
Abstract
Gliomas, especially glioblastomas, represent one of the most aggressive and difficult-to-treat human brain tumors. In the last few decades, clinical immunotherapy has been developed and has provided exceptional achievements in checkpoint inhibitors and vaccines for cancer treatment. Immunization with cellular vaccines has the advantage of containing specific antigens and acceptable safety to potentially improve cancer therapy. Based on T cells, dendritic cells (DC), tumor cells and natural killer cells, the safety and feasibility of cellular vaccines have been validated in clinical trials for glioma treatment. For TAA engineered T cells, therapy mainly uses chimeric antigen receptors (IL13Rα2, EGFRvIII and HER2) and DNA methylation-induced technology (CT antigen) to activate the immune response. Autologous dendritic cells/tumor antigen vaccine (ADCTA) pulsed with tumor lysate and peptides elicit antigen-specific and cytotoxic T cell responses in patients with malignant gliomas, while its pro-survival effect is biased. Vaccinations using autologous tumor cells modified with TAAs or fusion with fibroblast cells are characterized by both effective humoral and cell-mediated immunity. Even though few therapeutic effects have been observed, most of this therapy showed safety and feasibility, asking for larger cohort studies and better guidelines to optimize cellular vaccine efficiency in anti-glioma therapy.
Similar content being viewed by others
Background
Arising from supporting glial cells, gliomas represent over 36% of malignant primary central nervous system (CNS) tumors [1]. Gliomas vary in aggressiveness from low-grade to highly malignant, with 5 year overall survival no greater than 35% [2]. According to the pathological features and clinical outcomes, the World Health Organization (WHO) grades gliomas on a scale of I to IV. The most benign brain tumor is designated grade I, has distinct boundaries, grows slowly, rarely spreads and typically arises in childhood. The most common glioma in adults is glioblastoma (GBM), an astrocytoma designated by the WHO as grade IV. GBMs remain among the most difficult brain tumors to treat, with a median survival of less than 2 years, despite surgical resection, radiation, and chemotherapy [3]. Over the past decades, an explosion in the understanding of treatment strategies of gliomas has progressed beyond the standard of care and consists of complete resection followed by radiotherapy and pharmacological treatment with chemotherapeutic agents, such as temozolomide. For example, on the basis of mutations in the genes encoding the isocitrate dehydrogenases IDH1 and IDH2, co-deletion of 1p19q (oligodendroglioma) and methylation of O6-Methylguanine-DNA methyltransferase (MGMT) have been subclassified as molecular diagnostics and prognostic markers for gliomas [4]. Unfortunately, limited therapeutic access to brain tumor and peritumoral tissue caused by the blood-brain barrier (BBB) still offers a new challenge to optimize glioma treatment, despite such efforts [5]. Considering the special anatomical position of gliomas, new cancer therapies need to be discovered to achieve optimally safer, less invasive and more effective treatment. On the basis of these observations, the clinical success of immunotherapy seems a predictable option in basic biological principles of glioma diagnosis and prognosis.
Cancer immunotherapy, also known as immuno-oncology, is a type of cancer treatment that uses the power of the body’s own immune system to prevent, control, and eliminate cancer [6, 7]. Recently, immunotherapy has been proposed against existing neoplasms, including breast cancer, lung cancer and even glioma, and may show significant promise where other approaches have failed [8]. Based on immune checkpoint inhibitors and vaccine-mediated immunization, previous studies have reported immunotherapeutic strategies that reverse immunosuppressive tumor environment, stimulate antigen presentation, and induce anti-tumor T cell responses [9]. For example, the dendritic cell (DC)-based vaccinations has been established as a promising approach for the immunotherapy of cancer [10,11,12]. In tumor cells, the physical modalities inducing immunogenic cell death (ICD), such as radiotherapy, UV light C, high hydrostatic pressure (HHP), hypericin-based photodynamic therapy (Hyp-PDT) or hyperthermia, are of particular interest in the development of DC-based vaccines for cancer immunotherapy [13]. Using an orthotopic HGG murine model, Garg et al.. observed that DC-based vaccines pulsed with Hyp-PDT-induced ICD provided strong anti-glioma survival benefit by inducing an immunostimulatory shift in the brain immune contexture from T regulatory cells (Tregs) to T helper 1 (Th1)/cytotoxic T lymphocyte (CTL)/Th17 cells. Meanwhile, a combination of ICD-based DC vaccines and temozolomide synergistically prolonged overall survival in malignant glioma-bearing mice, leading to ~ 50% long-term survivors [14]. Thus, therapeutic vaccines represent another valuable option for the management of glioma immunotherapy. Moreover, the demonstration of the efficacy of a vaccine for gliomas seems relatively straightforward to improve both anti-tumor innate and adaptive immune responses.
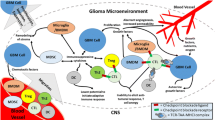
Surprisingly, cellular vaccines, which can be divided into autologous and allogeneic vaccines, have achieved significant levels of objective response in glioma treatment [15]. Theoretically derived from the patient’s own tumor, autologous vaccines have numerous potential advantages. The most important benefit is that autologous vaccines are likely to carry the unique tumor-associated antigens (TAA), which have been investigated as the specific immunological targets for immunotherapy in particular patients [16]. In a phase I/II trial in patients with recurrent malignant glioma, a DC-based multi-peptide vaccine derived from glioma-associated antigens (GAA) showed expected clinical efficacy in 22 patients, about 41% of vaccinated patients remained progression-free for ≥ 12 months [17]. More importantly, the safety and measurable immune response of the autologous ICT-107 vaccines produced from TAA-pulsed DC were confirmed for patients with newly diagnosed GBM in a phase I clinical trial [18]. However, even though vaccine efficacy has been demonstrated, the difficulty to produce and harvest enough vaccine doses still exists and diminishes its ultimate availability for clinical usage as an autologous vaccine. Unlike autologous cell therapy, allogeneic vaccines are composed of intact or modified cells harboring shared antigens found on a large percentage of gliomas from other patients or healthy donors. Chimeric antigen receptors (CARs) are artificial fusion proteins that incorporate an extracellular ligand recognition domain, a transmembrane domain, and an intracellular signaling domain to induce T cell activation upon antigen binding [19, 20]. Genetic engineering of allogeneic T cells to express CARs directed against specific antigens has explored a new way to optimize personalized glioma therapy [21]. In this review, we summarize the safety, feasibility, and immune/tumor response of cellular vaccines based on T cells, DC cells, tumor cells and natural killer (NK) cells in preclinical and clinical trials for glioma immunotherapy, and discuss the future prospects to optimize vaccine-mediated immunity according to current findings (Fig. 1).

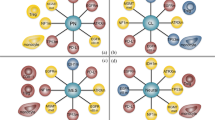
Cellular vaccine with TAA-engineered T cells in anti-glioma clinical trial. In glioma treatment, particularly, recurrent GBM, therapy with TAA-engineered T cells mainly uses chimeric antigen receptors (CARs) and DNA methylation-induced technology. For CAR-engineered T cells, single-chain variable fragments (scFv) of cell-surface receptor-engineered T cells mainly include IL13Rα2, EGFRvIII and HER2 selective ligands. In addition, treatment with DNA-demethylating agents, such as 5-aza-2’-deoxycytidine, facilitates CT antigen expression and constitutes an attractive immunological target for cancer immunotherapy. E13Y, IL13Ra2-selective ligand IL13; FRP5, HER2-specific MAb; CD4tm, CD4 transmembrane domain; CD8tm, CD8 transmembrane domain; 4-1BB, CD137 cytoplasmic signaling domain; CD3ζ, cytoplasmic signaling domain sequences
Current strategy of cellular vaccine in glioma immunotherapy
High-grade glioma, especially recurrent GBM, is almost inevitable, and the prognosis remains poor, with a median survival of 12–15 months [22]. Immunotherapies using autologous vaccines are now considered promising approaches for treating patients with malignant gliomas. The microenvironment of brain tumors contains unique tissue-resident cell types, in addition to cancer cells, including microglia, astrocytes, neurons and immune cells. The normal brain is considered an “immune privileged” organ that is sheltered from immune cell entry through the integrity of the BBB, which physically protects the brain from inflammatory factors that can be cytotoxic and cause neurodegeneration [23, 24]. On the other hand, peripheral immune cells infiltrating from the circulation can cross the BBB and enter the CNS after injury and, in disease, then join resident immune cells to modulate neuroprotective lymphocyte responses and brain function via their interactions with glia [25]. Indeed, current data indicate that therapeutic vaccines based on T cells, DC cells, tumor cells and NK cells are feasible and generally well tolerated. Furthermore, their clinical efficacy has been demonstrated in several randomized clinical trials.
