
Abstract
Eph receptors and the corresponding Eph receptor-interacting (ephrin) ligands jointly constitute a critical cell signaling network that has multiple functions. The tyrosine kinase EphA2, which belongs to the family of Eph receptors, is highly produced in tumor tissues, while found at relatively low levels in most normal adult tissues, indicating its potential application in cancer treatment. After 30 years of investigation, a large amount of data regarding EphA2 functions have been compiled. Meanwhile, several compounds targeting EphA2 have been evaluated and tested in clinical studies, albeit with limited clinical success. The present review briefly describes the contribution of EphA2-ephrin A1 signaling axis to carcinogenesis. In addition, the roles of EphA2 in resistance to molecular-targeted agents were examined. In particular, we focused on EphA2’s potential as a target for cancer treatment to provide insights into the application of EphA2 targeting in anticancer strategies. Overall, EphA2 represents a potential target for treating malignant tumors.
Similar content being viewed by others
Introduction
Ephrin receptors (Eph) represent the most important class of receptor tyrosine kinases (RTKs) [1]. EphA1, the firstly described Eph receptor, was identified in liver cancer cells while screening for RTKs in 1987 [2]. Nowadays, there are 14 Eph receptors and 8 related ligands (ephrins) [3]. Eph receptor signaling contributes to multiple biological events, mostly causing cell-cell repulsion or adhesion. Therefore, Eph receptors and the corresponding ligands have essential functions in tissue patterning, neuronal targeting, and blood vessel development in the embryo [4, 5]. Meanwhile, Eph proteins are found in high levels in multiple malignancies, with such overexpression significantly contributing to carcinogenesis [6].
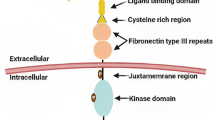
Eph receptors are single transmembrane proteins with extra- (N-terminal) and intracellular domains with ligand-binding and intrinsic enzymatic activities, respectively [7, 8]. Eph receptors are grouped into A and B categories according to their extracellular domains, which determine the binding affinity for ligands (Eph receptor-interacting proteins or ephrins) [9, 10]. Nine EphA and five EphB receptors are found in humans [11]. The ligands for Eph receptors, ephrins, are anchored to the cell membrane; they also comprise two subcategories, including ephrin A (ephrin A1-5) and ephrin B (ephrin B1-3) [12, 13].
Some Eph receptors, especially EphA2, attract increasing attention because of demonstrated or hypothesized contributions to modulatory processes controlling carcinogenesis and tumor progression (Fig. 1). The present manuscript reviewed the clinical associations and biological and cellular consequences of EphA2 overexpression in cancer. Potential opportunities for therapeutic intervention based on EphA2 targeting are particularly discussed.

Historical development and breakthroughs in targeting EphA2 in cancer
EphA2-ephrin A1 signaling
The EphA2 receptor is a 130-kDa transmembrane glycoprotein with 976 amino acids [14]. The EphA2 gene in humans is found on chromosome 1p36. Its initial detection occurred in 1990 while screening a HeLa cell cDNA library comprising degenerate oligonucleotides engineered to interact with highly conserved domains of tyrosine kinases [12]. EphA2 was originally termed epithelial cell kinase (eck) since it was detected in most epithelial cells.
EphA2 interacts with any of the eight different ephrin A-family ligands, with overt preference to ephrin A1 [13, 15]. Ephrin A1 represents a GPI-anchored protein containing 205 amino acids (apparent molecular weight, 22 kDa) [16]. The human ephrin A1 gene is located on 1q21-q22. This TNF-α early-inducible gene product was firstly described in human umbilical vein endothelial cells (HUVECs) three decades ago [17], and shown to bind EphA in 1994 [18]. Ephrin A1’s expression pattern in cancer seems to differ from that of EphA2, with attenuation in a variety of aggressive tumors, particularly those overexpressing EphA2 [16].
