
Abstract
NEDDylation, a post-translational modification through three-step enzymatic cascades, plays crucial roles in the regulation of diverse biological processes. NEDD8-activating enzyme (NAE) as the only activation enzyme in the NEDDylation modification has become an attractive target to develop anticancer drugs. To date, numerous inhibitors or agonists targeting NAE have been developed. Among them, covalent NAE inhibitors such as MLN4924 and TAS4464 currently entered into clinical trials for cancer therapy, particularly for hematological tumors. This review explains the relationships between NEDDylation and cancers, structural characteristics of NAE and multistep mechanisms of NEDD8 activation by NAE. In addition, the potential approaches to discover NAE inhibitors and detailed pharmacological mechanisms of NAE inhibitors in the clinical stage are explored in depth. Importantly, we reasonably investigate the challenges of NAE inhibitors for cancer therapy and possible development directions of NAE-targeting drugs in the future.
Similar content being viewed by others

Background
Neuronal precursor cell-expressed developmentally down-regulated protein 8 (NEDD8) shares about 60% of the amino acids with ubiquitin, which covalently binds to substrate proteins by generating an isopeptide chain between the lysine residue of substrates and the glycine residue of NEDD8 [1,2,3,4,5]. NEDDylation is a biochemical process of post-translational modification that conjugates NEDD8 to substrate proteins through the successive enzymatic cascades [6,7,8,9,10]. In the initial stage of NEDDylation, the precursor of NEDD8 is hydrolyzed to mature NEDD8 by the precursor processing enzymes [11,12,13,14]. Next, the mature NEDD8 is activated in the presence of adenosine triphosphate (ATP) by the E1 NEDD8-activating enzyme (NAE) consisting of amyloid protein-binding protein 1 (APPBP1) and ubiquitin-like modifier activating enzyme 3 (UBA3) [15,16,17]. Then, the activated NEDD8 is transferred to NEDD8-conjugating enzyme E2s (UBC12 and UBE2F) via a trans-thiolation process [18,19,20,21,22,23]. Finally, NEDD8 is transferred from NEDD8-conjugating enzyme E2s to a specific lysine residue of substrates through the catalytic action of NEDD8 E3 ligases [24,25,26]. In recent years, homogeneous time-resolved fluorescence [27], AlphaScreen [28], in vitro NEDD8 conjugation assay [29], cellular thermal shift assay [30], co-immunoprecipitation [31], pull-down assay [32] and ATP kinetics assay [33] are commonly used to identify NEDDylation process.
The main substrates for NEDDylation pathway are members of the cullin family (cullin1, 2, 3, 4A, 4B and 5), which are the core components of Cullin-RING ligases (CRLs) [34,35,36]. The activation of CRLs requires NEDD8 to be conjugated to lysine residues at C-terminus of cullins, thereby inducing conformational changes in the CRLs complex and eventually regulating ubiquitylation process [37, 38]. As the most important ubiquitin ligase family in E3, CRLs significantly control a variety of basic biological functions by heightening the activity of ubiquitylation and subsequent degradation of key regulatory proteins [39]. Although cullins are well-characterized substrates in NEDDylation pathway, some non-cullins have also been identified as substrate proteins of the NEDDylation modification, such as p21, p53, p73, caspase7, XIAP, RhoA, HuR and ATF4 [40]. The characteristics of cullin and non-cullin substrates determine the significant role of NEDDylation in regulating biological processes and controlling multiple diseases, primarily neurodegenerative diseases and human cancers [41].
The discovery of the covalent NAE inhibitor MLN4924 in 2009 set a landmark, demonstrating that targeting NEDDylation is an effective anticancer strategy [42]. The inhibition of NEDDylation by NAE inhibitors leads to the decrease of CRLs levels during ubiquitination process and ultimately results in the degradation of proteins that play significant roles in cell proliferation, DNA damage, cell cycle, stress responses and signal transduction [43,44,45,46,47,48,49]. In addition, targeting NAE for cancer therapy induces the abnormal NEDDylation modification of cullin and non-cullin substrates [50]. The relationships between NAE and cancers are summarized in Fig. 1. Thus, NAE as the only activation enzyme in NEDDylation process has been a promising target for the treatment of hematological tumors, solid tumors, etc. [51,52,53,54,55].

Targeting NAE as a promising therapeutic approach for cancer
Structure of NAE
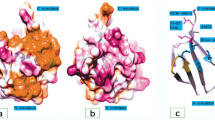
Canonical E1 enzymes including UAE, SAE and NAE have a common structure consisting of two pseudosymmetric adenylation domains: the active adenylation domain (AAD) and the inactive adenylation domain (IAD) [56,57,58,59,60,61]. As shown in Fig. 2A, NAE contains an insertion within the IAD, which is known as the catalytic cysteine (CC) domain in APPBP1 [62]. Furthermore, NAE also contains a ubiquitin fold domain (UFD) at the C-terminal of the AAD in UBA3 and a conserved CYS domain inserted within the AAD [63,64,65]. In the heterodimeric NAE, APPBP1 subunit corresponds to the N-terminal of the single-chain NAE, and UBA3 as the other subunit corresponds to the C-terminal of the single-chain NAE [66,67,68]. The biological function of NAE is to activate NEDD8 through the formation of NEDD8-NAE thioester conjugate, thereby completing the NEDDylation process of substrate proteins [69,70,71]. The crystal structure of NEDD8 is shown in Fig. 2B (PDB code: 1NDD, resolution: 1.60 Å) [72]. Furthermore, the crystal structure of NAE-NEDD8 complex is shown in Fig. 2C (PDB code: 1R4M, resolution: 3.00 Å).

Structure of NAE and its complex. A Primary structure of NAE. B Crystal structure of NEDD8. C Crystal structure of NAE-NEDD8 complex; APPBP1 part is wathet blue, UBA3 part is green and NEDD8 part is red
X-ray structures of NAE and NAE complex with the high-resolution can be available in protein data bank (https://www.rcsb.org/). In Table 1, we summarize the resolution and macromolecule contents of these X-ray structures. Total structure weight of NAE as a single enzyme is 220.58 kDa (PDB code: 1YOV, resolution: 2.60 Å). The NAE complexes, including 1R4N, 1R4M, 1TT5, 2NVU, 3DBR, 3DBL and 3DBH, demonstrate the structural basis for the activation of NEDD8 by NAE. The co-crystal structure of APPBP1/UBA3/NEDD8 with a small molecule inhibitor provides an extremely important tool for the rational design and efficient virtual screening of NAE-targeting agents (PDB code: 3GZN, resolution: 3.00 Å).
Multistep mechanisms of NEDD8 activation by NAE and approaches to discover NAE inhibitors
The multistep mechanisms of NEDD8 activation by NAE are summarized in Fig. 3. In the first reaction (i), NAE binds NEDD8 and ATP to form a high-energy NEDD8-AMP intermediate and release PPi in the presence of magnesium [73, 76, 78]. In the second reaction (ii), the catalytic cysteine of NAE reacts with the high-energy NEDD8-AMP intermediate to form a thioester bond between NAE and NEDD8 and release AMP [79]. In the third reaction (iii), NAE-NEDD8 intermediate binds NEDD8 and ATP to catalyze formation of a NEDD8-NAE-NEDD8-AMP complex and release PPi in the presence of magnesium [80]. In the final reaction (iv), a thioester-linked E2-NEDD8 is formed from the NEDD8-NAE-NEDD8-AMP complex in the presence of E2 via a transthioesterification reaction [81].

Detailed mechanisms of NEDD8 activation by NAE
The structural studies of NAE and detailed mechanisms of NEDD8 activation by NAE provide at least four approaches to discover potential NAE inhibitors. Approach (1) is the discovery of ATP-competitive NAE inhibitors for blocking the binding of ATP to the active adenylation site in NAE [82,83,84]. Approach (2) is the discovery of AMP analogues as covalent NAE inhibitors for the formation of NEDD8-compound covalent adducts to prevent the subsequent NEDD8 activating steps [42]. Approach (3) is the discovery of novel NAE inhibitors targeting the cysteine active-site for blocking the formation of NAE-NEDD8 thioesters [75]. Approach (4) is the discovery of novel NAE inhibitors disrupting NAE-E2 interactions for blocking the transferation of NEDD8 to NEDD8-conjugating enzyme E2s [74]. Most of the current NAE inhibitors are reported to exert their inhibitory effects through the first two modes. Currently, most reported inhibitors display the inhibitory activity against NAE via the first two modes, while NAE inhibitors with latter two modes have not been studied deeply.
Preclinical studies and pharmacokinetics of covalent NAE inhibitors
Up to now, design strategies, synthetic methods and anticancer mechanisms of various NAE-targeting agents have been developed [85,86,87,88,89]. Compared with non-covalent NAE inhibitors and NAE agonists, developments of covalent NAE inhibitors are relatively mature. All reported covalent inhibitors contain a sulfanilamide group to form the covalent bond with NAE. Chemical structures, enzymatic activity and cellular activity of reported covalent NAE inhibitors 1–9 are summarized in Table 2. In 2009, MLN4924 (compound 1) as the first NAE inhibitor was reported by Millennium Pharmaceuticals, Inc [42]. MLN4924 displays a potent inhibitory activity against NAE with an IC50 value of 4 nM. However, it has relatively poor activity against UBA6, SAE, UAE and ATG7 with IC50 values of 1.8 μM, 8.2 μM, 1.5 μM and > 10 μM, respectively. The selective inhibition of NAE activity has been an effective strategy for cancer therapy, and MLN4924 as a selective NAE inhibitor exhibits anticancer effects against various cancer types, including leukemia [90], endometrial carcinoma [91], renal cell carcinoma [92], urothelial carcinoma [93], liver cancer [94], colorectal cancer [95], ovarian cancer [96], glioblastoma [97], pancreatic cancer [98], cervical cancer [99], lung cancer [100], breast cancer [101], head and neck squamous cell carcinoma [102], nasopharyngeal carcinoma [103], uveal melanoma [104], gastric cancer [105], malignant melanoma [106], retinoblastoma [107], multiple myeloma [108], prostate cancer [109], osteosarcoma [110], malignant rhabdoid tumors [111], ewing sarcoma [112] and lymphoma [113].
Compared with covalent NAE inhibitors 1–6, TAS4464 (compound 7) displays more potent inhibitory activity against NAE with an IC50 value of 0.96 nM [114]. Moreover, TAS4464 shows inhibitory effects against SAE, UAE and carbonic anhydrase with IC50 values of 1280 nM, 449 nM and 730 nM, suggesting that TAS4464 is a selective NAE inhibitor. TAS4464 has potent growth-inhibitory effects against various kinds of cancers, including chronic lymphocytic leukemia, T cell acute lymphoblastic leukemia, mantle cell lymphoma, clear cell sarcoma, colon carcinoma, follicular lymphoma, small cell lung cancer, multiple myeloma, acute myeloid leukemia and diffuse large B cell lymphoma [115,116,117]. The antiproliferative activity of TAS4464 is 3–64 times more potent than that of MLN4924 in 240 cell lines derived from human tumor tissues. It display potent antiproliferative activity with IC50 values of 1.60–460.00 nM, 0.70–4223.00 nM and 0.2 nM against patient-derived AML, DLBCL and SCLC cells. Moreover, TAS4464 can obviously inhibit tumor regression in CCRF-CEM, GRANTA-519 MCL, SU-CCS-1 and LU5266 xenograft models.
More potent NAE inhibitors 8 and 9 containing a pyrimidotriazole framework are designed and synthesized via a scaffold hopping strategy in Ao Zhang’s group [123, 124]. Structure–activity relationship studies reveal that the pyrimidotriazole framework plays an important role for inhibitory effects against NAE and cancer cells. Compounds 8 and 9 significantly inhibit tumor growth in leukemia MV-4-11 and colon cancer HCT-116 xenograft models. All reported pharmacokinetic parameters of covalent NAE inhibitors are summarized in Table 3. Although TAS4464 exhibits potent anticancer effects in vitro and in vivo, its detailed pharmacokinetic data are not shown [117]. Following intravenous injection (1 mg/kg), compound 9 demonstrates a 4.46-fold increment in drug exposure with the AUClast value of 2500 h·ng/mL and a 5.57-fold decrease in systemic plasma clearance with the CL value of 5.33 mL/min/kg compared with MLN4924.