TAA engineered T cells
Adoptive T cell transfer can overcome the in vivo progression of gliomas by using autologous cells with engineered receptor specificities for TAAs [26]. Antigen-specific T cells can be expanded ex vivo for subsequent administration, which produces the cytokines that are essential for T cell expansion and sustained anti-tumor activity [27]. CAR-engineered T cell (CAR T cell) therapy is a promising therapeutic approach genetically generated with modified T cells to express recombinant protein CARs that may be effectively and safely applied to GBMs to reduce recurrence rates [28, 29]. Several cell surface proteins, such as interleukin 13 receptor α2 (IL13Rα2), epidermal growth factor receptor variant III (EGFRvIII), ephrin type-A receptor 2 (EphA2), and human epidermal growth factor receptor 2 (HER2), have been found to actively target CAR T cell therapy in preclinical models [30,31,32,33], but only a few of these cell-surface receptors have been validated in clinical trials. Accordingly, a phase I/II clinical study of adoptive immunotherapy suggests that anti-EGFRvIII CAR-engineered T cells effectively produced the effector cytokines and interferon-γ, contributing to lyse the antigen-expressing glioma cells [34]. Meanwhile, another completed phase I clinical trial program (NCT01109095) reveals that anti-HER2 CAR CMV-specifc T cells seem to be able to inhibit HER2 + glioma growth [35]. Here, to improve anti-glioma responses, we discuss the use of TAA-engineered T cells through their clinical strategies and outcomes under investigation.
IL13Rα2-engineered T cells
IL13Rα2, a cell-surface receptor positively expressed in 82% of GBM samples and > 70% of glioma stem-like cancer initiating cells [36, 37], was previously thought to be directly associated with increased mesenchymal signature gene expression and poor patient survival [38]. For the treatment of recurrent GBM, Christine et al. showed the first-in-human clinical experience for CAR-engineered IL13Rα2-specific CD8+ CTL and observed significant tumor regression. Briefly, for autologous IL13-zetakine+ CD8+ CTL manufacturing, the peripheral blood mononuclear cells (PBMCs) were stimulated with anti-CD3 antibody, followed by DNA electroporation, drug selection and ex vivo expansion using OKT3 and irradiated feeders. In three patients with recurrent GBM, the feasibility of repetitive intracranial administration of first-generation IL13Rα2-specific CD8+ CAR T cells was demonstrated and transient anti-tumor activity for some patients was reported in the absence of serious adverse events, such as occlusion, malfunction, or infection [30]. Building on these results, the modified IL13Rα2-targeted CAR T cells were further reported to improve anti-tumor potency and T cell persistence by 4-1BB co-stimulation and IgG4-Fc linker mutation [39]. A patient with recurrent multifocal GBM who received treatment with modified IL13Rα2-targeted CAR T cells had regression of all intracranial and spinal tumors, along with significant increases in the levels of cytokines C-X-C motif chemokine ligand 9 (CXCL9) and CXCL10, as well as immune cells in the cerebrospinal fluid [28]. Comparing the ability to abrogate tumor growth at local and distant sites, Christine et al. suggested intraventricular administration of CAR T cells is better than intracavitary therapy for the treatment of malignant brain tumors. Nevertheless, the above evidence of the safety and anti-tumor activity of IL13Rα2-targeted CAR T cell immunotherapy still needs to be evaluated in a larger cohort of patients.
EGFRvIII-engineered T cells
Negative prognostic indicator EGFRvIII is expressed in about 25–33% of all patients with GBMs [40] and is the most commonly mutated gene among the EGFR family in glioma [41]. In EGFRvIII-expressing newly diagnosed GBM, a peptide vaccine targeting EGFRvIII (rindopepimut) was previously evaluated and found to be well tolerated, providing immune responses with prolonged progression-free survival [42, 43]. More recently, O’Rourke et al. conducted a phase I safety study of autologous CAR T cells targeted to EGFRvIII (CART-EGFRvIII) in 10 patients with recurrent GBMs. Intravenous infusion of a single dose of CART-EGFRvIII cells was found to be feasible and safe, without off-tumor toxicity or cytokine release syndrome [44]. For vaccine delivery, CART-EGFRvIII cells were detected transient expansion in peripheral blood. Trafficking of CART-EGFRvIII cells were also found in regions of active GBM in 7 patients with surgical intervention. Compared to pre-CART-infusion, tumors had markedly induced expression of immunosuppressive molecules (IDO1 and FoxP3) post-infusion. However, marked tumor regression was not observed by MRI over 18 months of follow-up after CART infusion. It is possible that this invalid clinical benefit of CART-EGFRvIII, which can be definitively determined from a large sample size, may improve the responsiveness of tumors through intraventricular therapy.
HER2-engineered T cells
Elevated HER2 expression has been reported in 41% of primary GBM samples and in 81.4% of GBM primary cell lines and were correlated with impaired survival [45, 46]. In preclinical models of GBM, bispecific CAR molecules that incorporated 2 antigen recognition domains for HER2 and IL13Rα2 showed the functional superiority of T cell expressing antigens ex vivo and in an orthotopic GBM xenograft model [47]. However, the safety concerns of autologous CART-HER2 cells were raised by the report of a serious adverse event following the administration 1 × 1010 T cells of vaccine based on trastuzumab [48]. While administration of up to 1 × 106/m2 CART-HER2 cells showed no evident toxicities, unfortunately, the expansion and persistence of CART-HER2 cells was limited [49]. To treat HER2-positive GBM, Nabil Ahmed et al. developed HER2-specific CAR-modified virus-specific T cells (CAR VS Ts-HER2) with an FRP5-based exodomain and a CD28.ζ endodomain [50]. Up to 1 × 108/m2 CAR VS Ts-HER2 were infused intravenously without dose-limiting toxic effects in 17 patients with progressive GBM. After the infusion, CAR VS Ts-HER2 was detectable in the peripheral blood for up to 12 months, with no observed expansion in peripheral blood (but expansion at GBM sites). Of 16 evaluable patients, 50% of patients had clinical benefit, as defined by a partial response (N = 1, over 9 months) and stable disease (N = 7, 8 weeks–29 months). Despite the feasibility and safety of CAR VS Ts-HER2, a clinical strategy using it alone or in combination with other immunomodulatory approaches is warranted to improve the anti-GBM activity of CAR VSTs-HER2 by augmenting their expansion and persistence.
DNA methylation-induced T cells
In cancer cells, epigenetic regulation by DNA methylation facilitates cancer/testis (CT) antigen expression and constitutes an attractive immunological target for cancer immunotherapy [51,52,53]. Recently, a novel approach was reported based on the adoptive transfer of cytotoxic lymphocytes with specificity for antigens induced by a DNA demethylating agent [54]. In this report, Kirkin et al.. found that after treatment with 5-aza-2’-deoxycytidine, a DNA-demethylating agent, activated CD4+ T helper cells from the patients with recurrent GBM could express a broad repertoire of endogenous CT antigens and serve as antigen-presenting cells to generate autologous CTL and NK cell responses. This group also revealed that injections of tumor-reactive lymphocytes generated by this DNA demethylation-mediated procedure into peripheral vein of GBM patients generated persistent anti-tumor immune response without treatment-related adverse effects. Treatment of cancer cells with 5-aza-2’-deoxycytidine also leads to the increased levels of MHC and costimulatory molecules (CD80, CD86, and CD40) necessary for the antigen-presenting function of DC cells [55, 56]. In addition, as an important determinant of anti-tumor immune response, DNA methylation aberration implicates epigenetic modulation as a combination regimen for potential precision immunotherapy with checkpoint blockade [57].
Overall, TAA-engineered T cells, along with DNA-demethylated T helper cells against recurrent GBMs, initially confirm the safety and anti-tumor activity of autologous T cell immunotherapy, providing a minimally invasive strategy for glioma treatment and prevention.