Under normal conditions, EphA2 interacts with ephrin A1 on the neighboring cell and induce diverse signaling networks following cell-to-cell contact. As membrane proteins, ephrins are engaged in both forward (termed ephrin:EphA2 forward) and reverse (called EphA2:ephrin reverse) signaling from ephrin ligands to EphA2 and vice versa; this is also known as ephrin-EphA2 bidirectional signaling [19, 20]. Forward signaling is often cell repulsive and promotes EphA2 oligomerization and phosphorylation, therefore enhancing kinase activity. The immediate biological consequences of EphA2 phosphorylation include decreased cell–extracellular matrix (ECM) attachment. Ephrin A1-associated EphA2 induction inhibits focal adhesion kinase (FAK), extracellular regulated protein kinases (ERK), and Akt phosphorylation to regulate motility, viability, and proliferation in multiple malignant cell lines [7, 21], whereas reverse signaling is more likely to be adhesive and is generally considered as kinase-independent, due to lacking enzyme activity in ephrin A1. However, the reverse signaling by ephrin A1 is largely poorly understood. In addition, EphA2 possesses ligand-independent kinase activity in cultured cancer cells, which might partially explain its malignant effects in the non-phosphorylated state [22, 23]. Actually, EphA2-ephrin A1 interaction or EphA2 ligand-independent kinase activity likely functions through multiple factors acting jointly, e.g., cell type and the microenvironment. Altogether, the EphA2-ephrin A1 signaling regulates multiple cellular processes (proliferation, survival, migration, morphology, cell-to-cell repulsion, and adhesion) in embryonic development, angiogenesis, and tumorigenesis [11] (Fig. 2).

Expression and biological pathways linked with EphA2. The interaction of cell-membrane-bound EphA2 with ephrin A1 induces forward or reverse signals in the corresponding cells. Under normal conditions, cell–cell contacts allow EphA2 to interact with ephrin A1, which induces EphA2 phosphorylation and activates its downstream signaling. Tyrosine phosphorylation of EphA2 promotes the generation of a complex with c-Cbl, subsequently induces EphA2 degradation. This leads to suppression of ECM attachment, cell proliferation, cell migration, and angiogenesis. In the malignant state, loss of cell–cell contacts induces receptor-ligand interaction and degradation of EphA2. In addition, tyrosine phosphorylation of EphA2 could be rapidly reversed by the phosphatase LMW-PTP, further leading to the overexpression and accumulation of unphosphorylated form of EphA2. This leads to promotion of ECM attachment, cell proliferation, cell migration, and angiogenesis
EphA2 in cancer
Different from the majority of Eph kinases that are mostly synthesized during the developmental process, EphA2 is mainly restricted to proliferating epithelial cells in adults [12]. EphA2 expression in the adult occurs in normal tissues only when they have highly proliferating epithelial cells [1], where its importance and function are not well understood. However, an accumulating body of evidence suggests human EphA2 is abundantly expressed in diverse cancers such as prostate [24], lung [25], esophageal [26], colorectal [27], cervical [28], ovarian [29], and breast [30] and skin cancers [31]. EphA2 is upregulated at the gene and protein levels in human tumor tissue specimens and established cancer cell lines [9, 16]. In particular, most elevated EphA2 expression is consistently detected in cells with highest malignancy [16]. In addition, EphA2 expression has associations with poor prognosis, elevated metastatic potential, and reduced survival of tumor patients [32, 33]. Moreover, EphA2 is not simply a biomarker of malignant character, but also an active participant in malignant progression [26, 28]. Consequently, EphA2’s expression patterns and functional relevance in malignancies make this protein an attractive therapeutic target in cancer.
There is considerable interest in the mechanisms that govern EphA2 expression and in understanding how these mechanisms are subverted in cancer. Emerging evidence links high EphA2 protein amounts with EphA2 regulation at the mRNA level as well as protein stability, although the precise mechanisms governing EphA2 upregulation in cancer remain largely undefined [16, 34].
EphA2 mRNA is tightly regulated. To date, a few somatic mutations of EphA2 have been reported [35,36,37]. In addition, EphA2 amplification detected in only a low percentage of cases (1 in 33 pancreatic cancer samples) [38]. The EphA2 promoter comprises DNA damage-responsive p53-binding sites, and this receptor is upregulated by ultravioletray (UV) treatment [39]. EphA2 is overexpressed in Ras-transformed cells and transgenic mice overexpressing Ras, suggesting EphA2 as a direct transcriptional target of rat sarcoma (Ras)–(rapidly accelerated fibrosarcoma) Raf–ERK signaling [22, 40]. EphA2 gene expression is also reduced by multiple stimuli such as signaling by the c-Myc and estrogen receptor [41]. These observations are intriguing given that EphA2 consistently shows highest expression in breast tumor cells with most pronounced aggressiveness and no expression of estrogen receptor (ER)-α [41, 42]. Thus, it is tempting to speculate that EphA2 overexpression in breast cancer might be linked to the loss of hormone dependence that frequently arises in advanced stages of the disease.
