DC, Allen-Mersh TG. Serum tryptophan decrease correlates with immune activation and impaired quality of life in colorectal cancer. Br J Cancer. 2002;86(11):1691–6.
Weinlich G, Murr C, Richardsen L, Winkler C, Fuchs D. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology. 2007;214(1):8–14.
Suzuki Y, Suda T, Furuhashi K, Suzuki M, Fujie M, Hahimoto D, et al. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer. 2010;67(3):361–5.
Masaki A, Ishida T, Maeda Y, Suzuki S, Ito A, Takino H, et al. Prognostic Significance of Tryptophan Catabolism in Adult T-cell Leukemia/Lymphoma. Clin Cancer Res. 2015;21(12):2830–9.
Engin AB, Ozkan Y, Fuchs D, Yardim-Akaydin S. Increased tryptophan degradation in patients with bronchus carcinoma. Eur J Cancer Care (Engl). 2010;19(6):803–8.
Sperner-Unterweger B, Neurauter G, Klieber M, Kurz K, Meraner V, Zeimet A, et al. Enhanced tryptophan degradation in patients with ovarian carcinoma correlates with several serum soluble immune activation markers. Immunobiology. 2011;216(3):296–301.
de Jong RA, Nijman HW, Boezen HM, Volmer M, Ten Hoor KA, Krijnen J, et al. Serum tryptophan and kynurenine concentrations as parameters for indoleamine 2,3-dioxygenase activity in patients with endometrial, ovarian, and vulvar cancer. Int J Gynecol Cancer. 2011;21(7):1320–7.
Creelan BC, Antonia S, Bepler G, Garrett TJ, Simon GR, Soliman HH. Indoleamine 2,3-dioxygenase activity and clinical outcome following induction chemotherapy and concurrent chemoradiation in Stage III non-small cell lung cancer. Oncoimmunology. 2013;2(3):e23428.
Zhai L, Dey M, Lauing KL, Gritsina G, Kaur R, Lukas RV, et al. The kynurenine to tryptophan ratio as a prognostic tool for glioblastoma patients enrolling in immunotherapy. J Clin Neurosci. 2015;22(12):1964–8.
Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res. 2011;17(22):6985–91.
Hornyak L, Dobos N, Koncz G, Karanyi Z, Pall D, Szabo Z, et al. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front Immunol. 2018;9:151.
Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137–43.
Prendergast GC. Cancer: Why tumours eat tryptophan. Nature. 2011;478(7368):192–4.
Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017;77(24):6795–811.
Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy - Challenges and Opportunities. Trends Pharmacol Sci. 2018;39(3):307–25.
Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–42.
Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. 2012;1(9):1460–8.
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203.
Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11(3):312–9.
Gaspari P, Banerjee T, Malachowski WP, Muller AJ, Prendergast GC, DuHadaway J, et al. Structure-activity study of brassinin derivatives as indoleamine 2,3-dioxygenase inhibitors. J Med Chem. 2006;49(2):684–92.
Pereira A, Vottero E, Roberge M, Mauk AG, Andersen RJ. Indoleamine 2,3-dioxygenase inhibitors from the Northeastern Pacific Marine Hydroid Garveia annulata. J Nat Prod. 2006;69(10):1496–9.
Brastianos HC, Vottero E, Patrick BO, Van Soest R, Matainaho T, Mauk AG, et al. Exiguamine A, an indoleamine-2,3-dioxygenase (IDO) inhibitor isolated from the marine sponge Neopetrosia exigua. J Am Chem Soc. 2006;128(50):16046–7.
Banerjee T, Duhadaway JB, Gaspari P, Sutanto-Ward E, Munn DH, Mellor AL, et al. A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene. 2008;27(20):2851–7.
Carr G, Chung MK, Mauk AG, Andersen RJ. Synthesis of indoleamine 2,3-dioxygenase inhibitory analogues of the sponge alkaloid exiguamine A. J Med Chem. 2008;51(9):2634–7.
Kumar S, Jaller D, Patel B, LaLonde JM, DuHadaway JB, Malachowski WP, et al. Structure based development of phenylimidazole-derived inhibitors of indoleamine 2,3-dioxygenase. J Med Chem. 2008;51(16):4968–77.
Lob S, Konigsrainer A, Rammensee HG, Opelz G, Terness P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer. 2009;9(6):445–52.