Molecular docking studies of NAE inhibitors in the clinical stage
Among all reported NAE inhibitors, only MLN4924 and TAS4464 as covalent NAE inhibitors currently entered into clinical trials for cancer therapy, particularly for AML [116, 125,126,127]. Chemical structures of MLN4924 and TAS4464 are shown in Fig. 4A. The co-crystal structure of NAE and MLN4924 (PDB: 3GZN, resolution: 3.00 Å) was reported in 2010 and shown in Fig. 4B, C [77]. G79 and R111 of NAE form a hydrogen bond with the sulfonamide group of MLN4924 (1.7 Å and 2.7 Å). The hydroxyl group on the ribose unit generates two hydrogen bonds with K124 (2.3 Å) and D100 (3.5 Å), respectively. The nitrogen atom of secondary amine group produces a hydrogen bond with Q149 (3.3 Å), and 7H-pyrrolo[2,3-d]pyrimidine unit forms two hydrogen bonds with I148 (2.2 Å) and D100 (2.7 Å). The most important binding force for the inhibitory effect is the covalent bond between the carbonyl carbon of G76 and the sulfonamide group of MLN4924.

NAE inhibitors in clinical trials. A Chemical structures of NAE inhibitors in the clinical stage; B Surface map of MLN4924 in NAE (PDB: 3GZN); C Three-dimensional binding model of MLN4924 in NAE; D Surface map of TAS4464 in NAE (PDB: 3GZN); E Three-dimensional binding model of TAS4464 in NAE; F Overlay of TAS4464 (wathet blue) and MLN4924 (yellow) in complex with NAE; G The formation of NEDD8-compound covalent adduct
Molecular docking studies of TAS4464 are also investigated and shown in Fig. 4D, E (PDB: 3GZN, resolution: 3.00 Å). The sulfonamide group of TAS4464 generates three hydrogen bonds with D167 (1.9 Å), K124 (2.3 Å) and Q112 (2.4 Å). The tetrahydrofuran unit also forms three hydrogen bonds with G78 (3.0 Å), G165 (2.3 Å) and D167 (2.2 Å). The amino group attaching 7H-pyrrolo[2,3-d]pyrimidine produces two hydrogen bonds with Q149 (2.6 Å) and M101 (3.1 Å), and 7H-pyrrolo[2,3-d]pyrimidine fragment forms a hydrogen bond with I148 (2.2 Å). The oxygen atom between ethyl group and phenyl group forms a hydrogen bond with Q149 (2.8 Å). As shown in Fig. 4F, TAS4464 occupies the same active pocket of NAE with MLN4924 and takes a similar binding pose. MLN4924 and TAS4464, which have a similar structure to adenosine 5'-monophosphate (AMP) and attack the thioester bond of NAE-NEDD8 intermediate to form NEDD8-compound covalent adducts for blocking the subsequent NEDD8 activating processes (Fig. 4G).
Pharmacological mechanisms of MLN4924 in cancers
Induction of apoptosis
Targeting NAE with MLN4924 induces the intrinsic apoptosis and extrinsic apoptosis through the inactivation of cullin NEDDylation [128,129,130,131,132]. The expression levels of cleaved caspase3 and cleaved caspase7 are increased, and the expression levels of Bcl-2 and Bcl-XL are decreased in a concentration-dependent manner with the treatment of MLN4924 against different cancer cells. Apoptosis induced by pharmaceutical inhibition of NEDDylation suppresses tumor growth in leukemia [133,134,135], colorectal cancer [136], ewing sarcoma [137], urothelial carcinoma [138], head and neck squamous cell carcinoma and intrahepatic cholangiocarcinoma [128, 139]. MLN4924-induced apoptosis includes enhancing the accumulation of CDT1 [140], inducing the dramatic accumulation of CRL E3 substrate I-kappa-B-alpha (IKB-α) [141] and increasing the expression of NOXA [142]. The NOXA-dependent apoptosis triggered by MLN4924 is activated by the elevated expression levels of c-Myc and transcription factor ATF-4. The combination of TNF-related apoptosis-inducing ligand (TRAIL) and MLN4924 synergistically causes apoptosis against head and neck squamous cell carcinoma (HNSCC) cells through enhancing the degradation of c-FLIP [143].
Alterations in mitochondrial functions and energy metabolism
MLN4924 generates mitochondrial fission-to-fusion conversion by the inactivation of SCFβ-TrCP E3 ligase and the prevention of mitochondrial translocation against breast cancer cells [144,145,146,147]. MLN4924 changes mitochondrial shape in breast cancer cells, lung adenocarcinoma cells and bronchial epithelial cells. MFN1, MFN2 and the ectopic expression of DRP1 play dominant roles in MLN4924-induced mitochondrial fission-to-fusion conversion. The inhibition of SCFβ-TrCP E3 ligase promotes the polyubiquitylation and subsequent accumulation of MFN1. The effects of NEDDylation blockage by MLN4924 on cell metabolism are investigated by a mass spectrometry-based metabolic profiling, and MLN4924 perturbs metabolites of mitochondrial tricarboxylic acid cycle (TCA) and glycolysis. The decrease of ATP production, the reduction of mitochondrial depolarization and an increase of mitochondrial ROS levels are induced by MLN4924 in concentration-dependent and time-dependent manners. The combined treatment of MLN4924 with inhibitors of glycolysis or mitochondrial oxidative phosphorylation system (OXPHOS) can remarkably enhance antitumor effects in vitro and in vivo.
Increase of G2 cell cycle arrest, induction of DNA damage and inducement of radiosensitization
MLN4924 sensitizes resistant cancer cells to ionizing radiation, which demonstrates that MLN4924 can be used as a novel radiosensitizing agent [102, 148,149,150,151]. The accumulation of p21/p27/WEE1 as three substrates of CRL induces the G2 cell cycle arrest against cancer cells with the treatment of MLN4924, and the co-treatment of MLN4924 with radiation enhances the induction of G2 cell cycle by the increased accumulation of p21/p27/WEE1 [152,153,154,155,156]. The turnover of p21/p27/WEE1 can be obviously delayed in prostate cancer PC3 and Du145 cells with the treatment of MLN4924-radiation. MLN4924 increases the expression levels of γ-H2AX and p-H2AX in a concentration-dependent manner, suggesting that MLN4924 causes DNA damage [157]. MLN4924-radiation combination also enhances the induction of DNA damage via the increased accumulation of ORC1 and CDT1 as CRL substrates. Radiosensitization by MLN4924 is attributable to the increase of G2 cell cycle arrest and induction of DNA damage. With the combination of radiation and MLN4924, antitumor effects in xenograft tumor models are remarkably enhanced.
Activation of autophagy
Autophagy as a lysosomal degradation process has significant roles in the development and metastasis of cancers [158,159,160,161,162,163]. MLN4924 triggers the conversion of LC3-I to LC3-II and up-regulates the accumulation of LC3-II in a time-dependent manner, indicating that it activates autophagy [164,165,166,167]. With the treatment of MLN4924, the expression level of DEPTOR as a substrate of SCFβTrCP E3 is increased in a cell line-dependent manner. MLN4924 concentration-dependently inactivates Cullin2 NEDDylation and subsequently induces a significant increase of HIF1α as a substrate of Cullin2. HIF1α-REDD1-TSC1 axis and DEPTOR are effectively involved in MLN4924-mediated mTORC1 suppression and autophagy inducement. Specifically, HIF1α triggers REDD1-TSC1 signal axis to suppress mTORC1 and DEPTOR directly binds to mTORC1, resulting in the activation of autophagy. Bafilomycin A1 as a well-known inhibitor of autophagy remarkably increases MLN4924-induced apoptosis by the up-regulated expression levels of cleaved PARP and cleaved caspase3/7. This combination of an autophagy inhibitor and a NAE inhibitor effectively increases the inhibitory effects of tumor growth through the strengthening of apoptosis.
Trigger of senescence
Cellular senescence is an irreversible arrest of cell growth, and bioactive compound targeting tumor cell senescence has been an effective strategy for cancer therapy [168,169,170,171,172]. MLN4924 induces cellular senescence by inactivating CRL/SCF E3s and thus exerts antitumor effects in vivo and in vitro against osteosarcoma, colorectal cancer, lung cancer, glioblastoma, gastric cancer, lymphoma, melanoma and laryngeal cancer [173,174,175,176,177]. MLN4924 triggers cellular senescence in p53-null H1299 cells, indicating that MLN4924-induced senescence is not related to p53. MLN4924 increases the expression level of p21 in cancer cells, and p21 as a substrate of CRL/SCF is responsible for MLN4924-induced senescence. The senescence-like morphology induced by MLN4924 is not changed after the removal of this drug, demonstrating that the trigger of senescence by MLN4924 is irreversible.
Suppression of tumor angiogenesis
The development of new blood vessels is called angiogenesis [178,179,180,181], and tumor angiogenesis is a potential target for the treatment of cancer [182,183,184]. With the intervention of MLN4924 in human microvascular endothelial cells, the branch count, junction count, area and skeleton length of the vascular network are obviously reduced [185]. MLN4924 potently suppresses the growth of new blood vessel in the rat aortic ring assay, significantly inhibits the formation of capillary vessels in the chick embryo chorioallantoic membrane assay and strongly reduces the density of microvessel in the matrigel plug assay [186]. These results from above angiogenic assays suggest that MLN4924 displays remarkably inhibitory effects on tumor angiogenesis. Importantly, MLN4924 induces the suppression of tumor angiogenesis in footpad xenograft and orthotopic models bearing pancreatic cancer cells. MLN4924 up-regulates the expression level of RhoA, which is a substrate of CRL E3. Knockdown of RhoA obviously restores the inhibitory effects of MLN4924 on the formation of capillary tube in human umbilical vein endothelial cells. Many studies demonstrate that induction of apoptosis and cell cycle arrest by MLN4924 can also suppress tumor angiogenesis [187,188,189]. In uveal melanoma cells, MLN4924 exerts antiangiogenic effects by impairing the secretion of VEGF-C [104].
Regulation of tumor microenvironment
Due to important roles of the tumor microenvironment (TME) in dynamically regulating tumor progression, therapeutic strategy targeting TME has become an attractive approach for cancer therapy [190,191,192,193]. The TME is the complicated multicellular environment that mainly comprises tumor-derived factors, cancer-associated fibroblasts, tumor-associated endothelial cells, natural killer cells, dendritic cells, T lymphocytes, tumor-associated macrophages and the extracellular matrix [194]. Inactivation of NEDDylation by MLN4924 affects the functions of several significant components of the TME [195]. MLN4924 activates the accumulation of the migration-related substrate RhoA, apoptosis-related substrates ATF4/NOXA and cell cycle-related substrates p21/p27/WEE1 in endothelial cells. MLN4924 regulates differentially expressed genes in fibroblasts isolated from hepatocellular carcinoma tissues, and these genes are involved in DNA replication, cell cycle, cytokine–cytokine receptor interaction and TNF signaling pathway. MLN4924 at 100 nM or 500 nM decreases the transcriptional activity of nuclear factor κB (NF-κB) induced by lipopolysaccharides in macrophage cells [196]. The activation of Erk is reduced in CD4+ T cells with the treatment of MLN4924 at 100 nM for 16 h [197]. NEDDylation accelerates the differentiation of follicular helper T cells through modulation the degradation of Foxo1 [198]. In dendritic cells, MLN4924 triggers the accumulation of DEPTOR and inhibits the activation of NF-κB [199, 200]. Therefore, MLN4924 exerts significantly antitumor effects in a variety of cancer models by regulating apoptosis, mitochondrial fission-to-fusion conversion, cell cycle, DNA damage, radiosensitization, autophagy, senescence, angiogenesis and tumor microenvironment (Fig. 5).

Pharmacological mechanisms of MLN4924 in cancers
Pharmacological mechanisms of TAS4464 in cancers
Induction of apoptosis and sub-G1 cell cycle arrest
TAS4464 decreases the expression level of Cullin-NEDD8 and increases the expression levels of CRL substrate proteins in a concentration-dependent and time-dependent manner [115]. TAS4464 increases expression levels of cleaved caspase 2/3/6/7/8/9/10 and cleaved PARP in leukemia HL60 and THP1 cell lines, demonstrating that TAS4464 can activate extrinsic and intrinsic apoptotic pathways [116]. With the treatment of TAS4464 at 100 nM in HL60 and THP1 cells, the mRNA transcriptional level of NOXA is increased and the mRNA transcriptional level of c-FLIP is decreased. In human AML xenograft models, TAS4464 at 100 mg kg−1 per day remarkably inhibits cancer cell growth through the increased expression level of NOXA and the decreased expression level of c-FLIP accompanied by the activation of c-Myc. TAS4464 induces the accumulation of p21/p27/CDT1 in concentration-dependent and time-dependent manners and subsequently arrests cell cycle at sub-G1 phase.
Inactivation of NF-κB pathways
With the treatment of TAS4464 for 4 h against myeloma KMS-11, MM.1S, KMS-26 and KMS-12-BM cells, the expression levels of NF-κB regulators p-p100 and p-IκBα are increased [115]. The DNA binding activity of RelB and p65 in KMS-26 and MM.1S cells is inhibited by TAS4464 in a concentration-dependent manner. Furthermore, TAS4464 suppresses the transcription levels of NF-κB-targeted genes in these myeloma cells. In xenograft mouse models bearing myeloma cells, TAS4464 down-regulates the NF-κB pathways and increases the expression levels of several apoptosis-related factors (cleaved caspase 3/8 and cleaved PARP). With the combination of chemotherapy drugs, TAS4464 synergistically improves the antitumor effects against multiple myeloma. The pharmacological mechanisms of TAS4464 regarding its therapeutic efficacy in cancers are shown in Fig. 6.