Autologous dendritic cell/tumor antigen vaccine (ADCTA)
DC therapy is a safe and well tolerated immunotherapeutic method, and its clinical effectiveness has been validated in melanoma, prostate cancer, malignant glioma, and renal cell carcinoma [58]. Previous studies have demonstrated that microglia are the predominant antigen-presenting cells (APCs) in the brain [59], while DC vaccines are likewise gaining significant clinical attention as a complementary strategy to stimulate T cell responses [60]. For instance, DC derived from blood monocytes could further enhance tumor-specific CD8+ T cell polyfunctionality in vivo when administered as a vaccine [61]. Recently, the presence of human cytomegalovirus (HCMV) antigens has been specifically found in gliomas, but not surrounding normal brain tissue. These unique and immunogenic HCMV antigens provide an attractive opportunity to leverage HCMV antigens as tumor-specific immunotherapy targets whilst minimizing toxicity [62]. To prove this conjecture, a randomized pilot trial in patients with newly diagnosed GBM was conducted to demonstrate that HCMV pp65 RNA-loaded DC vaccination experienced enhanced tumor-specific CD8+ T cell polyfunctionality and significantly increased overall survival [63]. The initial results obtained from clinical trials of autologous antigen or peptide-pulsed DC appear to be encouraging for a variety of tumors [64, 65]. The clinical trials testing DC vaccines after modified or adjuvant treatment with novel chemotherapy for gliomas defined as malignant or special subtype are underway at the NIH Clinical Center (Table 1). Here, we mainly review the feasibility and safety of autologous DC vaccines in phase I-III clinical trials and discuss recent progress in immunotherapy using autologous DC-based vaccination (Fig. 2).

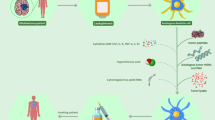
Autologous dendritic cell/tumor antigen vaccine (ADCTA) in anti-glioma clinical trial. Vaccination with tumor lysate-pulsed dendritic cells (DC) elicits antigen-specific, CD4+/CD8+ cytotoxic T cell responses and induces IFN γ secretion in patients with malignant glioma. The peptide modified DC with cocktail (WT-1, HER2, MAGE-A3, and MAGE-A1 or gp100) had a positively response in HLA-A24+ glioma patients
Fusions of autologous DC and glioma cells
DC can sample tumor antigens through capturing the ‘eat me’ or ‘do not eat me’ signal of tumor cells [66]. Molecules such as milk fat globule-EGF factor 8 (MFG-E8) bridge the phosphatidylserine of dying cells with integrin αvβ3 of DC, then link to downstream signals through integrin receptors on the phagocyte [67]. Tumor lysates include poorly identified high-grade glioma-specific tumor antigens, which have practicality in terms of personalized medicine [68]. Previous studies have indicated that vaccination with glioma cell lysate-pulsed DC elicited a stronger specific CTL response, thereby preventing glioma formation in C57BL/6 mice model [69]. More importantly, combinatorial treatment of tumor lysate-pulsed DC vaccines and other therapeutic strategies, such as checkpoint inhibitors [70], angiogenesis inhibitors [71] and cytokine gene therapy [72], conferred a greater survival advantage and significantly increased the therapeutic anti-glioma efficacy. In addition, counteracting the immunosuppressive environment before vaccination is requisite to facilitate the long-term anti-glioma immune responses [9]. Nowadays, using DC from the peripheral blood of patients pulsed with an autologous tumor lysate is currently being evaluated in phase I/II clinical trials for gliomas [73], but the objective response rate still has been relatively low in some research.
Vaccination with tumor lysate-pulsed DC elicits antigen-specific and CTL responses in patients with malignant glioma. The percentage of NK cells, such as CD16+ and CD56+ cells, in peripheral blood lymphocytes were safely increased after immunization [73, 74]. Accordingly, a significant positive correlation was observed between activated NK cell populations and overall survival in patients administrated with autologous tumor lysate-pulsed DC vaccination [75]. For newly diagnosed GBM, autologous vaccination with tumor lysate-pulsed DC enhanced the precursor frequency of CD4+ T and CD4+ interferon (IFN) γ-producing cells, suggesting an induced tumor-specific CTL response [76]. A recent phase I study was conducted to evaluate the safety and bioactivity of vaccination with tumor lysate-pulsed DCs in patients with GBM and anaplastic astrocytoma. In response to tumor lysate after vaccination, most of patients displayed robust systemic cytotoxicity as indicated by peripheral IFN γ accumulation and intratumoral CD8+ T cell infiltrate. Furthermore, no clinical evidence of autoimmune diseases was detected, suggesting that tumor lysate-pulsed DC vaccination was safe [77, 78]. By comparing specific immune responses before or after vaccination, up-regulation and/or cytoplasmic accumulation of chemoresistance-associated peptides, including Wilms tumor protein (WT1), glycoprotein 100 (gp100), and MAGE family member A3 (MAGEA3), were noted in tumors being treated with immunotherapy [79]. Yu et al. noted significant expansion in CD8+ antigen-specific T cell clones against TAAs, including melanoma-associated antigen (MAGE) 1, gp100, and HER2, and the CD8+ T cell intratumoral infiltrate was increased in 50% of patients [77]. Moreover, tumor lysate and IL-18 loaded DC vaccines can elicit a specific CD8+ cytotoxic T lymphocyte response in GBM patients. The cytotoxic responses were augmented by transfecting DC with the gene for IL-18, but significantly inhibited by anti-human leukocyte antigen (HLA) class I antibody [80].
In addition, several small size studies showed biased results with or without a pro-survival effect from tumor lysate-pulsed DC vaccination in glioma patients. Ryuya et al. showed the maturation of DC with OK-432, granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 that were pulsed with autologous tumor lysate showed better survival in 24 patients with recurrent GBMs. Also, patients injected with both intratumoral and intradermal administrations had longer survival times as compared with intradermal administration only [81]. A phase II trial demonstrated 53% of GBM patients (N = 32) exhibited over a 1.5-fold increase in vaccine-enhanced cytokine (IFN γ) and had significantly longer times to tumor progression and survival [82]. Furthermore, intradermal administration of fusion cells and subcutaneous injection of recombinant human interleukin 12 (rhIL-12) at the same site showed a greater than 50% reduction in tumor size in some patients without adverse effects and safely induced clinical anti-tumor effects with malignant gliomas [83]. Accounting for the narrow therapeutic index of rhIL-12 [84], the alteration of chemoresistance-associated peptides may achieve susceptibility of TAA-expressing glioma cells to the specific immune response in DC-based immunotherapy. However, vaccination with Audencel, a tumor lysate-charged autologous DC vaccine, failed to improve progression-free survival and median overall survival, although no severe toxicity was observed in those newly diagnosed with GBM (N = 76) [85]. The factors dictating the efficacy of DC vaccines may represent a viable strategy to improve anti-tumor immunotherapy. The enzyme-linked immune absorbent spot (ELISPOT) is one of the most commonly used to understand the frequency of cytokine-secreting cells at the single-cell level [86]. Increased ELISPOT and delayed-type hypersensitivity responses after vaccination could provide good laboratory markers to predict the clinical outcome of patients receiving DC vaccination [73, 81]. The content of tumor-infiltrating lymphocytes (TILs) and T cell receptor (TCR) repertoire in brain tumors and peripheral blood have been proved to be correlated with improved clinical outcome in GBM patients [87]. TCR repertoires are widely shared sequences that have been suggested to be over-represented due to their potential immune functionality or their ease of generation by V(D)J recombination [88]. After treatment, tracking TCR repertoire shifts in tumors and peripheral blood can be used to monitor the treatment-associated immune response without the need to know the specificity of receptors [89]. Hsu and colleague revealed a statistically significant correlation between higher degrees of TCR repertoires and prolonged overall survival in autologous tumor lysate-pulsed DC vaccines-treated GBM patients [87]. In a prospective case control study that enrolled 47 GBM patients with DC vaccine adjuvant therapy, better outcomes were predicted with younger age and a lower programmed cell death protein 1 (PD-1)+/CD8+ ratio in TILs and PBMCs [90]. Further analysis of the immune system factors demonstrated that the patients with an immune system equipped with favorable pre-existing or post-vaccination anti-tumor capabilities, such as IFN γ secretion and CD8+ cells, are more likely to live longer [91]. Also, another clinical trial protocol of DC vaccination, including inclusion/exclusion criteria, was proposed and may increase the immune response and safety in pediatric and adult subjects [92, 93]. Thus, even DC immunotherapy against GBM has some exciting outcomes, and the bias caused by sample size, mode of administration and non-specificity of vaccine-target interactions indicate that investigating combination therapies or developing meaningful biomarkers should be studied in further phase II/III clinical trials.