Koblish HK, Hansbury MJ, Bowman KJ, Yang G, Neilan CL, Haley PJ, et al. Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and the growth of IDO-expressing tumors. Mol Cancer Ther. 2010;9(2):489–98.
Huang Q, Zheng M, Yang S, Kuang C, Yu C, Yang Q. Structure-activity relationship and enzyme kinetic studies on 4-aryl-1H-1,2,3-triazoles as indoleamine 2,3-dioxygenase (IDO) inhibitors. Eur J Med Chem. 2011;46(11):5680–7.
Yang S, Li X, Hu F, Li Y, Yang Y, Yan J, et al. Discovery of tryptanthrin derivatives as potent inhibitors of indoleamine 2,3-dioxygenase with therapeutic activity in Lewis lung cancer (LLC) tumor-bearing mice. J Med Chem. 2013;56(21):8321–31.
Yamamoto S, Hayaishi O. Tryptophan pyrrolase of rabbit intestine. D- and L-tryptophan-cleaving enzyme or enzymes. J Biol Chem. 1967;242(22):5260–6.
Hwu P, Du MX, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol. 2000;164(7):3596–9.
Sugimoto H, Oda S, Otsuki T, Hino T, Yoshida T, Shiro Y. Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc Natl Acad Sci U S A. 2006;103(8):2611–6.
Swart M, Verbrugge I, Beltman JB. Combination Approaches with Immune-Checkpoint Blockade in Cancer Therapy. Front Oncol. 2016;6:233.
<Incyte and Merck Provide Update on Phase 3 Study of Epacadostat in Combination with KEYTRUDA® (pembrolizumab) in Patients with Unresectable or Metastatic Melanoma.pdf > https://investor.incyte.com/news-releases/news-release-details/incyte-and-merck-provide-update-phase-3-study-epacados. Published 2018. Accessed.
Iversen TZ, Andersen MH, Svane IM. The targeting of indoleamine 2,3 dioxygenase -mediated immune escape in cancer. Basic Clin Pharmacol Toxicol. 2015;116(1):19–24.
Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, et al. Molecular Pathways: Targeting IDO1 and Other Tryptophan Dioxygenases for Cancer Immunotherapy. Clin Cancer Res. 2015;21(24):5427–33.
Brochez L, Chevolet I, Kruse V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur J Cancer. 2017;76:167–82.
Mitchell TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Olszanski AJ, et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J Clin Oncol. 2018:JCO2018789602.
Daud A, Saleh MN, Hu J, Bleeker JS, Riese MJ, Meier R, et al. Epacadostat plus nivolumab for advanced melanoma: Updated phase 2 results of the ECHO-204 study. J Clin Oncol. 2018;36(15_suppl):9511–1.
Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20(8):1083–97.
Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115(17):3520–30.
Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3.
Werfel TA, Elion DL, Rahman B, Hicks DJ, Sanchez V, Gonzales-Ericsson PI, et al. Treatment-Induced Tumor Cell Apoptosis and Secondary Necrosis Drive Tumor Progression in the Residual Tumor Microenvironment through MerTK and IDO1. Cancer Res. 2019;79(1):171–82.
Georganaki M, Ramachandran M, Tuit S, Nunez NG, Karampatzakis A, Fotaki G, et al. Tumor endothelial cell up-regulation of IDO1 is an immunosuppressive feed-back mechanism that reduces the response to CD40-stimulating immunotherapy. Oncoimmunology. 2020;9(1):1730538.
Brown ZJ, Yu SJ, Heinrich B, Ma C, Fu Q, Sandhu M, et al. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol Immunother. 2018;67(8):1305–15.
Richards T, Brin E. Cell based functional assays for IDO1 inhibitor screening and characterization. Oncotarget. 2018;9(56):30814–20.
Yue EW, Douty B, Wayland B, Bower M, Liu X, Leffet L, et al. Discovery of potent competitive inhibitors of indoleamine 2,3-dioxygenase with in vivo pharmacodynamic activity and efficacy in a mouse melanoma model. J Med Chem. 2009;52(23):7364–7.
Lewis-Ballester A, Pham KN, Batabyal D, Karkashon S, Bonanno JB, Poulos TL, et al. Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat Commun. 2017;8(1):1693.