Pharmacological mechanisms of TAS4464 in cancers
Clinical trials of NAE inhibitors for cancer therapy
To date, 42 clinical trials for MLN4924 and TAS4464 have been registered (Table 4). Among them, five clinical trials at phase I of MLN4924 in patients with AML (ClinicalTrials.gov Identifier: NCT00911066, NCT03459859, NCT02610777, NCT02782468 and NCT03814005) have been completed [201,202,203,204,205,206,207]. For elderly patients with AML considered unfit for conventional chemotherapy, the combination therapy with MLN4924 and azacitidine was generally well tolerated (ClinicalTrials.gov Identifier: NCT01814826) [208]. In addition, six clinical trials at phase I of MLN4924 in patients with nonhematologic malignancies or solid tumors (ClinicalTrials.gov Identifier: NCT00677170, NCT01862328, NCT02122770, NCT03057366, NCT03330106 and NCT03486314) have also been completed [209,210,211,212]. For pediatric patients with recurrent or refractory solid tumors, MLN4924 in combination with temozolomide and irinotecan is well tolerated (ClinicalTrials.gov Identifier: NCT03323034) [213]. In this clinical trial, a dose of 25 mg/m2 dose on day 8 had a half-life of 5–6 h and a mean clearance of 20 L/h/m2.
Because of favorable results from phase I clinical trials for MLN4924, 15 clinical trials at phase II (ClinicalTrials.gov Identifier: NCT02610777, NCT03228186, NCT03238248, NCT03745352, NCT03709576, NCT04175912, NCT03965689, NCT04800627, NCT04985656, NCT04712942, NCT04266795, NCT03862157, NCT03330821, NCT01415765 and NCT03319537) and 2 clinical trials at phase III (ClinicalTrials.gov Identifier: NCT03268954 and NCT04090736) have been conducted. A phase I/II clinical trial of MLN4924 in combination with cytarabine and idarubicin was performed to investigate the therapeutic effects for patients with AML (ClinicalTrials.gov Identifier: NCT03330821). A phase II trial was first posted in 2019 to study the overall response rate, overall survival, progression-free survival and safety profile of MLN4924 in combination with paclitaxel and carboplatin for patients with advanced non-small cell lung cancer (ClinicalTrials.gov Identifier: NCT03965689). A phase III study was first posted in 2019 and last update posted in 2022 to compare the difference in overall survival between azacitidine alone and azacitidine plus MLN4924 in patients with AML (ClinicalTrials.gov Identifier: NCT04090736) [214].
TAS4464 is a potent and selective NAE inhibitor with broad-spectrum antitumor activity against a variety of cancer cell lines [117]. A phase I clinical trial of TAS4464 was conducted in 2016 to study the appropriate dose, pharmacogenomics, pharmacokinetics, pharmacodynamics and safety of TAS4464 for patients with multiple myeloma or lymphoma (ClinicalTrials.gov Identifier: NCT02978235). However, this clinical trial of TAS4464 was terminated in 2021 due to cases of drug-induced liver injury. In 2021, a phase I clinical trial was performed to explore the safety, clinical response, pharmacokinetics, pharmacogenomics and pharmacodynamics of TAS4464 in patients with advanced/metastatic solid tumors (Trial number: JapicCTI-173,488). The most common treatment-related adverse events in this trail are the increase of alanine aminotransferase (68.8%), the increase of aspartate aminotransferase (62.5%) and nausea (43.8%).
Challenges of NAE inhibitors for cancer therapy
Low response rate for AML/MDS and hepatotoxicity
Although the developments of NAE inhibitors have achieved some positive results, NAE inhibitors also face several expected or unexpected challenges. A phase I clinical trial of MLN4924 in patients with AML and myelodysplastic syndromes (MDS) has been successfully completed (ClinicalTrials.gov Identifier: NCT00911066). However, MLN4924 as a monotherapy in this trail only displayed an overall response rate of 17% [215]. In order to improve the efficacy, the structural optimization or the combined pharmacotherapy of MLN4924 is investigated. In 2021, Zhang's group performed a structural optimization of MLN4924 by a scaffold hopping strategy to design a pyrimidotriazole derivative with improved overall PK properties and good safety in vivo [123]. Although TAS4464 is a selective NAE inhibitor with potent antitumor effects, it generates drug-induced liver injury for patients with multiple myeloma or lymphoma in the phase I trial (ClinicalTrials.gov Identifier: NCT02978235). Thus, it is necessary to design novel NAE inhibitors with high potency and low hepatotoxicity.
Resistance
In 2011, research demonstrated that the loss of phosphatase and tension homolog on chromosome ten (PTEN) in melanoma cells is a driver of drug resistance [216]. Similarly, protein levels of PTEN are decreased by 30-40% in breast cancer and the absence of PTEN also contributes to MLN4924 resistance in breast cancer cells [217]. With the absence of PTEN, the antitumor effects of MLN4924 in breast cancer cells are significantly decreased. The activity of Akt pathway is positively correlated with NEDDylation pathway in patients with high PTEN expression, but not in patients with low PTEN expression. PTEN plays an indispensable role for supression of PI3K/Akt pathway by MLN4924 in breast cancer cells.
Sphere formation and ciliogenesis
Recently, Sun’s group reported that MLN4924 has the side effects of tumor sphere formation and ciliogenesis inhibition through the dimerization of EGFR [218, 219]. MLN4924 at 0.1 μM significantly stimulates the tumorigenesis of nude mice bearing H1299 cells and enhances the activation of EGFR and the downstream RAS/RAF/MEK/ERK signaling pathways. Further mechanistic studies demonstrate that MLN4924 does not directly bind to EGFR but instigates the dimerization of EGFR in a time-dependent manner. MLN4924 at 0.3 μM obviously inhibits cilia initiation and effectively promotes cilia disassembly in BEAS2B cells, and MLN4924 at 30 mg/kg suppresses hair re-growth in C57BL/6 mice. Protein kinase B (AKT1) plays a key role in the regulation of cilia initiation and disassembly induced by MLN4924. Although these studies raise some concerns for cancer therapy, they also provide an opportunity for future development of MLN4924 as a potential treatment for patients with abnormal ciliogenesis.
Glycolysis
Pyruvate kinase isoform M2 (PKM2) exhibits significant roles in cancer cell growth and metabolic reprogramming, and it is overexpressed in cancer cells to catalyze glycolysis [220]. MLN4924 promotes glycolysis through the up-regulation of PKM2 tetramers levels in a concentration-dependent manner against breast cancer MDA-MB-231 cells. Glycolysis triggered by MLN4924 confers survival to breast cancer cells, and 2-deoxy-D-glucose as a glycolysis inhibitor coupled with MLN4924 can exhibit much better effects of growth inhibition against breast cancer cells in xenograft models [144]. Therefore, the tetramerization of PKM2 to increase glycolysis and cancer cell growth has been a side effect of MLN4924 to treat breast cancer [221]. The challenges and related mechanisms of NAE inhibitors are summarized in Fig. 7.

The challenges and related mechanisms of NAE inhibitors
Potential development directions of NAE-targeting drugs in the future
Combined pharmacotherapy or multi-target drugs
Although a number of NAE inhibitors have been developed, only AMP analogues MLN4924 and TAS4464 have formally entered the clinic trails to treat various cancers. Some NAE inhibitors demonstrate moderate antitumor effects, potential hepatotoxicity and resistance against cancer cells in vitro and in vivo. In order to improve antitumor potency and reduce side effects, development of combined pharmacotherapy or multi-target drugs might be promising approaches in the future directions [222,223,224,225]. Because of the complex pathogenesis of cancer, combined pharmacotherapy can synergistically exhibit better therapeutic effects by intervening in multiple targets or signal pathways in cancer cells [226,227,228]. Furthermore, combined pharmacotherapy or multi-target drugs can reduce the toxic side effects and decrease the therapeutic dose [229]. To date, there is growing evidence that MLN4924 coupled with different anticancer drugs can also overcome drug resistance and improve anticancer effects [230,231,232,233]. Up to now, there are more than 30 clinical trials investigating the combined pharmacotherapy of MLN4924. Recent studies investigated that NAE inhibitor MLN4924 and HDAC inhibitor belinostat could interact synergistically by reciprocally disabling the DNA damage response in AML/MDS cells [234, 235]. Guo’s group reported that the combinational of celecoxib as a cyclooxygenase-2 inhibitor with MLN4924 synergistically inhibits the survival of human urothelial carcinoma cells by decreasing phosphorylation of AKT/ERK signaling pathways [236]. Additionally, combined pharmacotherapy of MLN4924 could enhance the antitumor activity with Chk1 inhibitor, Bcl-2 inhibitor, IAP antagonist, mTOR/PI3K inhibitor, MEK inhibitor, anti-PD-L1 antibody and LSD1 inhibitor (Fig. 8A) [237,238,239,240,241,242]. Compared with combined pharmacotherapy, multi-target drugs have therapeutic advantages due to their abilities to reduce the complexity of pharmacokinetics and drug-drug interactions [243,244,245]. More importantly, the successful combined pharmacotherapy in clinical trials might provide a solid experimental basis for the development of multi-target NAE inhibitors to treat cancer in the future.

Potential future directions of NAE-targeting drugs. A Combination strategies with NAE inhibitor for cancer therapy; B NAE agonist and its anticancer mechanisms; C Non-covalent NAE inhibitors; D Application for other diseases beyond cancer by targeting NAE
NAE agonists
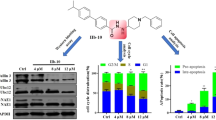
In 2020, we reported the world's first NAE agonist (compound 10) with IC50 values of 0.09 μM, 0.10 μM and 1.15 μM against MGC803, MCF7 and PC3 cells, respectively [246]. Structure–activity relationships reveal that its 3,4,5-trimethoxyphenyl ring exerts a crucial role for the antiproliferative activity against all cancer cell lines. Compound 10, a novel tertiary amide derivative, activated NEDDylation in a concentration-dependent manner through the direct interaction with APPBP1 in gastric cancer MGC803 cells. Moreover, it induces the degradation of c-IAP1 and the down-regulation of XIAP via the activation of NEDDylation (Fig. 8B). Compound 10 induces cell morphology changes, causes apoptosis and arrests cell cycle in G2/M phase against MGC803 cells. It increases expression levels of Fasl, FADD, cleaved caspase 8/9, PIDD and Bax and decreases expression levels of pro-caspase 2 and Bcl-2 in a concentration-dependent manner against MGC803 cells. Compound 10 at 50 mg/kg obviously suppresses tumor growth in MGC803 xenograft models via the activation of NAE-UBC12-Cullin1 NEDDylation. Therefore, NAE agonists may regulate the proliferation, apoptosis and cell cycle of tumor cells through the promotion of NEDDylation, and NAE agonists can be one of the future research directions of NAE-targeted drugs for cancer therapy [247, 248].
Non-covalent NAE inhibitors
So far, many reported NAE inhibitors inhibit the activity of NAE through covalent binding [121]. All these covalent NAE inhibitors contain a sulfamate group to enable the formation of NEDD8-compound covalent adducts with the carboxyl group of NEDD8. Although two covalent NAE inhibitors MLN4924 and TAS4464 entered into clinical trials for cancer therapy, there are no non-covalent NAE inhibitors that have entered the clinical stage. Natural product 11 and 12 (Fig. 8C) as non-covalent NAE inhibitors exhibit anti-prostate cancer effects via the degradation of Skp2, and natural product analogue 13 and natural product 14 are also identified as non-covalent NAE inhibitors by enzymatic and cellular assays [249,250,251,252]. A rhodium(III) complex 15 targeting NAE by a non-covalent binding model displays potential anti-inflammatory activity in vivo [82]. Approved drugs 16 (candesartan cilexetic), 17 (mitoxantrone) and 18 (piperacillin) are discovered as non-covalent NAE inhibitors through the drug repositioning strategy [33, 253, 254]. N-heterocycles 19, 20 and 21 are discovered as non-covalent NAE inhibitors through the virtual screening strategy, and show the promising antitumor activity [255,256,257]. A indole analogue 22 is synthesized by us that induces apoptosis and cell cycle arrest at G2/M phase via suppression of NEDDylation and MAPK pathways [258]. Molecular docking studies reveal that non-covalent NAE inhibitors can block the interaction of NAE-NEDD8-ATP by targeting the ATP binding site of NAE. Most of reported non-covalent NAE inhibitors have not been evaluated for their antitumor effects at the animal level due to moderate antiproliferative activity against cell lines. Some inhibitors lack sufficient experiments (e.g., cellular thermal shift, co-immunoprecipitation, biolayer interferometry, isothermal titration calorimetry, or protein pull-down assays) to investigate their on-target effects and the selectivity against NAE. Therefore, there is great scope to develop non-covalent NAE inhibitors for the treatment of cancer.