Standard therapy for GBMs post-surgery includes radiotherapy and chemotherapy with temozolomide. For DC vaccination as adjuvant therapy, up to now, most of the clinical trial showed no serious vaccine-related adverse events, and it may extend survival [76]. Early insight into tumor lysate-pulsed DC vaccines was provided in a phase III randomized, double-blinded, placebo-controlled clinical trial (N = 331) and suggested the addition of the vaccination to standard therapy is feasible and safe [94]. Only 2.1% of patients experienced any grade 3 or 4 adverse events that were at least possibly related to treatment with tumor lysate-pulsed DC vaccines. Autologous DC vaccines benefit patients with malignant glioma but may cause transient and reversible elevation of serum AST/ALT levels [95].
Peptide modified DC
DC are the most potent professional APCs capable of engulfing foreign antigens from invading pathogens. Through activation of pathogen recognition receptors (PRRs), mature and activated DC via complex downstream signaling pathways (such as cell surface co-stimulatory molecules) assemble the antigen peptide-major histocompatibility complex (MHC) and promote antigen-specific T cell expansion [96]. In early phase I trials, activated DC vaccines showed good safety, but had a weak anti-tumor response, which was not impressive compared with chemotherapeutic regimens. The α-type-1 polarized DC activated by maturation reagents (tumor necrosis factor α, IL 1β, IFN α, IFN γ and polyinosinic acid-polycytidylic acid) and pulsed with cocktail of 5 synthetic peptides (WT-1, HER2, MAGE-A3, and MAGE-A1 or gp100) were well tolerated, except for transient liver dysfunction. Long-term recurrence-free and positive immunological responses were only observed in 1.3% of HLA-A24+ glioma patients with stable disease (SD) [97]. The valuable target antigens of immunogenic synthetic peptides that are in existence in tumor lysate still seem to have an advantage to improve the vaccine-induced benefits and relapse-free period and optimal combinations need to be developed.
Vaccine generated by tumor cells
Vaccination using autologous tumor cells benefit existing humoral and cell-mediated immunity to antigenic epitopes, as well stimulates polyclonal immune attack against multiple, even undetected, TAAs. In separate reports, preclinical evidence shows that tumor cell immunotherapy can enhance anti-glioma immunity and can be effective in intracranial glioma models [98, 99] (Fig. 3). Autologous tumor-derived peptides from GBM can be used to safely immunize patients with recurrent GBM. After vaccination, brain biopsies revealed significant infiltration of focal CD4+, CD8+, CD56+ and IFN γ producing T cells against autologous tumor-derived peptides bound to HSP-96 [100]. Additionally, the NIH Clinical Center provided the promising attempt of administration of tumor lysate vaccine with a potent immune response modifier, such as imiquimod or poly-polyinosinic-polycytidilic acid (ICLC) adjuvant, to stimulate cell-mediated immune responses (Table 2). However, even though the feasibility and safety of vaccination were noted, the relatively weak anti-tumor activity of vaccination with irradiated autologous glioma has been demonstrated in recent clinical trials.

Vaccination using autologous tumor cells in anti-glioma clinical trial
Vaccines generated by TAA modified tumor cells with GM-CSF or TGF-β2 induce cellular and humoral anti-tumor immune responses by increasing CD4+ T cells with CTLA4, PD-1, 4-1BB, and OX40 expression and CD8+ T cells with PD-1 and 4-1BB expression. Using autologous glioma cells and IL-4 gene transfected fibroblasts increases IL-12 p70 level and infiltration of CD4+ and CD8+ T cells in glioma patients
TAA modified tumor vaccine
Following the observance of the effective anti-tumor ability of GM-CSF in vitro and in vivo, the US Food and Drug Administration has approved GM-CSF for use with dose-intensive chemotherapy. A strategy referred to as “GVAX”, composed of GM-CSF-engineered irradiated autologous tumor cells, has demonstrated an immunostimulatory effect related to GM-CSF in extensive preclinical results [101]. Using autologous tumor vaccine plus GM-CSF as an adjuvant, 89% of the vaccinated patients developed an autologous tumor-associated delayed type hypersensitivity (DTH) response; 42% of patients showed radiological evidence of a response, while only 26% patients showed clinical improvement after vaccination [102]. In a phase I study of recurrent malignant glioma (N = 10), irradiated autologous glioma cells were mixed with irradiated K562 cells, which over-expressed GM-CSF, to strengthen its DTH responses and humoral immunity. Vaccination revealed activation of T-lymphocytes, with increasing cytotoxic T-lymphocyte-associated protein 4 (CTLA4), PD-1, 4-1BB, and OX40 expressed CD4+ T cells, as well as PD-1 and 4-1BB expressed CD8+ T cells. No dose-limiting toxicities were seen when vaccinating patients with subcutaneous and intradermal injections of irradiated autologous glioma cells [103]. Transforming growth factor-beta 2 (TGF-β2) is a secreted protein known as an immunosuppressive molecule, whose dysregulated signaling contributes to the initiation and progression of many cancers, including glioma [104]. Preclinical study demonstrated that inhibition of TGF-β2 expression significantly enhances tumor-cell immunogenicity in eliminating previously implanted tumors [105]. Administration of TGF-β2 modified tumor cells showed recoverable and low-grade treatment-related toxicities and may be safe for glioma patients. In a phase I clinical trial, injection of TGF-β2 antisense-modified autologous tumor vaccine increased survival in some patients and induced generation of cellular and humoral anti-tumor immune responses in stage IV astrocytoma [106].
Vaccination-generated Abs may contribute to the in vivo immunogenicity of selected tumor antigens and may contribute to the therapeutic efficacy of cellular vaccines mainly designed to induce/up-regulate a tumor-specific CTL response. Recently, therapeutic vaccines based on genetically modified allogeneic tumor cells have been evaluated in clinical trial, regarding its advantage of acceptable construction costs, and the host of special antigen presenting. Clinical support for an allogeneic cellular vaccine approach has been validated in melanoma and has shown the extent of immunization against selected tumor antigens [107], but such is not the case in glioma. These findings support the clinical application of autologous tumor vaccines comprised of systemic immunosuppression antigens in glioma. Further randomized trials with allogeneic tumor cell immunotherapy may take a chance to establish efficacy as an alternative approach.
Fusion of autologous tumor and fibroblast cells
The risk of local recurrence occurs highly at and around the site of injury after surgical removal of the tumor [108]. Previous studies demonstrated fibroblasts may be applicable to cancer-targeting gene therapy for local control of the tumor around the injured tissue [109]. Using autologous glioma cells and IL-4 gene transfected fibroblasts, the feasibility and safety of adjuvant vaccinations were evaluated in patients with newly diagnosed and recurrent GBM, and it demonstrated encouraging immunological and clinical responses without allergic encephalitis [110]. In recurrent GBM, IL-4 dose-dependent infiltration of CD4+ and CD8+ T cells was observed at local vaccine sites. Vaccinations in HLA-A2+ patients demonstrated systemic T cell responses against an HLA-A2-restricted GAA epitope, EphA2883-89. In newly diagnosed GBM, monocyte-derived DC produce high levels of IL-12 p70 followed by two intradermal vaccinations with transfected fibroblasts admixed with DC loaded with autologous tumor lysate. However, detectable IFN γ post-vaccine responses or prolonged progression-free survival was not observed in these participants. To improve the intensity of the vaccine regimen, future studies will need to identify the potential vaccine intensification approach to enhance therapeutic efficacy without detrimental counterbalancing of autoimmunity.
Autologous natural killer cell therapy
NK cells are highly efficient in the cellular immune response against diseases, including malignancies. The past several years have seen tremendous advances in the selection and expansion of NK cells, and they have been used in clinical trials as adaptive immunotherapy for cancer [111]. Current NK cell-based cancer immunotherapy was designed to improve NK cell paralysis using several approaches, which are more practical for quality control and large-scale production by using stable autologous NK cell lines, for adoptive cellular immunotherapy [112]. In a phase I clinical trial, autologous NK cell therapy was demonstrated to be safe and partially effective in patients with recurrent GBM. The NK cell-rich effecter cells were manufactured from PBMCs through co-culturing with irradiated human feeder cell line (HFWT) and IFN β [113]. To a certain degree, autologous NK cell therapy could not yet exhibit their full cytotoxic capacity in vivo due to MHC class I expression in cancer patients that suppresses autologous NK cells [114]. A previous study demonstrated NK cell cytotoxicity is up-regulated by killer cell immunoglobulin-like receptors (KIRs), which interact with HLA class I ligands [115], while this finding was not tested in phase I clinical trial in human patients with glioma.