Fang K, Dong G, Wang H, He S, Wu S, Wang W, et al. Improving the Potency of Cancer Immunotherapy by Dual Targeting of IDO1 and DNA. ChemMedChem. 2018;13(1):30–6.
Fu R, Zhang YW, Li HM, Lv WC, Zhao L, Guo QL, et al. LW106, a novel indoleamine 2,3-dioxygenase 1 inhibitor, suppresses tumour progression by limiting stroma-immune crosstalk and cancer stem cell enrichment in tumour micro-environment. Br J Pharmacol. 2018;175(14):3034–49.
Feng X, Shen P, Wang Y, Li Z, Bian J. Synthesis and in vivo antitumor evaluation of an orally active potent phosphonamidate derivative targeting IDO1/IDO2/TDO. Biochem Pharmacol. 2019;168:214–23.
Song X, Sun P, Wang J, Guo W, Wang Y, Meng LH, et al. Design, synthesis, and biological evaluation of 1,2,5-oxadiazole-3-carboximidamide derivatives as novel indoleamine-2,3-dioxygenase 1 inhibitors. Eur J Med Chem. 2020;189:112059.
Beatty GL, O’Dwyer PJ, Clark J, Shi JG, Bowman KJ, Scherle PA, et al. First-in-Human Phase I Study of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase-1 Epacadostat (INCB024360) in Patients with Advanced Solid Malignancies. Clin Cancer Res. 2017;23(13):3269–76.
Shi JG, Bowman KJ, Chen X, Maleski J, Leopold L, Yeleswaram S. Population Pharmacokinetic and Pharmacodynamic Modeling of Epacadostat in Patients With Advanced Solid Malignancies. J Clin Pharmacol. 2017;57(6):720–9.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47.
Kristeleit R, Davidenko I, Shirinkin V, El-Khouly F, Bondarenko I, Goodheart MJ, et al. A randomised, open-label, phase 2 study of the IDO1 inhibitor epacadostat (INCB024360) versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)-only epithelial ovarian cancer, primary peritoneal carcinoma, or fallopian tube cancer. Gynecol Oncol. 2017;146(3):484–90.
Gibney GT, Hamid O, Lutzky J, Olszanski AJ, Mitchell TC, Gajewski TF, et al. Phase 1/2 study of epacadostat in combination with ipilimumab in patients with unresectable or metastatic melanoma. J Immunother Cancer. 2019;7(1):80.
Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol. 2019;41(1):41–8.
Eynde BJVd B, Nv, Baurain J-F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annual Review of Cancer Biology. 2020;4(1):241–56.
Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12(9):870–8.
Albini E, Rosini V, Gargaro M, Mondanelli G, Belladonna ML, Pallotta MT, et al. Distinct roles of immunoreceptor tyrosine-based motifs in immunosuppressive indoleamine 2,3-dioxygenase 1. J Cell Mol Med. 2017;21(1):165–76.
FDA-NIH Biomarker Working Group. In: BEST (Biomarkers, EndpointS, and other Tools) Resource. edn. Silver Spring (MD); 2016.
Theate I, van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld JC, et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res. 2015;3(2):161–72.
Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114(2):280–90.
Riesenberg R, Weiler C, Spring O, Eder M, Buchner A, Popp T, et al. Expression of indoleamine 2,3-dioxygenase in tumor endothelial cells correlates with long-term survival of patients with renal cell carcinoma. Clin Cancer Res. 2007;13(23):6993–7002.
Ishio T, Goto S, Tahara K, Tone S, Kawano K, Kitano S. Immunoactivative role of indoleamine 2,3-dioxygenase in human hepatocellular carcinoma. J Gastroenterol Hepatol. 2004;19(3):319–26.
Pan K, Wang H, Chen MS, Zhang HK, Weng DS, Zhou J, et al. Expression and prognosis role of indoleamine 2,3-dioxygenase in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134(11):1247–53.
Kapil A, Meier A, Zuraw A, Steele KE, Rebelatto MC, Schmidt G, et al. Deep Semi Supervised Generative Learning for Automated Tumor Proportion Scoring on NSCLC Tissue Needle Biopsies. Sci Rep. 2018;8(1):17343.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20.
Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064–74.
Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–7.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther. 2017;16(11):2598–608.
Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. 2019;12(1):54.