Application for other diseases beyond cancer
Currently, MLN4924 is the most reported NAE inhibitor and it has potential therapeutic effects for other diseases besides cancer (Fig. 8D). Interleukin-17A (IL-17A) plays critical roles in inflammatory diseases and MLN4924 can suppress IL-17A-induced pulmonary inflammation in vivo through the inhibition of ACT1-mediated signaling [259]. NEDDylation is overactivated after ischemic stroke and MLN4924 is a therapeutic candidate for ischemic stroke by maintaining the integrity of blood–brain barrier and reducing neutrophil extravasation [260]. MLN4924 displays potent cardioprotective effects against myocardial ischemia–reperfusion injury by the induction of autophagic flux and the up-regulation of Nrf2 dependent on sirt1 [261]. MLN4924 also exhibits neuroprotective effects against oxidative stress injury, and the inhibition of NEDDylation induced by MLN4924 play significant roles in various neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, Huntington’s disease, fragile X syndrome and Machado-Joseph disease [262]. MLN4924 activates the formation of choroidal neovascularization lesion by the up-regulation of autophagy and the suppression of hedgehog pathway [263]. Therefore, the development of NAE-targeting drugs to treat additional diseases beyond cancer is also a new direction for future research. These future research directions of NAE-targeting drugs are summarized in Fig. 8.
Conclusions and perspectives
Since the NAE inhibitor MLN4924 was reported in 2009, researches on NAE-targeting compounds have rapidly developed in the field of medicinal chemistry. Many preclinical studies have revealed that a variety of NAE inhibitors and NAE agonists exhibit good antitumor effects in vitro and in vivo. MLN4924 and TAS4464 as covalent NAE inhibitors currently undergo clinical assessment to treat various tumors, especially hematological tumors. These findings demonstrate that NAE is a novel and effective therapeutic target for the treatment of cancers. The approaches to discover NAE inhibitors in this review provide a unique perspective to develop various NAE-targeting agents. Chemical structures, antitumor efficacy and detailed pharmacological mechanisms of reported NAE-targeting agents discussed in this review offer insights into the design of candidate compounds for clinical drug development. The overview of single medication or combined pharmacotherapy of MLN4924 and TAS4464 in clinical trials strongly encourages researchers to explore the therapeutic potential of NAE inhibitors in order to propose new treatments for cancers.
Even though preclinical studies and clinical trials have achieved many successes, some challenges and limitations associated with NAE inhibitors are arising. For patients with AML or MDS, the overall response rate for MLN4924 is only 17% (ClinicalTrials.gov Identifier: NCT00911066). Due to the structural similarity with AMP, the selectivity against NAE and the toxicity against normal cells of covalent NAE inhibitors may be limited. The clinical trial in phase I of TAS4464 was terminated due to hepatotoxicity (ClinicalTrials.gov Identifier: NCT02978235). Meanwhile, several side effects (tumor sphere formation, ciliogenesis and glycolysis) also limit the development of NAE inhibitors. Therefore, design of novel NAE-targeting agents requires greater consideration on the selectivity to maintain the balance of anticancer efficacy and safety.
At present, only two covalent NAE inhibitors enter into clinical trials and the development of non-covalent NAE inhibitors needs to be further deepened. Although the antitumor effects of NAE agonists illustrate the feasibility of NAE activation to design anticancer agents, the research of NAE agonists has just begun. Through the systematic summary of NAE-targeting agents, we found that combined pharmacotherapy, multi-target NAE inhibitors and developing other disease applications of NAE-targeting agents might also be future directions in this field. Development of NAE-targeting drugs may lead to some meaningful breakthroughs for disease therapy in the future.
Availability of data and materials
Not applicable.
Abbreviations
- NAE:
-
NEDD8-activating enzyme
- NEDD8:
-
Neuronal precursor cell-expressed developmentally down-regulated protein 8
- ATP:
-
Adenosine triphosphate
- APPBP1:
-
Amyloid protein-binding protein 1
- UBA3:
-
Ubiquitin-like modifier activating enzyme 3
- CRLs:
-
Cullin-RING ligases
- AAD:
-
Active adenylation domain
- IAD:
-
Inactive adenylation domain
- CC:
-
Catalytic cysteine
- UFD:
-
Ubiquitin fold domain
- AML:
-
Acute myeloid leukemia
- T 1/2 :
-
Half-life
- AUC:
-
Area under the plasma concentration–time curve
- Vss:
-
Volume of distribution
- CL:
-
Clearance
- C max :
-
Peak concentration
- IKB-α:
-
I-kappa-B-alpha
- AMP:
-
Adenosine 5′-monophosphate
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- HNSCC:
-
Head and neck squamous cell carcinoma
- TCA:
-
Tricarboxylic acid cycle
- OXPHOS:
-
Oxidative phosphorylation system
- TME:
-
Tumor microenvironment
- NF-κB:
-
Nuclear factor κB
- MM:
-
Multiple myeloma
- HM:
-
Hematologic malignancies
- HL:
-
Hodgkin lymphoma
- MS:
-
Myelodysplastic syndromes
- ALL:
-
Acute lymphoblastic leukemia
- CLL:
-
Chronic lymphocytic leukemia
- CMML:
-
Chronic myelomonocytic leukemia
- MPN:
-
Myeloproliferative neoplasm
- MDS:
-
Myelodysplastic syndromes
- PTEN:
-
Chromosome ten
- AKT1:
-
Protein kinase B
- PKM2:
-
Pyruvate kinase isoform M2
- IL-17A:
-
Interleukin-17A
References
Zubiete-Franco I, Fernández-Tussy P, Barbier-Torres L, Simon J, Fernández-Ramos D, Lopitz-Otsoa F, et al. Deregulated neddylation in liver fibrosis. Hepatology. 2017;65:694–709.
Jiang Y, Li L, Li Y, Liu G, Hoffman RM, Jia L. Neddylation regulates macrophages and implications for cancer therapy. Front Cell Dev Biol. 2021;9:681186.
He X, Zhu A, Feng J, Wang X. Role of neddylation in neurological development and diseases. Biotechnol Appl Biochem. 2022;69:330–41.
Zhou Q, Zheng Y, Sun Y. Neddylation regulation of mitochondrial structure and functions. Cell Biosci. 2021;11:55.
Yu Q, Jiang Y, Sun Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm Sin B. 2020;10:746–65.
Stuber K, Schneider T, Werner J, Kovermann M, Marx A, Scheffner M. Structural and functional consequences of NEDD8 phosphorylation. Nat Commun. 2021;12:5939.
Castagnoli L, Mandaliti W, Nepravishta R, Valentini E, Mattioni A, Procopio R, et al. Selectivity of the CUBAN domain in the recognition of ubiquitin and NEDD8. FEBS J. 2019;286:653–77.
Schwechheimer C. NEDD8-its role in the regulation of Cullin-RING ligases. Curr Opin Plant Biol. 2018;45:112–9.
Mohanty P, Chatterjee KS, Das R. NEDD8 deamidation inhibits cullin RING ligase dynamics. Front Immunol. 2021;12:695331.
Baek K, Krist DT, Prabu JR, Hill S, Klügel M, Neumaier L-M, et al. NEDD8 nucleates a multivalent cullin-RING-UBE2D ubiquitin ligation assembly. Nature. 2020;578:461–6.
Ribet D, Cossart P. Ubiquitin, SUMO, and NEDD8: key targets of bacterial pathogens. Trends Cell Biol. 2018;28:926–40.
Kostrhon S, Prabu JR, Baek K, Horn-Ghetko D, von Gronau S, Klügel M, et al. CUL5-ARIH2 E3–E3 ubiquitin ligase structure reveals cullin-specific NEDD8 activation. Nat Chem Biol. 2021;17:1075–83.
Zhao B, Zhang K, Villhauer EB, Bhuripanyo K, Kiyokawa H, Schindelin H, et al. Phage display to identify nedd8-mimicking peptides as inhibitors of the Nedd8 transfer cascade. ChemBioChem. 2013;14:1323–30.
Watson IR, Irwin MS, Ohh M. NEDD8 pathways in cancer, sine quibus non. Cancer Cell. 2011;19:168–76.
Schmidt MHH, Dikic I. Ubiquitin and NEDD8: brothers in arms. Sci STKE. 2006;2006:pe50.
Kamitani T, Kito K, Nguyen HP, Yeh ETH. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem. 1997;272:28557–62.
Assumpção ALFV, Lu Z, Marlowe KW, Shaffer KS, Pan X. Targeting NEDD8-activating enzyme is a new approach to treat canine diffuse large B-cell lymphoma. Vet Comp Oncol. 2018;16:606–15.
Li L, Kang J, Zhang W, Cai L, Wang S, Liang Y, et al. Validation of NEDD8-conjugating enzyme UBC12 as a new therapeutic target in lung cancer. EBioMedicine. 2019;45:81–91.
Wang S, Xian J, Li L, Jiang Y, Liu Y, Cai L, et al. NEDD8-conjugating enzyme UBC12 as a novel therapeutic target in esophageal squamous cell carcinoma. Signal Transduct Target Ther. 2020;5:123.
Huang DT, Paydar A, Zhuang M, Waddell MB, Holton JM, Schulman BA. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8’s E1. Mol Cell. 2005;17:341–50.
Zhou W, Xu J, Tan M, Li H, Li H, Wei W, et al. UBE2M is a stress-inducible dual E2 for neddylation and ubiquitylation that promotes targeted degradation of UBE2F. Mol Cell. 2018;70:1008–24.
Zhou L, Lin X, Zhu J, Zhang L, Chen S, Yang H, et al. NEDD8-conjugating enzyme E2s: critical targets for cancer therapy. Cell Death Discov. 2023;9:23.
Zheng Y-C, Guo Y-J, Wang B, Wang C, Mamun MAA, Gao Y, et al. Targeting neddylation E2s: a novel therapeutic strategy in cancer. J Hematol Oncol. 2021;14:57.
Chew E-H, Hagen T. Substrate-mediated regulation of cullin neddylation. J Biol Chem. 2007;282:17032–40.
Zhou H, Lu J, Liu L, Bernard D, Yang C-Y, Fernandez-Salas E, et al. A potent small-molecule inhibitor of the DCN1-UBC12 interaction that selectively blocks cullin 3 neddylation. Nat Commun. 2017;8:1150.
Scott Daniel C, Sviderskiy Vladislav O, Monda Julie K, Lydeard John R, Cho Shein E, Harper JW, et al. Structure of a RING E3 trapped in action reveals ligation mechanism for the ubiquitin-like protein NEDD8. Cell. 2014;157:1671–84.
Xirodimas DP, Sundqvist A, Nakamura A, Shen L, Botting C, Hay RT. Ribosomal proteins are targets for the NEDD8 pathway. EMBO Rep. 2008;9:280–6.
Yan Z-H, Burkhardt A, Loke H-K, Chen J, Xu Q, Brauer P, et al. Quantifiable analysis of cellular pathway inhibition of a Nedd8-activating enzyme inhibitor, MLN4924, using AlphaScreen. Anal Biochem. 2013;439:109–15.
Li X, Yokoyama NN, Zhang S, Ding L, Liu H-M, Lilly MB, et al. Flavokawain A induces deNEDDylation and Skp2 degradation leading to inhibition of tumorigenesis and cancer progression in the TRAMP transgenic mouse model. Oncotarget. 2015;6:41809–24.
Hammill JT, Scott DC, Min J, Connelly MC, Holbrook G, Zhu F, et al. Piperidinyl ureas chemically control defective in cullin neddylation 1 (DCN1)-mediated cullin neddylation. J Med Chem. 2018;61:2680–93.
Zhao J, Zhang B, Lai G, Xu R, Chu G, Zhao Y. 20-Hydroxyeicosatetraenoic acid regulates the expression of Nedd4-2 in kidney and liver through a neddylation modification pathway. Mol Med Rep. 2017;16:9671–7.
Zhang X, Zhang Y-L, Qiu G, Pian L, Guo L, Cao H, et al. Hepatic neddylation targets and stabilizes electron transfer flavoproteins to facilitate fatty acid β-oxidation. Proc Natl Acad Sci USA. 2020;117:2473–83.
Wu K-J, Zhong H-J, Li G, Liu C, Wang H-MD, Ma D-L, et al. Structure-based identification of a NEDD8-activating enzyme inhibitor via drug repurposing. Eur J Med Chem. 2018;143:1021–7.