Discussion
Among all the therapies that have demonstrated significant safety and feasibility for gliomas in clinical trials, including radiation, chemotherapy (temozolomide and PCV [procarbazine, lomustine, vincristine]) and targeted therapies (bevacizumab) [116], the impact of cellular vaccine therapies has been most modest in glioma. For unequivocal clinical benefit, improvements should be achieved to maximize the vaccine-induced T cells with optimal amplitude, specificity and effector profile [117]. Combination with immune response modifiers in glioma promises to boost the true power of cellular vaccines and potentially offer long-term protection from tumor recurrence. The cytokine IL-12 is a potent inducer of anti-tumor activity in a variety of preclinical models [118]. By inflammation regulation, IL-12 has been proven to establish a link between innate and adaptive immunity that involves different immune effector cells and cytokines [119]. The inducible expressed IL-12-armored CART cells have broadened the application of CART-based immunotherapy and might provide an alternative therapeutic strategy for cancer treatment [120]. For recurrent glioma, results of a phase I trial have revealed IL-12 increased IFN γ and PD-1 with acceptable tolerability in TILs [121]. Additionally, the U.S. Food and Drug Administration (FDA) has approved synthetic immunomodulatory agents, such as resiquimod and imiquimod, that act as vaccine adjuvants, enhancing cytokine production and skewing immunity towards a Th1/cytotoxic response [122, 123]. Targeting tumor-specific T cells, therapeutic interceptions that inhibit receptors, including PD1 (pembrolizumab, nivolumab), CTLA4 (ipilimumab, tremelimumab) and LAG3 (BMS-986,016), have been approved or are in clinical trials for the treatment of various cancer types [124]. In practice, however, the challenges the BBB poses for glioma therapy and how these immune response modifiers activate the local brain tumor immune system and enhance cytolytic effects should be further discussed in future clinical models.
So far, there is solid evidence that intrinsic factors (sex, age and comorbidities) and vaccine-related factors (adjuvants and vaccination schedule), as well as the immune system (such as innate and adaptive responses), strongly influence vaccine efficacy [125]. Recent progress in the molecular subtypes of histologic-based glioma classification suggests some potential reasons for the indistinctive effort of the cellular vaccine. For example, given the spectrum of aggressive phenotypes, tumor marker-based classification, such as IDH mutation, 1p/19q co-deletion, and TERT promoter mutations, predict favorable prognosis in gliomas [126,127,128]. In IDH-wild type GBM, a gene-based signature could be a potential prognostic biomarker [129]. Thus, based on molecular markers, educated and advisable vaccine use for a cellular vaccine may be critical for safety and promotable anti-tumor effects in glioma treatment. Typical GBM alterations, such as IDH mutation, NF1 inactivation, and CDK4-MARCH9 locus amplification, characterize tumor-associated immunosuppression [130]. IDH1 mutations caused down-regulation of leukocyte chemotaxis, resulting in reduced immune infiltrates that may contribute, in part, to differences in the aggressiveness of mutant type gliomas [131]. The immune features of B7-H3, an immune checkpoint member found to positively correlate with the grade of malignancy, may become an attractive target for IDH-wild type glioma immunotherapy [132]. These findings encourage researchers to further confirm the tumor response of cellular vaccines based on specific molecular subtypes in the ongoing larger randomized trials, such as IDH1 R132H-DC vaccine (NCT02771301), which may provide the hope to optimize cellular vaccines in gliomas.
To a large extent, cancers are particularly evolutionary events owing to their genetic heterogeneity [133, 134]. An understanding of the heterogeneous characteristics of cancer clones allows us to well address the individual tumor behavior and therapeutic response [135]. Indeed, such a framework could also be applied to explore the patient-specific neoantigens in the course of tumor evolution for successful vaccine immunotherapy. The availability of neoantigen-based personalized vaccines further provides a powerful aid to elucidate inter-individual variability and intra-tumor heterogeneity [136]. In patients with gliomas, intratumour heterogeneity of IDH1 mutations can be considered as a favorable independent prognostic biomarker. Schumacher and colleague demonstrated the promising function of IDH1 mutation–specific vaccination for glioma treatment [137]. In this report, the neopeptides carrying IDH1 R132H p123-142 mutated region were produced to interact with transgenic human MHC-II molecules in glioma mouse model. Peptide vaccination resulted in efficient mutation-specific antitumor immunity in the mouse model with IDH1 R132H-mutated gliomas. The synthetic neopeptides encompassing histone 3 variant H3.3K27M mutation could be recognized by TCR. After then, TCR-transduced T cells specifically lysed the H3.3K27M+ glioma cells [138] and prolonged overall survival in patients with diffuse midline gliomas [139]. In addition, Duperret et al. presented a pre-clinical study about utilizing nucleic acid vaccine platform to target tumor multi-neoantigens [140]. These nucleic acid vaccines induced predominantly MHC I-restricted, CD8+ T cell responses and subsequently killed tumor cells. Recently, by used personalized neoantigen-targeting vaccines to immunize glioma patients, Keskin et al. found that neoantigen-based vaccines had the potential to evoke the neoantigen-specific CD4+ and CD8+ T cell responses [141]. Therefore, considering the biological importance of neoantigens in cancer immunotherapies [142], it is reasonable to suppose that the neoantigen-based therapeutic strategies might have a viable future in anti-glioma immune responses.
Up to now, the clinical relevance of immune regulation in glioma research and treatment remains debated. However, emerging studies about the immunological processes participated in glioma tumorigenesis have yielded a basis for clinical translation of glioma-associated vaccination strategies. Even though these immunotherapy strategies have yielded increasingly good outcomes for glioma patients, challenges with vaccine-based immunotherapy still inevitably remain. The first conceptual challenge is the choice of individualized neoantigen to be targeted and the accurate status assessment of such targets [143]. As more and more tumor neoantigens have been identified, the future fields of investigation should focus on the effective neoepitope dosage without tolerization and finding the best neoepitope delivery system [144]. In BALB/c mice, using a potent immunostimulatory adjuvant and delivery system for neoepitope immunization might be of great benefit to induce the CD8 + T cells-mediated tumor rejection response [145]. The immunosuppression and T-cell exhaustion in tumor microenvironment pose additional engineering challenges. Thus, effective treatment of solid tumors with vaccines needs to generate the activated CAR T cells that can function in the immunosuppressive tumor microenvironment [146]. Another apparent limitation is that if used for cancer prevention, vaccines must elicit effective long-term memory in order to avoid causing autoimmunity [147]. In addition, immunotherapy should be carefully considered to be integrated into the existing standard therapies. The immunotherapeutic agents, including vaccine-based therapies, limited its therapeutic potential when used alone [148]. Oppositely, the combination of vaccine therapy with other conventional methods provides obviously synergistic effects on the eradication of cancer cells [149]. Overall, with continued research addressed these limitation, such as clarifying immune-cancer interactions and discovering innovative vaccine targets, we can design novel strategies and technologies to better optimize vaccine-mediated immunity to further improve the outcomes for glioma patients.
Conclusions
To date, it is well-known that cellular vaccines can be considered as a promising therapeutic strategy for glioma patients. Increasing studies have demonstrated that therapeutic vaccines based on T cells, DC cells, tumor cells and NK cells are feasible and generally well tolerated. More importantly, clarifying the functional roles of cellular vaccines in glioma immunotherapy may bear potential implications for apparent therapeutic advantages and clinical benefits. Even so, in the future, forthcoming investigations are needed to comprehensively explore their unique characteristics involving in anti-glioma immune responses. The in-depth understanding of cellular vaccines will contribute to uncover the detailed mechanisms and biological functions in glioma research and treatment. Furthermore, in view of the non-negligible roles of cellular vaccines, more preclinical and clinical trials can be conducted to focus on their combination with current treatment regimens.
Availability of data and materials
Not applicable.
Abbreviations
- ADCTA:
-
Autologous dendritic cells/tumor antigen vaccine
- CNS:
-
Central nervous system
- WHO:
-
World Health Organization
- GBM:
-
Glioblastoma
- BBB:
-
blood-brain barrier
- MGMT:
-
O6-Methylguanine-DNA methyltransferase
- Tregs:
-
T regulatory cells
- Th1:
-
T helper 1
- GAAs:
-
Glioma-associated antigens
- TAA:
-
Tumor-associated antigen
- CARs:
-
Chimeric antigen receptors
- NK:
-
Natural killer
- CAR T cell:
-
CAR-engineered T cell
- IL13Rα2:
-
Interleukin 13 receptor α2
- EGFRvIII:
-
Epidermal growth factor receptor variant III
- EphA2:
-
Ephrin type-A receptor 2
- HER2:
-
Human epidermal growth factor receptor 2
- CTL:
-
Cytotoxic T lymphocytes
- PBMCs:
-
Peripheral blood mononuclear cells
- CXCL9:
-
C-X-C motif chemokine ligand 9
- APCs:
-
Antigen-presenting cells
- MFG-E8:
-
Milk fat globule-EGF factor 8
- WT1:
-
Wilms tumor protein
- gp100:
-
Glycoprotein 100
- MAGE:
-
Melanoma-associated antigen
- MAGEA3:
-
MAGE family member A3
- HLA:
-
Human leukocyte antigen
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- KIRs:
-
Killer cell immunoglobulin-like receptors
- PD-1:
-
Programmed cell death protein 1
- TILs:
-
Tumor-infiltrating lymphocytes
- TCR:
-
T cell receptor
- HCMV:
-
Human cytomegalovirus
References
Adamson DC, Rasheed BA, McLendon RE, Bigner DD. Central nervous system. Cancer biomarkers: section A of Disease markers. 2010;9(1–6):193–210. .
Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018;392(10145):432–46.
Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nature immunology. 2019;20(9):1100–9.
Chen X, Yan Y, Zhou J, Huo L, Qian L, Zeng S, et al. Clinical prognostic value of isocitrate dehydrogenase mutation, O-6-methylguanine-DNA methyltransferase promoter methylation, and 1p19q co-deletion in glioma patients. Annals of translational medicine. 2019;7(20):541.
Dai T, Jiang K, Lu W. Liposomes and lipid disks traverse the BBB and BBTB as intact forms as revealed by two-step Forster resonance energy transfer imaging. Acta pharmaceutica Sinica B. 2018;8(2):261–71.
Akinleye A, Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J Hematol Oncol. 2019;12(1):92.
Wei J, Long Y, Guo R, Liu X, Tang X, Rao J, et al. Multifunctional polymeric micelle-based chemo-immunotherapy with immune checkpoint blockade for efficient treatment of orthotopic and metastatic breast cancer. Acta pharmaceutica Sinica B. 2019;9(4):819–31.
Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Investig. 2015;125(9):3335–7.
Grauer OM, Sutmuller RP, van Maren W, Jacobs JF, Bennink E, Toonen LW, et al. Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. International journal of cancer. 2008;122(8):1794–802.
Berger TG, Schultz ES. Dendritic cell-based immunotherapy. Curr Top Microbiol Immunol. 2003;276:163–97.
Hlavackova E, Pilatova K, Cerna D, Selingerova I, Mudry P, Mazanek P, et al. Dendritic Cell-Based Immunotherapy in Advanced Sarcoma and Neuroblastoma Pediatric Patients: Anti-cancer Treatment Preceding Monocyte Harvest Impairs the Immunostimulatory and Antigen-Presenting Behavior of DCs and Manufacturing Process Outcome. Frontiers in oncology. 2019;9:1034.
Fedorova L, Mudry P, Pilatova K, Selingerova I, Merhautova J, Rehak Z, et al. Assessment of Immune Response Following Dendritic Cell-Based Immunotherapy in Pediatric Patients With Relapsing Sarcoma. Frontiers in oncology. 2019;9:1169.
Adkins I, Fucikova J, Garg AD, Agostinis P, Spisek R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology. 2014;3(12):e968434.
Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW, et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Science translational medicine. 2016;8(328):328ra27.
Sondak VK, Sabel MS, Mule JJ. Allogeneic and autologous melanoma vaccines: where have we been and where are we going? Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12(7 Pt 2):2337s-41 s.
Smith C, Lineburg KE, Martins JP, Ambalathingal GR, Neller MA, Morrison B, et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. The Journal of clinical investigation. 2020.
Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8 + T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29(3):330–6.
Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuno MA, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer immunology immunotherapy: CII. 2013;62(1):125–35.
Srivastava S, Riddell SR. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015;36(8):494–502.
Wu C, Zhang L, Brockman QR, Zhan F, Chen L. Chimeric antigen receptor T cell therapies for multiple myeloma. J Hematol Oncol. 2019;12(1):120.
Bagley SJ, Desai AS, Linette GP, June CH, O’Rourke DM. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neurooncology. 2018;20(11):1429–38.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96.
Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nature reviews Neuroscience. 2006;7(1):41–53.
Braun DJ, Bachstetter AD, Sudduth TL, Wilcock DM, Watterson DM, Van Eldik LJ. Genetic knockout of myosin light chain kinase (MLCK210) prevents cerebral microhemorrhages and attenuates neuroinflammation in a mouse model of vascular cognitive impairment and dementia. GeroScience. 2019;41(5):671–9.
Greenhalgh AD, David S, Bennett FC. Immune cell regulation of glia during CNS injury and disease. Nature reviews Neuroscience. 2020;21(3):139–52.
Choi BD, Maus MV, June CH, Sampson JH. Immunotherapy for Glioblastoma: Adoptive T-cell Strategies. Clinical cancer research: an official journal of the American Association for Cancer Research. 2019;25(7):2042–8.
Ilyas S, Yang JC. Landscape of Tumor Antigens in T Cell Immunotherapy. Journal of immunology. 2015;195(11):5117–22.
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016;375(26):2561–9.
Han X, Wang Y, Wei J, Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol. 2019;12(1):128.
Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8 + T Cells in Patients with Recurrent Glioblastoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21(18):4062–72.
Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Science translational medicine. 2015;7(275):275ra22.
Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Molecular therapy: the journal of the American Society of Gene Therapy. 2013;21(3):629–37.
Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16(2):474–85.
Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Human gene therapy. 2012;23(10):1043–53.
Badhiwala J, Decker WK, Berens ME, Bhardwaj RD. Clinical trials in cellular immunotherapy for brain/CNS tumors. Expert Rev Neurother. 2013;13(4):405–24.
Joshi BH, Plautz GE, Puri RK. Interleukin-13 receptor alpha chain: a novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer research. 2000;60(5):1168–72.
Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, et al. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(8):2199–209.
Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC, et al. Glioma IL13Ralpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PloS one. 2013;8(10):e77769.
Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Molecular therapy: the journal of the American Society of Gene Therapy. 2015;23(4):757–68.
Yang J, Yan J, Liu B. Targeting EGFRvIII for glioblastoma multiforme. Cancer letters. 2017;403:224–30.
An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. 2018;37(12):1561–75.
Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neurooncology. 2015;17(6):854–61.
Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28(31):4722–9.
O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Science translational medicine. 2017;9(399):eaaa0984.
Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer research. 2004;64(14):4980–6.
Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P, et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. Journal of the National Cancer Institute. 2016;108:5. .
Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Investig. 2016;126(8):3036–52.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Molecular therapy: the journal of the American Society of Gene Therapy. 2010;18(4):843–51.
Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015;33(15):1688–96.
Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA oncology. 2017;3(8):1094–101.
Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunological reviews. 2002;188:22–32.
Zhang W, Barger CJ, Link PA, Mhawech-Fauceglia P, Miller A, Akers SN, et al. DNA hypomethylation-mediated activation of Cancer/Testis Antigen 45 (CT45) genes is associated with disease progression and reduced survival in epithelial ovarian cancer. Epigenetics. 2015;10(8):736–48.
Rozman P. The potential of non-myeloablative heterochronous autologous hematopoietic stem cell transplantation for extending a healthy life span. GeroScience. 2018;40(3):221–42.
Kirkin AF, Dzhandzhugazyan KN, Guldberg P, Fang JJ, Andersen RS, Dahl C, et al. Adoptive cancer immunotherapy using DNA-demethylated T helper cells as antigen-presenting cells. Nature communications. 2018;9(1):785.
Dubovsky JA, McNeel DG, Powers JJ, Gordon J, Sotomayor EM, Pinilla-Ibarz JA. Treatment of chronic lymphocytic leukemia with a hypomethylating agent induces expression of NXF2, an immunogenic cancer testis antigen. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15(10):3406–15.
Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q, et al. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta pharmaceutica Sinica B. 2020;10(5):723–33.
Jung H, Kim HS, Kim JY, Sun JM, Ahn JS, Ahn MJ, et al. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nature communications. 2019;10(1):4278.
Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. The Lancet Oncology. 2014;15(7):e257-67.
Garcia-Cabezas MA, John YJ, Barbas H, Zikopoulos B. Distinction of Neurons, Glia and Endothelial Cells in the Cerebral Cortex: An Algorithm Based on Cytological Features. Front Neuroanat. 2016;10:107.
Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer cell. 2017;31(3):326–41.
Colleton BA, Huang XL, Melhem NM, Fan Z, Borowski L, Rappocciolo G, et al. Primary human immunodeficiency virus type 1-specific CD8 + T-cell responses induced by myeloid dendritic cells. Journal of virology. 2009;83(12):6288–99.