Decreased ligand-mediated receptor internalization and degradation, consequently enhancing protein stability, might help increase EphA2 amounts in malignant cells. An interesting consequence of EphA2 stimulation (by ligand or antibody) is EphA2 phosphorylation, internalization, and degradation [43,44,45]. After ligand-dependent induction, EphA2 aggregation occurs at the cell surface, followed by tyrosine phosphorylation, promoting the generation of a complex with c-Cbl, which is internalized into early endosomes for subsequent EphA2 degradation [46]. Studies have shown that c-Cbl overexpression decreases the levels of the EphA2 protein, likely by enhancing protein degradation. Tyrosine phosphorylation of EphA2 could also be rapidly reversed by low-molecular-weight protein phosphatase (LMW-PTP), a phosphatase binding to and dephosphorylating EphA2 [47]. Increased LMW-PTP expression functions to reduce EphA2 phosphotyrosine content, contributing to elevated EphA2 levels in cancer cells. Despite EphA2 overexpression in cancer, phosphorylated EphA2 is found in lower amounts in cancer cells in comparison with non-transformed epithelial cells [42]. Unlike many other receptor tyrosine kinases, the enzymatic activity of EphA2 does not depend on ligand interaction or receptor autophosphorylation [34, 39, 42]. It is considered that deficient cell-to-cell contact (commonly found in malignant cells) and insufficient levels of ephrin A1 on cancer cells reduce EphA2 phosphorylation [48].
Targeting EphA2 in cancer
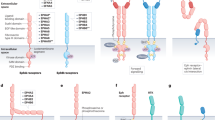
Overexpression and aggressive features of EphA2 in tumor cells and relatively low expression in most normal adult tissues make this protein a potential therapeutic target in cancer. The EphA2/ephrin A1 system could be targeted for cancer treatment at least via two mechanisms. First, EphA2’s oncogenic features could be inhibited, e.g., decreasing EphA2 expression, promoting EphA2 degradation, and blocking endogenous EphA2 activation. Alternatively, the EphA2 receptor could be employed to deliver therapeutics (exogenous drugs or endogenous immune cells) to cancer cells and associated vessels. Therapies targeting EphA2 in cancer are shown in Table 1 and Fig. 3.

Targeting EphA2 in cancer. EphA2’s expression patterns and functional relevance in malignancies make this protein an attractive therapeutic target in cancer. Accordingly, EphA2 overexpression has been targeted with several approaches such as decrease EPHA2 expression, promote EphA2 degradation, block endogenous EphA2 activation, EphA2 as drug delivery target, EphA2-based immunotherapy, and EphA2-based combination therapeutics
Inhibiting EphA2 expression
Given the positive association of EphA2 overexpression with aggressive clinical and pathological features in human cancers, investigators have examined the potential of downregulating EphA2 in preclinical models. Short interfering RNAs (siRNAs) for gene knockdown constitute a great tool for protein function assessment, gene discovery, and drug development [83, 84], and have been applied to silence EphA2 in human cancer cells. For example, in pancreatic adenocarcinoma-derived cells, sequence-specific siRNA targeting EphA2 suppresses EphA2 expression, retarding tumor growth in a nude mouse xenograft model [85]. In addition, treatment with EphA2-specific siRNA significantly reduces malignancy in glioma [86], non-small cell lung cancer (NSCLC) [70], and breast cancer cells [87]. However, despite the great success in in vitro knockdown, in vivo siRNA delivery is challenging [88, 89]. As a result, efficient and biocompatible delivery systems for systemic siRNA administration have been evaluated. For instance, EPHARNA, the 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) nanoliposomal EphA2-targeted therapeutic, has been developed [49]. In the nude mouse model administered ovarian tumors intraperitoneally, EPHARNA was shown to be taken up by the tumor, reducing EphA2 levels in the animals 48 h following single treatment [49]. This finding indicates that treatment with EPHARNA reduces tumor growth in the ovarian cancer mouse xenograft model. In addition, both signal dosing and multi-dosing of EPHARNA have an excellent safety profile in many mammalian species, including non-human primates [90].