Olaizola P, Lee-Law PY, Fernandez-Barrena MG, Alvarez L, Cadamuro M, Azkargorta M, et al. Targeting NAE1-mediated protein hyper-NEDDylation halts cholangiocarcinogenesis and impacts on tumor-stroma crosstalk in experimental models. J Hepatol. 2022;77:177–90.
Bornstein G, Ganoth D, Hershko A. Regulation of neddylation and deneddylation of cullin1 in SCFSkp2 ubiquitin ligase by F-box protein and substrate. Proc Natl Acad Sci U S A. 2006;103:11515–20.
Wu J-T, Lin H-C, Hu Y-C, Chien C-T. Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nat Cell Biol. 2005;7:1014–20.
Zhou L, Zhang W, Sun Y, Jia L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018;44:92–102.
Markmiller S, Fulzele A, Higgins R, Leonard M, Yeo GW, Bennett EJ. Active protein neddylation or ubiquitylation is dispensable for stress granule dynamics. Cell Rep. 2019;27:1356–63.
Zou T, Zhang J. Diverse and pivotal roles of neddylation in metabolism and immunity. FEBS J. 2021;288:3884–912.
Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16:30–44.
Ying J, Zhang M, Qiu X, Lu Y. Targeting the neddylation pathway in cells as a potential therapeutic approach for diseases. Cancer Chemother Pharmacol. 2018;81:797–808.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6.
Zhou L, Lin X, Zhang L, Chen S, Chen J, Zhou Z, et al. Neddylation pathway promotes myeloid-derived suppressor cell infiltration via NF-κB-mCXCL5 signaling in lung cancer. Int Immunopharmacol. 2022;113:109329.
Mittler F, Obeïd P, Haguet V, Allier C, Gerbaud S, Rulina AV, et al. Mechanical stress shapes the cancer cell response to neddylation inhibition. J Exp Clin Cancer Res. 2022;41:115.
Jayabalan AK, Sanchez A, Park RY, Yoon SP, Kang G-Y, Baek J-H, et al. NEDDylation promotes stress granule assembly. Nat Commun. 2016;7:12125.
Guo Z, Wang S, Xie Y, Han Y, Hu S, Guan H, et al. HUWE1-dependent DNA-PKcs neddylation modulates its autophosphorylation in DNA damage response. Cell Death Dis. 2020;11:400.
Guan J, Zheng X. NEDDylation regulates RAD18 ubiquitination and localization in response to oxidative DNA damage. Biochem Biophys Res Commun. 2019;508:1240–4.
Brown JS, Jackson SP. Ubiquitylation, neddylation and the DNA damage response. Open Biol. 2015;5:150018.
Li Z, Cui Q, Wang X, Li B, Zhao D, Xia Q, et al. Functions and substrates of NEDDylation during cell cycle in the silkworm, Bombyx mori. Insect Biochem Mol Biol. 2017;90:101–12.
Zhao Y, Morgan MA, Sun Y. Targeting neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid Redox Signal. 2014;21:2383–400.
Kittai A, Best SR, Rowland T, Bruss N, Okada C, Danilov AV. Pevonedistat, a small molecule inhibitor of NEDD8-activating enzyme (NAE), induces cell cycle deregulation, anaphase catastrophe, and apoptosis in T-cell lymphoma cells. Blood. 2018;132:1667.
McMillin DW, Jacobs HM, Delmore JE, Buon L, Hunter ZR, Monrose V, et al. Molecular and cellular effects of NEDD8-activating enzyme inhibition in myeloma. Mol Cancer Ther. 2012;11:942–51.
Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71:3042–51.
Godbersen C, Eastman A, Brown JR, Danilov AV. Targeting microenvironment-mediated NFκb activation with MLN4924, an inhibitor of the Nedd8-activating enzyme, in chronic lymphocytic leukemia B cells. Blood. 2013;122:2875.
Luo Z, Pan Y, Jeong LS, Liu J, Jia L. Inactivation of the cullin (CUL)-RING E3 ligase by the NEDD8-activating enzyme inhibitor MLN4924 triggers protective autophagy in cancer cells. Autophagy. 2012;8:1677–9.
Allan DC, Phillips JC. Evolution of the ubiquitin-activating enzyme Uba1 (E1). Physica A. 2017;483:456–61.
Bhogaraju S, Dikic I. Ubiquitination without E1 and E2 enzymes. Nature. 2016;533:43–4.
Lv Z, Yuan L, Atkison JH, Williams KM, Vega R, Sessions EH, et al. Molecular mechanism of a covalent allosteric inhibitor of SUMO E1 activating enzyme. Nat Commun. 2018;9:5145.
Barghout SH, Schimmer AD. E1 enzymes as therapeutic targets in cancer. Pharmacol Rev. 2021;73:1–58.
Burroughs AM, Iyer LM, Aravind L. Natural history of the E1-like superfamily: implication for adenylation, sulfur transfer, and ubiquitin conjugation. Proteins. 2009;75:895–910.
Lv Z, Yuan L, Atkison JH, Aldana-Masangkay G, Chen Y, Olsen SK. Domain alternation and active site remodeling are conserved structural features of ubiquitin E1. J Biol Chem. 2017;292:12089–99.
Walden H, Podgorski MS, Schulman BA. Insights into the ubiquitin transfer cascade from the structure of the activating enzyme for NEDD8. Nature. 2003;422:330–4.
Shah P, Chaumet A, Royle SJ, Bard FA. The NAE pathway: autobahn to the nucleus for cell surface receptors. Cells. 2019;8:915.
Yue Y, Ma Y, Qian P, Guo H. Catalytic mechanism of the ubiquitin-like NEDD8 transfer in RING E3–E2∼NEDD8-target complex from QM/MM free energy simulations. J Chem Inf Model. 2018;58:422–9.
Lim M, Newman JA, Williams HL, Masino L, Aitkenhead H, Gravard AE, et al. A ubiquitin-binding domain that binds a structural fold distinct from that of ubiquitin. Structure. 2019;27:1316–25.
Akimoto G, Fernandes AP, Bode JW. Site-specific protein ubiquitylation using an engineered, chimeric E1 activating enzyme and E2 SUMO conjugating enzyme Ubc9. ACS Cent Sci. 2022;8:275–81.
Miles JA, Frost MG, Carroll E, Rowe ML, Howard MJ, Sidhu A, et al. The Fanconi anemia DNA repair pathway is regulated by an interaction between ubiquitin and the E2-like fold domain of FANCL. J Biol Chem. 2015;290:20995–1006.
Wang J, Hu W, Cai S, Lee B, Song J, Chen Y. The intrinsic affinity between e2 and the Cys domain of E1 in ubiquitin-like modifications. Mol Cell. 2007;27:228–37.
Hill ZB, Pollock SB, Zhuang M, Wells JA. Direct proximity tagging of small molecule protein targets using an engineered NEDD8 ligase. J Am Chem Soc. 2016;138:13123–6.
Kurz T, Pintard L, Willis JH, Hamill DR, Gönczy P, Peter M, et al. Cytoskeletal regulation by the Nedd8 ubiquitin-like protein modification pathway. Science. 2002;295:1294–8.
Kamitani T, Kito K, Fukuda-Kamitani T, Yeh ETH. Targeting of NEDD8 and its conjugates for proteasomal degradation by NUB1. J Biol Chem. 2001;276:46655–60.
Wu K, Yamoah K, Dolios G, Gan-Erdene T, Tan P, Chen A, et al. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278:28882–91.
Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, Minor DL, et al. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol Cell. 2003;12:1427–37.
Huang DT, Miller DW, Mathew R, Cassell R, Holton JM, Roussel MF, et al. A unique E1–E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat Struct Mol Biol. 2004;11:927–35.
Huang DT, Hunt HW, Zhuang M, Ohi MD, Holton JM, Schulman BA. Basis for a ubiquitin-like protein thioester switch toggling E1–E2 affinity. Nature. 2007;445:394–8.
Souphron J, Waddell MB, Paydar A, Tokgöz-Gromley Z, Roussel MF, Schulman BA. Structural dissection of a gating mechanism preventing misactivation of ubiquitin by NEDD8’s E1. Biochemistry. 2008;47:8961–9.
Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37:102–11.
Lin C-M, Jiang Z, Gao Z, Arancillo M, Burgess K. Small molecules targeting the NEDD8·NAE protein–protein interaction. Chem Sci. 2021;12:1535–43.
Cappadocia L, Lima CD. Ubiquitin-like protein conjugation: structures, chemistry, and mechanism. Chem Rev. 2018;118:889–918.
Kitahara R, Yamaguchi Y, Sakata E, Kasuya T, Tanaka K, Kato K, et al. Evolutionally conserved intermediates between ubiquitin and NEDD8. J Mol Biol. 2006;363:395–404.
Huang DT, Zhuang M, Ayrault O, Schulman BA. Identification of conjugation specificity determinants unmasks vestigial preference for ubiquitin within the NEDD8 E2. Nat Struct Mol Biol. 2008;15:280–7.
Zhong H-J, Wang W, Kang T-S, Yan H, Yang Y, Xu L, et al. A Rhodium(III) complex as an inhibitor of neural precursor cell expressed, developmentally down-regulated 8-activating enzyme with in vivo activity against inflammatory bowel disease. J Med Chem. 2017;60:497–503.
Agius MP, Ko K, Johnson TK, Phadke S, Soellner MB. Conformation-tunable ATP-competitive kinase inhibitors. Chem Commun. 2022;58:3541–4.
Tang CP, Clark O, Ferrarone JR, Campos C, Lalani AS, Chodera JD, et al. GCN2 kinase activation by ATP-competitive kinase inhibitors. Nat Chem Biol. 2022;18:207–15.
Lu C, Lu P, Gong L, Zhu L-J, An Y, Wang Y. Rational design and development of novel NAE inhibitors for the treatment of pancreatic cancer. Med Chem Res. 2023;32:442–74.
Kim H-R, Jarhad DB, Sahu PK, Sung K, An D, Hyun YE, et al. Asymmetric synthesis of Fluoro-MLN4924 as a selective NEDD8-activating enzyme (NAE) inhibitor. Asian J Org Chem. 2019;8:1641–7.
Li Y, Plesescu M, Prakash SR. Synthesis of two isotopically labeled versions of NEDD8-activating enzyme (NAE) inhibitor. Tetrahedron Lett. 2011;52:1807–10.
Li Y, Wang C, Xu T, Pan P, Yu Q, Xu L, et al. Discovery of a small molecule inhibitor of cullin neddylation that triggers ER stress to induce autophagy. Acta Pharm Sin B. 2021;11:3567–84.
Zhong H-J, Yang H, Chan DS-H, Leung C-H, Wang H-M, Ma D-L. A metal-based inhibitor of NEDD8-activating enzyme. PLoS ONE. 2012;7:e49574.
Xu GW, Toth JI, da Silva SR, Paiva S-L, Lukkarila JL, Hurren R, et al. Mutations in UBA3 confer resistance to the NEDD8-activating enzyme inhibitor MLN4924 in human leukemic cells. PLoS ONE. 2014;9:e93530.
Li Y, Niu J-H, Wang Y. Machine learning-based neddylation landscape indicates different prognosis and immune microenvironment in endometrial cancer. Front Oncol. 2023;13:1084523.
Tong S, Si Y, Yu H, Zhang L, Xie P, Jiang W. MLN4924 (Pevonedistat), a protein neddylation inhibitor, suppresses proliferation and migration of human clear cell renal cell carcinoma. Sci Rep. 2017;7:5599.
Ho IL, Kuo K-L, Liu S-H, Chang H-C, Hsieh J-T, Wu J-T, et al. MLN4924 synergistically enhances cisplatin-induced cytotoxicity via JNK and Bcl-xL pathways in human urothelial carcinoma. Sci Rep. 2015;5:16948.
Chen P, Hu T, Liang Y, Jiang Y, Pan Y, Li C, et al. Synergistic inhibition of autophagy and neddylation pathways as a novel therapeutic approach for targeting liver cancer. Oncotarget. 2015;6:9002–17.
Sun Y, Baechler SA, Zhang X, Kumar S, Factor VM, Arakawa Y, et al. Targeting neddylation sensitizes colorectal cancer to topoisomerase I inhibitors by inactivating the DCAF13-CRL4 ubiquitin ligase complex. Nat Commun. 2023;14:3762.
Hong X, Li S, Li W, Xie M, Wei Z, Guo H, et al. Disruption of protein neddylation with MLN4924 attenuates paclitaxel-induced apoptosis and microtubule polymerization in ovarian cancer cells. Biochem Biophys Res Commun. 2019;508:986–90.
Brandt B, Németh M, Berta G, Szünstein M, Heffer M, Rauch TA, et al. A promising way to overcome temozolomide resistance through inhibition of protein neddylation in glioblastoma cell lines. Int J Mol Sci. 2023;24:7929.