Wen L, Zhao F, Qiu Y, Cheng S, Sun JY, Fang W, et al. Human cytomegalovirus DNA and immediate early protein 1/2 are highly associated with glioma and prognosis. Protein cell. 2020;11(7):525–33.
Reap EA, Suryadevara CM, Batich KA, Sanchez-Perez L, Archer GE, Schmittling RJ, et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer research. 2018;78(1):256–64.
Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nature medicine. 1996;2(1):52–8.
Carmi Y, Spitzer MH, Linde IL, Burt BM, Prestwood TR, Perlman N, et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature. 2015;521(7550):99–104.
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nature reviews Cancer. 2012;12(4):265–77.
Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. 2004;104(10):3257–66.
Berzofsky JA, Terabe M, Trepel JB, Pastan I, Stroncek DF, Morris JC, et al. Cancer vaccine strategies: translation from mice to human clinical trials. Cancer immunology immunotherapy: CII. 2018;67(12):1863–9.
Xu M, Yao Y, Hua W, Wu Z, Zhong P, Mao Y, et al. Mouse glioma immunotherapy mediated by A2B5 + GL261 cell lysate-pulsed dendritic cells. Journal of neuro-oncology. 2014;116(3):497–504.
Zhu S, Lv X, Zhang X, Li T, Zang G, Yang N, et al. An effective dendritic cell-based vaccine containing glioma stem-like cell lysate and CpG adjuvant for an orthotopic mouse model of glioma. International journal of cancer. 2019;144(11):2867–79.
Jiang XB, Lu XL, Hu P, Liu RE. Improved therapeutic efficacy using vaccination with glioma lysate-pulsed dendritic cells combined with IP-10 in murine glioma. Vaccine. 2009;27(44):6210–6.
Saito R, Mizuno M, Nakahara N, Tsuno T, Kumabe T, Yoshimoto T, et al. Vaccination with tumor cell lysate-pulsed dendritic cells augments the effect of IFN-beta gene therapy for malignant glioma in an experimental mouse intracranial glioma. International journal of cancer. 2004;111(5):777–82.
Yamanaka R, Abe T, Yajima N, Tsuchiya N, Homma J, Kobayashi T, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. British journal of cancer. 2003;89(7):1172–9.
Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, Ohno T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer immunology immunotherapy: CII. 2001;50(7):337–44.
Prins RM, Wang X, Soto H, Young E, Lisiero DN, Fong B, et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. Journal of immunotherapy. 2013;36(2):152–7.
Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, Gui J, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. Journal of immunotherapy. 2011;34(4):382–9.
Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer research. 2004;64(14):4973–9.
Gonzalo S, Coll-Bonfill N. Genomic instability and innate immune responses to self-DNA in progeria. GeroScience. 2019;41(3):255–66.
Akasaki Y, Kikuchi T, Homma S, Koido S, Ohkusa T, Tasaki T, et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer immunology immunotherapy: CII. 2016;65(12):1499–509.
Yamanaka R, Honma J, Tsuchiya N, Yajima N, Kobayashi T, Tanaka R. Tumor lysate and IL-18 loaded dendritic cells elicits Th1 response, tumor-specific CD8 + cytotoxic T cells in patients with malignant glioma. Journal of neuro-oncology. 2005;72(2):107–13.
Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, Kobayashi T, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11(11):4160–7.
Wheeler CJ, Black KL, Liu G, Mazer M, Zhang XX, Pepkowitz S, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer research. 2008;68(14):5955–64.
Kikuchi T, Akasaki Y, Abe T, Fukuda T, Saotome H, Ryan JL, et al. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. Journal of immunotherapy. 2004;27(6):452–9.
Lasek W, Zagozdzon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer immunology, immunotherapy. CII. 2014;63(5):419–35.
Buchroithner J, Erhart F, Pichler J, Widhalm G, Preusser M, Stockhammer G, et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers. 2018;10(10):372.
Slota M, Lim JB, Dang Y, Disis ML. ELISpot for measuring human immune responses to vaccines. Expert Rev Vaccines. 2011;10(3):299–306.
Hsu M, Sedighim S, Wang T, Antonios JP, Everson RG, Tucker AM, et al. TCR Sequencing Can Identify and Track Glioma-Infiltrating T Cells after DC Vaccination. Cancer immunology research. 2016;4(5):412–8.
Elhanati Y, Sethna Z, Callan CG Jr, Mora T, Walczak AM. Predicting the spectrum of TCR repertoire sharing with a data-driven model of recombination. Immunological reviews. 2018;284(1):167–79.
Emerson RO, Sherwood AM, Rieder MJ, Guenthoer J, Williamson DW, Carlson CS, et al. High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J Pathol. 2013;231(4):433–40.
Jan CI, Tsai WC, Harn HJ, Shyu WC, Liu MC, Lu HM, et al. Predictors of Response to Autologous Dendritic Cell Therapy in Glioblastoma Multiforme. Frontiers in immunology. 2018;9:727.
Erhart F, Buchroithner J, Reitermaier R, Fischhuber K, Klingenbrunner S, Sloma I, et al. Immunological analysis of phase II glioblastoma dendritic cell vaccine (Audencel) trial: immune system characteristics influence outcome and Audencel up-regulates Th1-related immunovariables. Acta neuropathologica communications. 2018;6(1):135.
Shah AH, Bregy A, Heros DO, Komotar RJ, Goldberg J. Dendritic cell vaccine for recurrent high-grade gliomas in pediatric and adult subjects: clinical trial protocol. Neurosurgery. 2013;73(5):863–7.
Okada H, Pollack IF, Lieberman F, Lunsford LD, Kondziolka D, Schiff D, et al. Gene therapy of malignant gliomas: a pilot study of vaccination with irradiated autologous glioma and dendritic cells admixed with IL-4 transduced fibroblasts to elicit an immune response. Human gene therapy. 2001;12(5):575–95.
Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. Journal of translational medicine. 2018;16(1):142.
Chang CN, Huang YC, Yang DM, Kikuta K, Wei KJ, Kubota T, et al. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. Journal of clinical neuroscience: official journal of the Neurosurgical Society of Australasia. 2011;18(8):1048–54.
Wu L, Zhang H, Jiang Y, Gallo RC, Cheng H. Induction of antitumor cytotoxic lymphocytes using engineered human primary blood dendritic cells. Proc Natl Acad Sci USA. 2018;115(19):E4453-E62.
Akiyama Y, Oshita C, Kume A, Iizuka A, Miyata H, Komiyama M, et al. alpha-type-1 polarized dendritic cell-based vaccination in recurrent high-grade glioma: a phase I clinical trial. BMC Cancer. 2012;12:623.
Jahan N, Talat H, Alonso A, Saha D, Curry WT. Triple combination immunotherapy with GVAX, anti-PD-1 monoclonal antibody, and agonist anti-OX40 monoclonal antibody is highly effective against murine intracranial glioma. Oncoimmunology. 2019;8(5):e1577108.
Agarwalla P, Barnard Z, Fecci P, Dranoff G, Curry WT. Jr. Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. Journal of immunotherapy. 2012;35(5):385–9.
Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(1):205–14.
Nemunaitis J. Vaccines in cancer: GVAX, a GM-CSF gene vaccine. Expert Rev Vaccines. 2005;4(3):259–74.
Sloan AE, Dansey R, Zamorano L, Barger G, Hamm C, Diaz F, et al. Adoptive immunotherapy in patients with recurrent malignant glioma: preliminary results of using autologous whole-tumor vaccine plus granulocyte-macrophage colony-stimulating factor and adoptive transfer of anti-CD3-activated lymphocytes. NeuroSurg Focus. 2000;9(6):e9.
Curry WT Jr, Gorrepati R, Piesche M, Sasada T, Agarwalla P, Jones PS, et al. Vaccination with Irradiated Autologous Tumor Cells Mixed with Irradiated GM-K562 Cells Stimulates Antitumor Immunity and T Lymphocyte Activation in Patients with Recurrent Malignant Glioma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22(12):2885–96.
Han J, Alvarez-Breckenridge CA, Wang QE, Yu J. TGF-beta signaling and its targeting for glioma treatment. American journal of cancer research. 2015;5(3):945–55.
Fakhrai H, Dorigo O, Shawler DL, Lin H, Mercola D, Black KL, et al. Eradication of established intracranial rat gliomas by transforming growth factor beta antisense gene therapy. Proc Natl Acad Sci USA. 1996;93(7):2909–14.