Promoting EphA2 degradation
Artificial ligands or antibodies interacting with EphA2 could suppress signaling by promoting internalization and degradation.
Soluble ephrin A1 and ephrin A1-Fc
Plasma membrane–bound ephrins and soluble ephrins with artificial clustering/dimerization associated with antibodies targeting COOH-terminal epitope tags or fusion to immunoglobulin g (IgG) Fc potently promote EphA2 phosphorylation and degradation [13].
Ephrin A1 has been demonstrated to be present at low levels and to possess tumor suppressing properties dependent on cell-to-cell contact in a variety of tumors [6, 30, 91]. Transfection with full-length human ephrin A1 into glioblastoma multiforme (GBM) cells exhibits a dramatic suppression of EphA2 and inhibits multiple malignant features, including impaired anchorage-independent growth, proliferation, and migration [50]. Of great interest, ephrin A1 was shown to be released as a soluble monomeric entity by GBM and breast cancer cells. This soluble ephrin A1 could function in a paracrine manner, induce EphA2 internalization and downregulation, elicit substantial alterations of cell morphology, and inhibit cell migration in treated GBM cells, in a juxtacrine interaction-independent manner [50]. Treatment with ephrin A1-conditioned media abolishes the phosphorylation of ERK induced by empty vector-conditioned media, which might contain growth factors. Moreover, treatment with a fusion protein of monomeric ephrin A1 (mEA1) also induced phosphorylation and degradation of in human breast cancer cells [51]. Thus, ephrin A1-associated tumor suppression might result from EphA2 downregulation as well as direct signaling through EphA2.
In addition to soluble ephrin A1, ephrin A1-Fc, obtained by fusing recombinant Ephrin A1 to human IgG Fc for dimerization, shows ephrin-like features and induces EphA2 phosphorylation [52]. Treatment with ephrin A1-Fc resulted in reduced amounts of membrane-associated EphA2 and inhibited cellular motility and invasion in pancreatic ductal adenocarcinoma cells [52]. In addition, proteasomal degradation was demonstrated to play a critical role in ephrin A1-Fc-associated EphA2 catabolism as the proteasome suppressor MG132 markedly inhibits ephrin A1-Fc-related EphA2 degradation. Likewise, ephrin A1-Fc increases EphA2 phosphorylation, decreases EphA2 protein expression, and inhibits growth in gastric cancer cells [53]. Dimeric ephrin A1-Fc suppresses Ras-mitogen-activated protein kinase (MAPK) signaling to reduce growth factor-associated ERK phosphorylation [92,93,94,95].
EphA2 monoclonal antibody
The large extracellular domain of EphA2 provides an antigen that is frequently upregulated on tumor cells [10, 96]. In addition, ligand stimulation is sufficient to induce EphA2 degradation. These evidences suggest that EphA2 could elicit a particularly attractive monoclonal antibody, and antibodies that mimic the actions of ephrin A1 would be expected to function similarly as the ligand.
Studies have shown that several agonist monoclonal antibodies raised against EphA2 induce its internalization and degradation, suppressing its malignant features. For example, Kinch et al. [45] isolated antibodies from mice after immunization with the pcDNA3-ecdEphA2-Fc expression plasmid, and identified EA1.2 that dose-dependently elevated phosphotyrosine amounts in EphA2. These authors demonstrated that EA1.2 inhibits more than 60% of soft agar-formed colonies in breast cancer cells compared with vehicle-treated controls. These findings indicate that the growth-suppressive effects of EphA2-specific antibodies correlate with their capability of stimulating EphA2 autophosphorylation and degradation. Coffman et al. [54] demonstrated that two antibodies, including EA2 and B233, promote EphA2 phosphorylation and degradation in cancer cells. The antibody EA2 (6 mg/kg) administered i.p. was shown to significantly decrease breast and lung cancer cell growth in vivo relative to the matched isotype controls (IgG1, 1A7). Goldgur et al. [55] isolated and characterized the anti-EphA2 single-chain antibody D2 scFv, which was highly specific to EphA2 and blocked ligand interaction in COS-7 cells. Indeed, treatment with D2 scFv induced apoptosis and reduced cell proliferation in the lymphoma cell line. In addition, Sakamoto et al. [43] showed that one of the EphA2 mAbs produced, SHM16, interacts with an EphA2 epitope differing from that affecting ephrin A1 binding to EphA2. SHM16 was clearly internalized in cells and inhibited malignant features in melanoma cells. However, SHM16 showed no effects on ephrin A1 interaction with EphA2 on the cell surface, while recognizing a different EphA2 epitope. SHM16 was shown to be clearly internalized by A375 cells.