Li J-A, Rong Y, Mao W, Zhang L, Kuang T, Lou W. Gene expression profiling reveals the genomic changes caused by MLN4924 and the sensitizing effects of NAPEPLD knockdown in pancreatic cancer. Cell Cycle. 2022;21:152–71.
Zhang H, He P, Zhou Q, Lu Y, Lu B. The potential oncogenic and MLN4924-resistant effects of CSN5 on cervical cancer cells. Cancer Cell Int. 2021;21:369.
Jiang Y, Cheng W, Li L, Zhou L, Liang Y, Zhang W, et al. Effective targeting of the ubiquitin-like modifier NEDD8 for lung adenocarcinoma treatment. Cell Biol Toxicol. 2020;36:349–64.
Chen Y, Du M, Yusuying S, Liu W, Tan Y, Xie P. Nedd8-activating enzyme inhibitor MLN4924 (Pevonedistat), inhibits miR-1303 to suppress human breast cancer cell proliferation via targeting p27Kip1. Exp Cell Res. 2020;392:112038.
Vanderdys V, Allak A, Guessous F, Benamar M, Read PW, Jameson MJ, et al. The neddylation inhibitor pevonedistat (MLN4924) suppresses and radiosensitizes head and neck squamous carcinoma cells and tumors. Mol Cancer Ther. 2018;17:368–80.
Xie P, Yang J-P, Cao Y, Peng L-X, Zheng L-S, Sun R, et al. Promoting tumorigenesis in nasopharyngeal carcinoma, NEDD8 serves as a potential theranostic target. Cell Death Dis. 2017;8:e2834.
Jin Y, Zhang P, Wang Y, Jin B, Zhou J, Zhang J, et al. Neddylation blockade diminishes hepatic metastasis by dampening cancer stem-like cells and angiogenesis in uveal melanoma. Clin Cancer Res. 2018;24:3741–54.
Lan H, Tang Z, Jin H, Sun Y. Neddylation inhibitor MLN4924 suppresses growth and migration of human gastric cancer cells. Sci Rep. 2016;6:24218.
Traore T, Mihollen M, Garnsey J, Berger A, Manfredi M, Cosmopolous K, et al. Antitumor activity of MLN4924, an investigational inhibitor of NEDD8-activating enzyme (NAE), in preclinical models of melanoma. J Clin Oncol. 2011;29:8594.
Aubry A, Yu T, Bremner R. Preclinical studies reveal MLN4924 is a promising new retinoblastoma therapy. Cell Death Discov. 2020;6:2.
El-Mesery M, Rosenthal T, Rauert-Wunderlich H, Schreder M, Stühmer T, Leich E, et al. The NEDD8-activating enzyme inhibitor MLN4924 sensitizes a TNFR1+ subgroup of multiple myeloma cells for TNF-induced cell death. Cell Death Dis. 2019;10:611.
Wang X, Li L, Liang Y, Li C, Zhao H, Ye D, et al. Targeting the neddylation pathway to suppress the growth of prostate cancer cells: therapeutic implication for the men’s cancer. Biomed Res Int. 2014;2014:974309.
Zhang S, Zhang J, Deng Z, Liu H, Mao W, Jiang F, et al. Circadian clock components RORα and Bmal1 mediate the anti-proliferative effect of MLN4924 in osteosarcoma cells. Oncotarget. 2016;7:66087–99.
Calandrini C, van Hooff SR, Paassen I, Ayyildiz D, Derakhshan S, Dolman MEM, et al. Organoid-based drug screening reveals neddylation as therapeutic target for malignant rhabdoid tumors. Cell Rep. 2021;36:109568.
Mackintosh C, García-Domínguez DJ, Ordóñez JL, Ginel-Picardo A, Smith PG, Sacristán MP, et al. WEE1 accumulation and deregulation of S-phase proteins mediate MLN4924 potent inhibitory effect on Ewing sarcoma cells. Oncogene. 2013;32:1441–51.
Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-κB-dependent lymphoma. Blood. 2010;116:1515–23.
Yoshimura C, Muraoka H, Ochiiwa H, Tsuji S, Hashimoto A, Kazuno H, et al. TAS4464, a highly potent and selective inhibitor of NEDD8-activating enzyme, suppresses neddylation and shows antitumor activity in diverse cancer models. Mol Cancer Ther. 2019;18:1205–16.
Muraoka H, Yoshimura C, Kawabata R, Tsuji S, Hashimoto A, Ochiiwa H, et al. Activity of TAS4464, a novel NEDD8 activating enzyme E1 inhibitor, against multiple myeloma via inactivation of nuclear factor κB pathways. Cancer Sci. 2019;110:3802–10.
Ochiiwa H, Ailiken G, Yokoyama M, Yamagata K, Nagano H, Yoshimura C, et al. TAS4464, a NEDD8-activating enzyme inhibitor, activates both intrinsic and extrinsic apoptotic pathways via c-Myc-mediated regulation in acute myeloid leukemia. Oncogene. 2021;40:1217–30.
Yamamoto N, Shimizu T, Yonemori K, Kitano S, Kondo S, Iwasa S, et al. A first-in-human, phase 1 study of the NEDD8 activating enzyme E1 inhibitor TAS4464 in patients with advanced solid tumors. Investig New Drugs. 2021;39:1036–46.
Chen JJ, Tsu CA, Gavin JM, Milhollen MA, Bruzzese FJ, Mallender WD, et al. Mechanistic studies of substrate-assisted inhibition of ubiquitin-activating enzyme by adenosine sulfamate analogues. J Biol Chem. 2011;286:40867–77.
Lukkarila JL, da Silva SR, Ali M, Shahani VM, Xu GW, Berman J, et al. Identification of NAE inhibitors exhibiting potent activity in leukemia cells: exploring the structural determinants of NAE specificity. ACS Med Chem Lett. 2011;2:577–82.
An H, Statsyuk AV. Development of activity-based probes for ubiquitin and ubiquitin-like protein signaling pathways. J Am Chem Soc. 2013;135:16948–62.
Zhang S, Tan J, Lai Z, Li Y, Pang J, Xiao J, et al. Effective virtual screening strategy toward covalent ligands: identification of novel NEDD8-activating enzyme inhibitors. J Chem Inf Model. 2014;54:1785–97.
An H, Statsyuk AV. An inhibitor of ubiquitin conjugation and aggresome formation. Chem Sci. 2015;6:5235–45.
Xiong C, Zhou L, Tan J, Song S, Bao X, Zhang N, et al. Development of potent NEDD8-activating enzyme inhibitors bearing a pyrimidotriazole scaffold. J Med Chem. 2021;64:6161–78.
Zhou L-N, Xiong C, Cheng Y-J, Song S-S, Bao X-B, Huan X-J, et al. SOMCL-19-133, a novel, selective, and orally available inhibitor of NEDD8-activating enzyme (NAE) for cancer therapy. Neoplasia. 2022;32:100823.
Khalife J, Radomska HS, Santhanam R, Huang X, Neviani P, Saultz J, et al. Pharmacological targeting of miR-155 via the NEDD8-activating enzyme inhibitor MLN4924 (Pevonedistat) in FLT3-ITD acute myeloid leukemia. Leukemia. 2015;29:1981–92.
Swords RT, Kelly KR, Smith PG, Gansey JJ, Mahalingam D, Padmanabhan S, et al. MLN4924, a novel first in class small molecule inhibitor of the Nedd8 activating enzyme (NAE), has potent activity in preclinical models of acute myeloid leukemia. Blood. 2009;114:1021.
Visconte V, Nawrocki ST, Espitia CM, Kelly KR, Possemato A, Beausoleil SA, et al. Comprehensive quantitative proteomic profiling of the pharmacodynamic changes induced by MLN4924 in acute myeloid leukemia cells establishes rationale for its combination with azacitidine. Leukemia. 2016;30:1190–4.
Zhang W, Liang Y, Li L, Wang X, Yan Z, Dong C, et al. The Nedd8-activating enzyme inhibitor MLN4924 (TAK-924/Pevonedistat) induces apoptosis via c-Myc-Noxa axis in head and neck squamous cell carcinoma. Cell Prolif. 2019;52:e12536.
Wu M-H, Lee C-Y, Huang T-J, Huang K-Y, Tang C-H, Liu S-H, et al. MLN4924, a protein neddylation inhibitor, suppresses the growth of human chondrosarcoma through inhibiting cell proliferation and inducing endoplasmic reticulum stress-related apoptosis. Int J Mol Sci. 2018;20:72.
Ai T-J, Sun J-Y, Du L-J, Shi C, Li C, Sun X-N, et al. Inhibition of neddylation by MLN4924 improves neointimal hyperplasia and promotes apoptosis of vascular smooth muscle cells through p53 and p62. Cell Death Differ. 2018;25:319–29.
Liu X, Jiang Y, Wu J, Zhang W, Liang Y, Jia L, et al. NEDD8-activating enzyme inhibitor, MLN4924 (Pevonedistat) induces NOXA-dependent apoptosis through up-regulation of ATF-4. Biophys Res Commun. 2017;488:1–5.
Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, et al. The Nedd8-activating enzyme inhibitor MLN4924 Thwarts microenvironment-driven NF-κB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. 2014;20:1576–89.
Bahjat M, de Wilde G, van Dam T, Maas C, Bloedjes T, Bende RJ, et al. The NEDD8-activating enzyme inhibitor MLN4924 induces DNA damage in Ph+ leukemia and sensitizes for ABL kinase inhibitors. Cell Cycle. 2019;18:2307–22.
Chen Y, Sun L. Inhibition of NEDD8 NEDDylation induced apoptosis in acute myeloid leukemia cells via p53 signaling pathway. Biosci Rep. 2022;42:BSR20220994.
Milhollen M, Narayanan U, Duffy J, Amidon B, Soucy TA, Berger AJ, et al. MLN4924, a potent and novel small molecule inhibitor of Nedd8 activating enzyme, induces DNA re-replication and apoptosis in cultured human tumor cells. Blood. 2008;112:3621.
Lv Y, Li B, Han K, Xiao Y, Yu X, Ma Y, et al. The Nedd8-activating enzyme inhibitor MLN4924 suppresses colon cancer cell growth via triggering autophagy. Korean J Physiol Pharmacol. 2018;22:617–25.
Rellinger EJ, Padmanabhan C, Qiao J, Appert A, Waterson AG, Lindsley CW, et al. ML327 induces apoptosis and sensitizes Ewing sarcoma cells to TNF-related apoptosis-inducing ligand. Biochem Biophys Res Commun. 2017;491:463–8.
Kuo K-L, Ho IL, Shi C-S, Wu J-T, Lin W-C, Tsai Y-C, et al. MLN4924, a novel protein neddylation inhibitor, suppresses proliferation and migration of human urothelial carcinoma: In vitro and in vivo studies. Cancer Lett. 2015;363:127–36.
Gao Q, Yu G-Y, Shi J-Y, Li L-H, Zhang W-J, Wang Z-C, et al. Neddylation pathway is up-regulated in human intrahepatic cholangiocarcinoma and serves as a potential therapeutic target. Oncotarget. 2014;5:7820–32.
Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 Elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70:10310–20.
Read MA, Brownell JE, Gladysheva TB, Hottelet M, Parent LA, Coggins MB, et al. Nedd8 modification of Cul-1 activates SCFβTrCP-dependent ubiquitination of IκBα. Mol Cell Biol. 2000;20:2326–33.
Zhou W, Xu J, Li H, Xu M, Chen ZJ, Wei W, et al. Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 linkage. Clin Cancer Res. 2017;23:1104–16.
Zhao L, Yue P, Lonial S, Khuri FR, Sun S-Y. The NEDD8-activating enzyme inhibitor, MLN4924, cooperates with TRAIL to augment apoptosis through facilitating c-FLIP degradation in head and neck cancer cells. Mol Cancer Ther. 2011;10:2415–25.
Zhou Q, Li H, Li Y, Tan M, Fan S, Cao C, et al. Inhibiting neddylation modification alters mitochondrial morphology and reprograms energy metabolism in cancer cells. JCI Insight. 2019;4:e121582.
Yu R, Liu T, Jin S-B, Ning C, Lendahl U, Nistér M, et al. MIEF1/2 function as adaptors to recruit Drp1 to mitochondria and regulate the association of Drp1 with Mff. Sci Rep. 2017;7:880.
Fonseca TB, Sánchez-Guerrero Á, Milosevic I, Raimundo N. Mitochondrial fission requires DRP1 but not dynamins. Nature. 2019;570:E34-42.
Yu R, Liu T, Ning C, Tan F, Jin S-B, Lendahl U, et al. The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J Bio Chem. 2019;294:17262–77.
Wan J, Zhu J, Li G, Zhang Z. Radiosensitization of human colorectal cancer cells by MLN4924: an inhibitor of NEDD8-activating enzyme. Technol Cancer Res Treat. 2015;15:527–34.