Fakhrai H, Mantil JC, Liu L, Nicholson GL, Murphy-Satter CS, Ruppert J, et al. Phase I clinical trial of a TGF-beta antisense-modified tumor cell vaccine in patients with advanced glioma. Cancer Gene Ther. 2006;13(12):1052–60.
Maio M, Fonsatti E, Lamaj E, Altomonte M, Cattarossi I, Santantonio C, et al. Vaccination of stage IV patients with allogeneic IL-4- or IL-2-gene-transduced melanoma cells generates functional antibodies against vaccinating and autologous melanoma cells. Cancer immunology immunotherapy: CII. 2002;51(1):9–14.
Enneking WF, Maale GE. The effect of inadvertent tumor contamination of wounds during the surgical resection of musculoskeletal neoplasms. Cancer. 1988;62(7):1251–6.
Yang W, Arii S, Mori A, Furumoto K, Nakao T, Isobe N, et al. sFlt-1 gene-transfected fibroblasts: a wound-specific gene therapy inhibits local cancer recurrence. Cancer research. 2001;61(21):7840–5.
Okada H, Lieberman FS, Walter KA, Lunsford LD, Kondziolka DS, Bejjani GK, et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. Journal of translational medicine. 2007;5:67.
Becker PS, Suck G, Nowakowska P, Ullrich E, Seifried E, Bader P, et al. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer immunology immunotherapy: CII. 2016;65(4):477–84.
Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol. 2013;10(3):230–52.
Ishikawa E, Tsuboi K, Saijo K, Harada H, Takano S, Nose T, et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer research. 2004;24(3b):1861–71.
Saetersmoen ML, Hammer Q, Valamehr B, Kaufman DS, Malmberg KJ. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin Immunopathol. 2019;41(1):59–68.
Cornillet M, Jansson H, Schaffer M, Hertwig L, Berglin L, Zimmer CL, et al. Imbalance of Genes Encoding Natural Killer Immunoglobulin-Like Receptors and Human Leukocyte Antigen in Patients With Biliary Cancer. Gastroenterology. 2019;157(4):1067–80 e9.
Touat M, Idbaih A, Sanson M, Ligon KL. Glioblastoma targeted therapy: updated approaches from recent biological insights. Annals of oncology: official journal of the European Society for Medical Oncology. 2017;28(7):1457–72.
Vermaelen K. Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Frontiers in immunology. 2019;10:8.
Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nature medicine. 2015;21(7):719–29.
Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, et al. New insights into IL-12-mediated tumor suppression. Cell death differentiation. 2015;22(2):237–46.
Liu Y, Di S, Shi B, Zhang H, Wang Y, Wu X, et al. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. Journal of immunology. 2019;203(1):198–207.
Chiocca EA, Yu JS, Lukas RV, Solomon IH, Ligon KL, Nakashima H, et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Science translational medicine. 2019;11(505):eaaw5680.
Rozenblit M, Hendrickx W, Heguy A, Chiriboga L, Loomis C, Ray K, et al. Transcriptomic profiles conducive to immune-mediated tumor rejection in human breast cancer skin metastases treated with Imiquimod. Scientific reports. 2019;9(1):8572.
Jansen MHE, Mosterd K, Arits A, Roozeboom MH, Sommer A, Essers BAB, et al. Five-Year Results of a Randomized Controlled Trial Comparing Effectiveness of Photodynamic Therapy, Topical Imiquimod, and Topical 5-Fluorouracil in Patients with Superficial Basal Cell Carcinoma. J Invest Dermatol. 2018;138(3):527–33.
Turnis ME, Andrews LP, Vignali DA. Inhibitory receptors as targets for cancer immunotherapy. Eur J Immunol. 2015;45(7):1892–905.
Zimmermann P, Curtis N. Factors That Influence the Immune Response to Vaccination. Clinical microbiology reviews. 2019;32:2. .
Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med. 2015;372(26):2499–508.
Cheng F, Guo D. MET in glioma: signaling pathways and targeted therapies. Journal of experimental clinical cancer research: CR. 2019;38(1):270.
Chen X, Hu L, Yang H, Ma H, Ye K, Zhao C, et al. DHHC protein family targets different subsets of glioma stem cells in specific niches. Journal of experimental clinical cancer research: CR. 2019;38(1):25.
Liu YQ, Wu F, Li JJ, Li YF, Liu X, Wang Z, et al. Gene Expression Profiling Stratifies IDH-Wildtype Glioblastoma With Distinct Prognoses. Frontiers in oncology. 2019;9:1433.
Luoto S, Hermelo I, Vuorinen EM, Hannus P, Kesseli J, Nykter M, et al. Computational Characterization of Suppressive Immune Microenvironments in Glioblastoma. Cancer research. 2018;78(19):5574–85.
Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017;31(8):774–86.
Zhang C, Zhang Z, Li F, Shen Z, Qiao Y, Li L, et al. Large-scale analysis reveals the specific clinical and immune features of B7-H3 in glioma. Oncoimmunology. 2018;7(11):e1461304.
Lim ZF, Ma PC. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J Hematol Oncol. 2019;12(1):134.
Wong K, Young GS, Makale M, Hu X, Yildirim N, Cui K, et al. Characterization of a human tumorsphere glioma orthotopic model using magnetic resonance imaging. Journal of neuro-oncology. 2011;104(2):473–81.
Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nature reviews Genetics. 2019;20(7):404–16.
Tureci O, Vormehr M, Diken M, Kreiter S, Huber C, Sahin U. Targeting the Heterogeneity of Cancer with Individualized Neoepitope Vaccines. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22(8):1885–96.
Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512(7514):324–7.
Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. The Journal of experimental medicine. 2018;215(1):141–57.
Mueller S, Taitt JM, Villanueva-Meyer JE, Bonner ER, Nejo T, Lulla RR, et al. Mass cytometry detects H3.3K27M-specific vaccine responses in diffuse midline glioma. The Journal of clinical investigation. 2020.
Duperret EK, Perales-Puchalt A, Stoltz R, Mandloi GHH, Barlow N. J, et al. A Synthetic DNA, Multi-Neoantigen Vaccine Drives Predominately MHC Class I CD8(+) T-cell Responses, Impacting Tumor Challenge. Cancer immunology research. 2019;7(2):174–82.
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–9.
Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei J, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. 2019;12(1):93.
Weller M, Roth P, Preusser M, Wick W, Reardon DA, Platten M, et al. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nature reviews Neurology. 2017;13(6):363–74.
Zhao Z, Zheng L, Chen W, Weng W, Song J, Ji J. Delivery strategies of cancer immunotherapy: recent advances and future perspectives. J Hematol Oncol. 2019;12(1):126.
Brennick CA, George MM, Corwin WL, Srivastava PK, Ebrahimi-Nik H. Neoepitopes as cancer immunotherapy targets: key challenges and opportunities. Immunotherapy. 2017;9(4):361–71.
Lohmueller J, Finn OJ. Current modalities in cancer immunotherapy: Immunomodulatory antibodies, CARs and vaccines. Pharmacol Ther. 2017;178:31–47.
Finn OJ. Cancer vaccines: between the idea and the reality. Nature reviews Immunology. 2003;3(8):630–41.
Osipov A, Murphy A, Zheng L. From immune checkpoints to vaccines: The past, present and future of cancer immunotherapy. Adv Cancer Res. 2019;143:63–144.
Chu Y, Liu Q, Wei J, Liu B. Personalized cancer neoantigen vaccines come of age. Theranostics. 2018;8(15):4238–46.
Acknowledgements
Not Applicable.
Funding
This work was supported by the Natural Science Foundation of Hunan Province, China (2019JJ50932, 2020JJ5934), the National Natural Science Foundation of China (81803035, 81703036), the China Postdoctoral Science Foundation (2020M672521, 2017M610510) and Dr. Yuanliang Yan is right now a Postdoctoral Fellow at the Center for Molecular Medicine, Xiangya Hospital, Central South University (248485).
Author information
Authors and Affiliations
Contributions
YL Yan wrote this review article. SS Zeng, ZJ Xu and ZC Gong performed technical and administrative support. ZJ Xu designed the study and contributed to manuscript preparation. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors give consent for the publication of manuscript in Journal of Experimental & Clinical Cancer Research.
Competing interests
We declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yan, Y., Zeng, S., Gong, Z. et al. Clinical implication of cellular vaccine in glioma: current advances and future prospects. J Exp Clin Cancer Res 39, 257 (2020). https://doi.org/10.1186/s13046-020-01778-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-020-01778-6