Liang H, Chen M, Qi F, Shi L, Duan Z, Yang R, et al. The proatherosclerotic function of indoleamine 2, 3-dioxygenase 1 in the developmental stage of atherosclerosis. Signal Transduct Target Ther. 2019;4:23.
Giil LM, Midttun O, Refsum H, Ulvik A, Advani R, Smith AD, et al. Kynurenine Pathway Metabolites in Alzheimer’s Disease. J Alzheimers Dis. 2017;60(2):495–504.
Weaver D, Gupta M, Meek A, Wang Y, Wu F. Alzheimer’s Disease as a Disorder of Tryptophan Metabolism (2745). Neurology. 2020; 94.
Badawy AA, Guillemin G. The Plasma [Kynurenine]/[Tryptophan] Ratio and Indoleamine 2,3-Dioxygenase: Time for Appraisal. Int J Tryptophan Res. 2019;12:1178646919868978.
Cavia-Saiz M, Muniz P, De Santiago R, Herreros-Villanueva M, Garcia-Giron C, Lopez AS, et al. Changes in the levels of thioredoxin and indoleamine-2,3-dioxygenase activity in plasma of patients with colorectal cancer treated with chemotherapy. Biochem Cell Biol. 2012;90(2):173–8.
Hascitha J, Priya R, Jayavelu S, Dhandapani H, Selvaluxmy G, Sunder Singh S, et al. Analysis of Kynurenine/Tryptophan ratio and expression of IDO1 and 2 mRNA in tumour tissue of cervical cancer patients. Clin Biochem. 2016;49(12):919–24.
Liu P, Xie BL, Cai SH, He YW, Zhang G, Yi YM, et al. Expression of indoleamine 2,3-dioxygenase in nasopharyngeal carcinoma impairs the cytolytic function of peripheral blood lymphocytes. BMC Cancer. 2009;9:416.
Ben-Haj-Ayed A, Moussa A, Ghedira R, Gabbouj S, Miled S, Bouzid N, et al. Prognostic value of indoleamine 2,3-dioxygenase activity and expression in nasopharyngeal carcinoma. Immunol Lett. 2016;169:23–32.
Wang YQ, Zhang Y, Jiang W, Chen YP, Xu SY, Liu N, et al. Development and validation of an immune checkpoint-based signature to predict prognosis in nasopharyngeal carcinoma using computational pathology analysis. J Immunother Cancer. 2019;7(1):298.
Fang W, Zhang J, Hong S, Zhan J, Chen N, Qin T, et al. EBV-driven LMP1 and IFN-gamma up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget. 2014;5(23):12189–202.
Du L, Xing Z, Tao B, Li T, Yang D, Li W, et al. Both IDO1 and TDO contribute to the malignancy of gliomas via the Kyn-AhR-AQP4 signaling pathway. Signal Transduct Target Ther. 2020;5(1):10.
D’Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS, et al. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015;75(21):4651–64.
Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frederick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109(7):2497–502.
Hoffmann D, Dvorakova T, Stroobant V, Bouzin C, Daumerie A, Solvay M, et al. Tryptophan 2,3-Dioxygenase Expression Identified in Human Hepatocellular Carcinoma Cells and in Intratumoral Pericytes of Most Cancers. Cancer Immunol Res. 2020;8(1):19–31.
Li H, Ning S, Ghandi M, Kryukov GV, Gopal S, Deik A, et al. The landscape of cancer cell line metabolism. Nat Med. 2019;25(5):850–60.
Acknowledgements
Not applicable.
Funding
This work was supported by the Key Biomedical Program of Shanghai (NO. 18431902600&17431902200).
Author information
Authors and Affiliations
Contributions
QY, YY and HL contributed to conceive, design and revision of the manuscript sections, YY and HL wrote the manuscript, XF, SNZ, ZKX, and LS collected the related paper. QY, BS and CXK supervised the manuscript by providing critical feedbacks and revisions. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and Consent to participate
Not applicable.
Consent for publication
All the authors consent for publication.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yao, Y., Liang, H., Fang, X. et al. What is the prospect of indoleamine 2,3-dioxygenase 1 inhibition in cancer? Extrapolation from the past. J Exp Clin Cancer Res 40, 60 (2021). https://doi.org/10.1186/s13046-021-01847-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-01847-4