Wei D, Li H, Yu J, Sebolt JT, Zhao L, Lawrence TS, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72:282–93.
Wang X, Zhang W, Yan Z, Liang Y, Li L, Yu X, et al. Radiosensitization by the investigational NEDD8-activating enzyme inhibitor MLN4924 (pevonedistat) in hormone-resistant prostate cancer cells. Oncotarget. 2016;7:38380–91.
Ding Z, Knipp GT, van Rijn RM, Chester JA, Watts VJ. The CUL3/neddylation inhibitor MLN4924 reduces ethanol-induced locomotor sensitization and inflammatory pain allodynia in mice. Behav Brain Res. 2021;399:113051.
Yang D, Tan M, Wang G, Sun Y. The p21-dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. PLoS ONE. 2012;7:e34079.
Wei D, Morgan MA, Sun Y. Radiosensitization of cancer cells by inactivation of cullin-RING E3 ubiquitin ligases. Transl Oncol. 2012;5:305–12.
Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239–53.
Godbersen C, Paiva C, Berger AJ, Brown JR, Danilov AV. Targeting Nedd8 activating enzyme induces DNA damage and cell cycle arrest and sensitizes chronic lymphocytic leukemia (CLL) B-cells to alkylating agents. Blood. 2014;124:4690.
Zhang Q, Hou D, Luo Z, Chen P, Lv B, Wu L, et al. The novel protective role of P27 in MLN4924-treated gastric cancer cells. Cell Death Dis. 2015;6:e1867.
Blank JL, Liu XJ, Cosmopoulos K, Bouck DC, Garcia K, Bernard H, et al. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. 2013;73:225–34.
Wollert T. Autophagy. Curr Biol. 2019;29:R671–7.
Yim WW-Y, Mizushima N. Lysosome biology in autophagy. Cell Discov. 2020;6:6.
Towers CG, Thorburn A. Therapeutic targeting of autophagy. EBioMedicine. 2016;14:15–23.
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–42.
Morel E, Mehrpour M, Botti J, Dupont N, Hamaï A, Nascimbeni AC, et al. Autophagy: a druggable process. Annu Rev Pharmacol Toxicol. 2017;57:375–98.
Santana-Codina N, Mancias JD, Kimmelman AC. The role of autophagy in cancer. Annu Rev Cancer Biol. 2017;1:19–39.
Serrano-Maciá M, Simón J, González-Rellan MJ, Azkargorta M, Goikoetxea-Usandizaga N, Lopitz-Otsoa F, et al. Neddylation inhibition ameliorates steatosis in NAFLD by boosting hepatic fatty acid oxidation via the DEPTOR-mTOR axis. Mol Metab. 2021;53:101275.
Zhao Y, Xiong X, Jia L, Sun Y. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 2012;3:e386.
Yang D, Zhao Y, Liu J, Sun Y, Jia L. Protective autophagy induced by RBX1/ROC1 knockdown or CRL inactivation via modulating the DEPTOR-MTOR axis. Autophagy. 2012;8:1856–8.
Zhao Y, Sun Y. Targeting the mTOR-DEPTOR pathway by CRL E3 ubiquitin ligases: therapeutic application. Neoplasia. 2012;14:360–7.
Walton CC, Begelman D, Nguyen W, Andersen JK. Senescence as an amyloid cascade: the amyloid senescence hypothesis. Front Cell Neurosci. 2020;14:129.
Lozano-Torres B, Estepa-Fernández A, Rovira M, Orzáez M, Serrano M, Martínez-Máñez R, et al. The chemistry of senescence. Nat Rev Chem. 2019;3:426–41.
Varela-Eirín M, Demaria M. Cellular senescence. Curr Biol. 2022;32:R448–52.
Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–53.
Ruiz de Galarreta M, Lujambio A. DNA sensing in senescence. Nat Cell Biol. 2017;19:1008–9.
Jia L, Li H, Sun Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia. 2011;13:561–9.
Wang Y, Luo Z, Pan Y, Wang W, Zhou X, Jeong LS, et al. Targeting protein neddylation with an NEDD8-activating enzyme inhibitor MLN4924 induced apoptosis or senescence in human lymphoma cells. Cancer Biol Ther. 2015;16:420–9.
Martínez-Zamudio RI, Robinson L, Roux P-F, Bischof O. SnapShot: cellular senescence in pathophysiology. Cell. 2017;170:1044.
Zhang Y, Shi C-C, Zhang H-P, Li G-Q, Li S-S. MLN4924 suppresses neddylation and induces cell cycle arrest, senescence, and apoptosis in human osteosarcoma. Oncotarget. 2016;7:45263–74.
Wood EA, Lu Z, Jia S, Assumpção ALFV, Van Hesteren MA, Huelsmeyer MK, et al. Pevonedistat targeted therapy inhibits canine melanoma cell growth through induction of DNA re-replication and senescence. Vet Comp Oncol. 2020;18:269–80.
Harjes U. Controlling nerves. Nat Rev Cancer. 2017;17:708.
Morse MA, Sun W, Kim R, He AR, Abada PB, Mynderse M, et al. The role of angiogenesis in hepatocellular carcinoma. Clin Cancer Res. 2019;25:912–20.
Griffioen AW, Dudley AC. The rising impact of angiogenesis research. Angiogenesis. 2022;25:435–7.
Strzyz P. Migrasomes promote angiogenesis. Nat Rev Mol Cell Biol. 2023;24:84.
Elfiky AA, Rosenberg JE. Targeting angiogenesis in bladder cancer. Curr Oncol Rep. 2009;11:244–9.
Yetkin-Arik B, Kastelein AW, Klaassen I, Jansen CHJR, Latul YP, Vittori M, et al. Angiogenesis in gynecological cancers and the options for anti-angiogenesis therapy. Biochim Biophys Acta Rev Cancer. 2021;1875:188446.
Rajabi M, Mousa SA. The role of angiogenesis in cancer treatment. Biomedicines. 2017;5:34.
Mao W, Zhang L, Rong Y, Kuang T, Wang D, Xu X, et al. NEDD8-activating enzyme inhibitor MLN4924 inhibits both the tumor stroma and angiogenesis in pancreatic cancer via Gli1 and REDD1. Dig Dis Sci. 2023;68:1351–63.
Yao WT, Wu JF, Yu GY, Wang R, Wang K, Li LH, et al. Suppression of tumor angiogenesis by targeting the protein neddylation pathway. Cell Death Dis. 2014;5:e1059.
Watson EC, Grant ZL, Coultas L. Endothelial cell apoptosis in angiogenesis and vessel regression. Cell Mol Life Sci. 2017;74:4387–403.
Folkman J. Angiogenesis and apoptosis. Semin Cancer Biol. 2003;13:159–67.
Kollmann K, Heller G, Schneckenleithner C, Warsch W, Scheicher R, Ott RG, et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2016;30:359–60.
Bejarano L, Jordāo MJC, Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021;11:933–59.
Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–8.
Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–54.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37.
Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. 2020;30:R921–5.
Zhou L, Jiang Y, Luo Q, Li L, Jia L. Neddylation: a novel modulator of the tumor microenvironment. Mol Cancer. 2019;18:77.
Chang F-M, Reyna SM, Granados JC, Wei S-J, Innis-Whitehouse W, Maffi SK, et al. Inhibition of neddylation represses lipopolysaccharide-induced proinflammatory cytokine production in macrophage cells. J Biol Chem. 2012;287:35756–67.
Jin H, Liao L, Park Y, Liu Y-C. Neddylation pathway regulates T-cell function by targeting an adaptor protein Shc and a protein kinase Erk signaling. Proc Natl Acad Sci USA. 2013;110:624–9.
Cheng Q, Liu J, Pei Y, Zhang Y, Zhou D, Pan W, et al. Neddylation contributes to CD4+ T cell-mediated protective immunity against blood-stage Plasmodium infection. PLOS Pathog. 2018;14: e1007440.
Cheng M, Hu S, Wang Z, Pei Y, Fan R, Liu X, et al. Inhibition of neddylation regulates dendritic cell functions via Deptor accumulation driven mTOR inactivation. Oncotarget. 2016;7:35643–54.
Mathewson N, Toubai T, Kapeles S, Sun Y, Oravecz-Wilson K, Tamaki H, et al. Neddylation plays an important role in the regulation of murine and human dendritic cell function. Blood. 2013;122:2062–73.
Handa H, Cheong J-W, Onishi Y, Iida H, Kobayashi Y, Kim H-J, et al. Pevonedistat in East Asian patients with acute myeloid leukemia or myelodysplastic syndromes: a phase 1/1b study to evaluate safety, pharmacokinetics and activity as a single agent and in combination with azacitidine. J Hematol Oncol. 2022;15:56.
Ades L, Watts JM, Radinoff A, Arnan M, Cerrano M, Font Lopez P, et al. Phase II study of pevonedistat (P) + azacitidine (A) versus A in patients (pts) with higher-risk myelodysplastic syndromes (MDS)/chronic myelomonocytic leukemia (CMML), or low-blast acute myelogenous leukemia (LB AML) (NCT02610777). J Clin Oncol. 2020;38:7506.
Zhou X, Mould DR, Zhao D, Sekeres MA, Adès L, Swords RT, et al. Model-based analysis to support dose selection of pevonedistat (PEV) combined with azacitidine (AZA) in patients (pts) with higher-risk myelodysplastic syndromes (MDS)/chronic myelomonocytic leukemia (CMML) and acute myeloid leukemia (AML). J Clin Oncol. 2021;39:7042.
Watts J, Adès L, Radinoff A, Sangerman MA, Cerrano M, Lopez PF, et al. MDS-336: phase 2 study of pevonedistat + azacitidine versus azacitidine in patients with higher-risk myelodysplastic syndromes (MDS)/chronic myelomonocytic leukemia (CMML) or low-blast acute myelogenous leukemia (LB-AML) (NCT02610777): subset analysis in higher-risk MDS. Cl Lymph Myelom Leuk. 2020;20:S323–4.
Zeidner JF, Mazerolle F, Bell JA, Cain LE, Faller DV, Dalal M, et al. Randomized phase 2 trial of pevonedistat plus azacitidine versus azacitidine in higher-risk myelodysplastic syndromes/chronic myelomonocytic leukemia or low-blast acute myeloid leukemia: exploratory analysis of patient-reported outcomes. Blood. 2020;136:39–40.
Smith BN, Cojocari D, Purkal JJ, Arrate M, Ramsey HE, Leverson JD, et al. Pevonedistat, a Nedd-8 activating enzyme inhibitor, upregulates NOXA to increase effectiveness of azacitidine and venetoclax in preclinical models of acute myelogenous leukemia. Blood. 2019;134:1380.
Cojocari D, Smith BN, Purkal JJ, Arrate MP, Huska JD, Xiao Y, et al. Pevonedistat and azacitidine upregulate NOXA (PMAIP1) to increase sensitivity to venetoclax in preclinical models of acute myeloid leukemia. Haematologica. 2021;107:825–35.
Swords RT, Savona MR, Maris MB, Erba HP, Berdeja JG, Foran JM, et al. Pevonedistat (MLN4924), an investigational, first-in-class NAE inhibitor, in combination with azacitidine in elderly patients with acute myeloid leukemia (AML) considered unfit for conventional chemotherapy: updated results from the phase 1 C15009 trial. Blood. 2014;124:2313.
Bauer TM, Harvey RD, Lee CB, Aggarwal C, Cohen RB, Sedarati F, et al. Investigational NEDD8-activating enzyme inhibitor pevonedistat (Pev) plus chemotherapy in patients (Pts) with solid tumors (Phase 1b study): Antitumor activity of pev plus carboplatin (Carbo)/paclitaxel (Pac). J Clin Oncol. 2016;34:2580.
Lockhart AC, Bauer TM, Aggarwal C, Lee CB, Harvey RD, Cohen RB, et al. Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Investig New Drugs. 2019;37:87–97.
Zhou X, Richardson DL, Dowlati A, Goel S, Sahebjam S, Strauss J, et al. Effect of pevonedistat, an investigational NEDD8-activating enzyme inhibitor, on the QTc interval in patients with advanced solid tumors. Clin Pharmacol Drug Dev. 2023;12:257–66.
Zhou X, Vaishampayan U, Mahalingam D, Harvey RD, Chung KY, Sedarati F, et al. Phase 1 study to evaluate the effects of rifampin on pharmacokinetics of pevonedistat, a NEDD8-activating enzyme inhibitor in patients with advanced solid tumors. Investig New Drugs. 2022;40:1042–50.
Foster J, Reid JM, Minard CG, Isikwei E, Liu X, Berg SL, et al. Phase 1 study of pevonedistat (MLN4924) a NEDD8 activating enzyme inhibitor, in combination with temozolomide (TMZ) and irinotecan (IRN) in pediatric patients with recurrent or refractory solid tumors (ADVL1615). J Clin Oncol. 2021;39:10019.
Sekeres MA, Fram RJ, Hua Z, Ades L. Phase 3 study of first line pevonedistat (PEV) + azacitidine (AZA) versus single-agent AZA in patients with higher-risk myelodysplastic syndromes (HR MDS), chronic myelomonocytic leukemia (CMML) or low-blast acute myelogenous leukemia (AML). J Clin Oncol. 2018;36:TPS7077.
Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, et al. Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol. 2015;169:534–43.
Paraiso KHT, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–60.
Du M, Peng Z, Gai W, Liu F, Liu W, Chen Y, et al. The absence of PTEN in breast cancer is a driver of MLN4924 resistance. Front Cell Dev Biol. 2021;9:667435.
Zhou X, Tan M, Nyati MK, Zhao Y, Wang G, Sun Y. Blockage of neddylation modification stimulates tumor sphere formation in vitro and stem cell differentiation and wound healing in vivo. Proc Natl Acad Sci USA. 2016;113:E2935–44.
Mao H, Tang Z, Li H, Sun B, Tan M, Fan S, et al. Neddylation inhibitor MLN4924 suppresses cilia formation by modulating AKT1. Protein Cell. 2019;10:726–44.
Keller KE, Tan IS, Lee YS. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science. 2012;338:1069–72.
Zhou Q, Sun Y. MLN4924: additional activities beyond neddylation inhibition. Mol Cell Oncol. 2019;6:e1618174.
Hollebecque A, Argiles G, Andre T, Cervantes A, Leger C, Valette A, et al. A phase I dose-escalation of trifluridine/tipiracil in combination with oxaliplatin in metastatic colorectal cancer. J Clin Oncol. 2017;35:3626.
Kostine M, Mauric E, Tison A, Barnetche T, Barre A, Nikolski M, et al. Baseline co-medications may alter the anti-tumoural effect of checkpoint inhibitors as well as the risk of immune-related adverse events. Eur J Cancer. 2021;157:474–84.
He S, Dong G, Wu S, Fang K, Miao Z, Wang W, et al. Small molecules simultaneously inhibiting p53-murine double minute 2 (MDM2) interaction and histone deacetylases (HDACs): discovery of novel multitargeting antitumor agents. J Med Chem. 2018;61:7245–60.
Da C, Zhang D, Stashko M, Vasileiadi E, Parker RE, Minson KA, et al. Data-driven construction of antitumor agents with controlled polypharmacology. J Am Chem Soc. 2019;141:15700–9.
Siu L, Brody J, Gupta S, Marabelle A, Jimeno A, Munster P, et al. Safety and clinical activity of intratumoral MEDI9197 alone and in combination with durvalumab and/or palliative radiation therapy in patients with advanced solid tumors. J Immunother Cancer. 2020;8:e001095.
Dogan I, Iribas A, Ekenel M, Basaran M. Efficacy of vincristine, irinotecan, and temozolomide (VIT) combination in adult patients with metastatic Ewing sarcoma. J Clin Oncol. 2021;39:e23510.
Nikanjam M, Liu S, Kurzrock R. Dosing de novo two-drug combinations based on 32,894 patients in phase I–III clinical trials. J Clin Oncol. 2016;34:2563.
Li K, Liu W, Zhao Q, Wu C, Fan C, Lai H, et al. Combination of tanshinone IIA and doxorubicin possesses synergism and attenuation effects on doxorubicin in the treatment of breast cancer. Phytother Res. 2019;33:1658–69.
Salaroglio IC, Belisario DC, Bironzo P, Ananthanarayanan P, Ricci L, Digiovanni S, et al. SKP2 drives the sensitivity to neddylation inhibitors and cisplatin in malignant pleural mesothelioma. J Exp Clin Cancer Res. 2022;41:75.
Megger DA, Abou-Eid S, Zülch B, Sitek B. Systematic analysis of synergistic proteome modulations in a drug combination of cisplatin and MLN4924. Mol Omics. 2018;14:450–7.
Nawrocki ST, Kelly KR, Smith PG, Keaton M, Carraway H, Sekeres MA, et al. The NEDD8-activating enzyme inhibitor MLN4924 disrupts nucleotide metabolism and augments the efficacy of cytarabine. Clin Cancer Res. 2015;21:439–47.
Czuczman NM, Barth MJ, Dwar R, Mavis C, Klener P, Czuczman MS, et al. Evaluation of the anti-tumor activity of MLN4924, a novel NEDD8 activating enzyme inhibitor, in pre-clinical models of rituximab chemotherapy-sensitive or -resistant B-cell lymphoma. Blood. 2012;120:2761.
Zhou L, Chen S, Zhang Y, Kmieciak M, Leng Y, Li L, et al. The NAE inhibitor pevonedistat interacts with the HDAC inhibitor belinostat to target AML cells by disrupting the DDR. Blood. 2016;127:2219–30.
Bhalla KN, Fiskus W. NEDD8 and HDACs: promising cotargets in AML. Blood. 2016;127:2168–70.
Xiong S, Huang W, Liu X, Chen Q, Ding Y, Huang H, et al. Celecoxib synergistically enhances MLN4924-induced cytotoxicity and EMT inhibition Via AKT and ERK pathways in human urothelial carcinoma. Cell Transplant. 2022;31:09636897221077921.
Li J, Song C, Rong Y, Kuang T, Wang D, Xu X, et al. Chk1 inhibitor SCH 900776 enhances the antitumor activity of MLN4924 on pancreatic cancer. Cell Cycle. 2018;17:191–9.
Knorr KLB, Schneider PA, Meng XW, Dai H, Smith BD, Hess AD, et al. MLN4924 induces Noxa upregulation in acute myelogenous leukemia and synergizes with Bcl-2 inhibitors. Cell Death Differ. 2015;22:2133–42.
Sumi H, Inazuka M, Morimoto M, Hibino R, Hashimoto K, Ishikawa T, et al. An inhibitor of apoptosis protein antagonist T-3256336 potentiates the antitumor efficacy of the Nedd8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924). Biochem Biophys Res Commun. 2016;480:380–6.
Cooper J, Xu Q, Zhou L, Pavlovic M, Ojeda V, Moulick K, et al. Combined inhibition of NEDD8-activating enzyme and mTOR suppresses NF2 loss-driven tumorigenesis. Mol Cancer Ther. 2017;16:1693–704.
Ishikawa Y, Nakayama K, Morimoto M, Mizutani A, Nakayama A, Toyoshima K, et al. Synergistic anti-AML effects of the LSD1 inhibitor T-3775440 and the NEDD8-activating enzyme inhibitor pevonedistat via transdifferentiation and DNA rereplication. Oncogenesis. 2017;6:e377.
Zhang S, You X, Xu T, Chen Q, Li H, Dou L, et al. PD-L1 induction via the MEK-JNK-AP1 axis by a neddylation inhibitor promotes cancer-associated immunosuppression. Cell Death Dis. 2022;13:844.
Liang T, Lu L, Song X, Qi J, Wang J. Combination of microtubule targeting agents with other antineoplastics for cancer treatment. Biochim Biophys Acta Rev Cancer. 2022;1877:188777.
Liu T, Wan Y, Xiao Y, Xia C, Duan G. Dual-target inhibitors based on HDACs: novel antitumor agents for cancer therapy. J Med Chem. 2020;63:8977–9002.
Arnst KE, Banerjee S, Chen H, Deng S, Hwang D-J, Li W, et al. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med Res Rev. 2019;39:1398–426.
Fu D-J, Song J, Zhu T, Pang X-J, Wang S-H, Zhang Y-B, et al. Discovery of novel tertiary amide derivatives as NEDDylation pathway activators to inhibit the tumor progression in vitro and in vivo. Eur J Med Chem. 2020;192:112153.
Song J, Liu Y, Yuan X-Y, Liu W-B, Li Y-R, Yu G-X, et al. Discovery of 1,2,4-triazine dithiocarbamate derivatives as NEDDylation agonists to inhibit gastric cancers. Eur J Med Chem. 2021;225:113801.
Liang Q, Liu M, Li J, Tong R, Hu Y, Bai L, et al. NAE modulators: a potential therapy for gastric carcinoma. Eur J Med Chem. 2022;231:114156.
Li X, Pham V, Tippin M, Fu D, Rendon R, Song L, et al. Flavokawain B targets protein neddylation for enhancing the anti-prostate cancer effect of Bortezomib via Skp2 degradation. Cell Commun Signal. 2019;17:25.
Pham V, Rendon R, Le VX, Tippin M, Fu D-J, Le TH, et al. Gartanin is a novel NEDDylation inhibitor for induction of Skp2 degradation, FBXW2 expression, and autophagy. Mol Carcinog. 2020;59:193–201.
Leung C-H, Chan DS-H, Yang H, Abagyan R, Lee SM-Y, Zhu G-Y, et al. A natural product-like inhibitor of NEDD8-activating enzyme. Chem Commun. 2011;47:2511–3.
Zhong H-J, Pui-Yan Ma V, Cheng Z, Shiu-Hin Chan D, He H-Z, Leung K-H, et al. Discovery of a natural product inhibitor targeting protein neddylation by structure-based virtual screening. Biochimie. 2012;94:2457–60.
Ni S, Chen X, Yu Q, Xu Y, Hu Z, Zhang J, et al. Discovery of candesartan cilexetic as a novel neddylation inhibitor for suppressing tumor growth. Eur J Med Chem. 2020;185:111848.
Zhong H-J, Liu L-J, Chan DS-H, Wang H-M, Chan PWH, Ma D-L, et al. Structure-based repurposing of FDA-approved drugs as inhibitors of NEDD8-activating enzyme. Biochimie. 2014;102:211–5.
Ma H, Zhuang C, Xu X, Li J, Wang J, Min X, et al. Discovery of benzothiazole derivatives as novel non-sulfamide NEDD8 activating enzyme inhibitors by target-based virtual screening. Eur J Med Chem. 2017;133:174–83.
Lu P, Liu X, Yuan X, He M, Wang Y, Zhang Q, et al. Discovery of a novel NEDD8 activating enzyme inhibitor with piperidin-4-amine scaffold by structure-based virtual screening. ACS Chem Biol. 2016;11:1901–7.
Lu P, Guo Y, Zhu L, Xia Y, Zhong Y, Wang Y. A novel NAE/UAE dual inhibitor LP0040 blocks neddylation and ubiquitination leading to growth inhibition and apoptosis of cancer cells. Eur J Med Chem. 2018;154:294–304.
Fu D-J, Cui X-X, Zhu T, Zhang Y-B, Hu Y-Y, Zhang L-R, et al. Discovery of novel indole derivatives that inhibit NEDDylation and MAPK pathways against gastric cancer MGC803 cells. Bioorg Chem. 2021;107:104634.
Hao R, Song Y, Li R, Wu Y, Yang X, Li X, et al. MLN4924 protects against interleukin-17A-induced pulmonary inflammation by disrupting ACT1-mediated signaling. Am J Physiol Lung Cell Mol Physiol. 2019;316:L1070–80.
Yu H, Luo H, Chang L, Wang S, Geng X, Kang L, et al. The NEDD8-activating enzyme inhibitor MLN4924 reduces ischemic brain injury in mice. Proc Natl Acad Sci USA. 2022;119:e2111896119.
Zhang J, Cui J, Zhao F, Yang L, Xu X, Shi Y, et al. Cardioprotective effect of MLN4924 on ameliorating autophagic flux impairment in myocardial ischemia-reperfusion injury by Sirt1. Redox Biol. 2021;46:102114.
Andérica-Romero AC, Hernández-Damián J, Vázquez-Cervantes GI, Torres I, Pedraza-Chaverri J. The MLN4924 inhibitor exerts a neuroprotective effect against oxidative stress injury via Nrf2 protein accumulation. Redox Biol. 2016;8:341–7.
Xie L, Ji X, Tu Y, Wang K, Zhu L, Zeng X, et al. MLN4924 inhibits hedgehog signaling pathway and activates autophagy to alleviate mouse laser-induced choroidal neovascularization lesion. Biomed Pharmacother. 2020;130:110654.
Acknowledgements
Not applicable.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities [Grant No. 2022-JYB-XJSJJ025 to Dong-Jun Fu]. This work was also funded by China Postdoctoral Science Foundation [Grant No. 2020M670239 to Dong-Jun Fu].
Author information
Authors and Affiliations
Contributions
D-JF collected the references, designed the outline, prepared all figures and wrote the manuscript. TW offered important guidance of manuscript reviewing and editing. D-JF and TW approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fu, DJ., Wang, T. Targeting NEDD8-activating enzyme for cancer therapy: developments, clinical trials, challenges and future research directions. J Hematol Oncol 16, 87 (2023). https://doi.org/10.1186/s13045-023-01485-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-023-01485-7