Antibody-dependent cellular cytotoxicity (ADCC) kills cells via perforin/granzyme, TRAIL, and FasL [97]. ADCC also affects adaptive tumor immunity, and its enhancement could remarkably alter the tumor microenvironment [98, 99]. DS-8895a, a newly developed humanized anti-EphA2 mAb afucosylated for ADCC enhancement, was generated by mouse immunization with recombinant human EphA2 and further humanized as human IgG1 [56]. Treatment with DS-8895a of EphA2-positive breast and gastric cancer cells was shown to partially inhibit ephrin A1-associated EphA2 phosphorylation. In agreement, treatment with DS-8895a inhibits tumor growth in EphA2-positive human breast and gastric cancer xenografts in mice [57]. Another EphA2 effector-enhanced agonist monoclonal antibody that exhibits ADCC activity is 3F2-3M [44]. 3F2-3M was obtained by fusing the mouse parental antibody B233 and the humanized antibody 3F2. 3F2-3M administration dose-dependently increased EphA2 phosphorylation in the breast cancer cell line, which was similar to that of the parental antibodies 3F2-WT and B233 [44]. 3F2-3M significantly inhibited ovarian, breast, and lung cancer cell lines, which were co-cultured with peripheral blood monocytes from a healthy donor. However, 3F2-3M was minimally toxic in the absence of NK cells. On the other hand, interaction with NKs was increased by 100–250-fold for 3F2-3M in comparison with 3F2-WT, with improved affinity to FcγRIIIa. Modifying the Fc portion of the EphA2 antibody resulted in enhanced interaction with FcγRIIIa. Administration of 3F2-3M significantly induced tumor growth inhibition in a breast cancer xenograft orthotopic model, compared with the isotype control antibody and phosphate buffer saline (PBS) groups.
Blocking EphA2 activation
Compounds binding to EphA2 or ephrin A1 could suppress signaling by direct antagonist effects.
Inhibiting Eph-ephrin interactions
Small molecules that block EphA2 could represent efficient alternatives to peptides and antibodies. Recently, small molecules disrupting the Eph-ephrin complex have been described, with most exerting pharmacological activities through targeting of the ligand-binding domain of EphA2, thereby acting as common protein-protein interaction (PPI) inhibitors. The ephrin binding site in Eph receptors allow high-affinity binding of small molecules [59, 100].
It was hypothesized that soluble receptors repress EphA signaling by suppressing the interactions of endogenous ephrins with EphA receptors. EphA2-Fc represents a soluble protein chimera involving the fusion of EphA2’s extracellular domain with human IgG1 Fc, preventing interactions of several ephrin A ligands with endogenous receptors and potently inhibiting EphA receptor activation in cultured cells. By interacting with ephrin A1, EphA2-Fc could induce ephrin-initiated reverse signaling. Treatment with EphA2-Fc was shown to dose-dependently inhibit EphA2 receptor phosphorylation and activity. In addition, EphA2-Fc strongly inhibited angiogenesis and microvessel growth in vitro as well as growth in pancreatic tumor xenografts [58]. Furthermore, soluble EphA2-Fc was demonstrated to inhibit endothelial cell migration upon 4 T1 mouse mammary adenocarcinoma tumor cell-induced angiogenesis in vitro. Moreover, EphA2-Fc inhibited 4 T1 tumor growth in vivo and reduced tumor vascular density and growth while increasing cell apoptosis.
Lithocholic acid (LCA, (3a,5b)-3-hydroxycholan-24-oic acid), a secondary bile acid produced by prokaryotic transformation of chenodeoxycholic acid, is considered an EphA2 antagonist. LCA interacts with the nuclear receptor farnesoid X receptor (FXR) and the G-protein-coupled receptor G-protein-coupled bile acid receptor 1 (GPBAR1, also called TGR5) under physiological conditions [101, 102]. Molecular modeling investigations revealed that LCA mimics ephrin A1 in interacting with EphA2 via insertion of its cyclopenta[a]-perhydrophenanthrene scaffold into the hydrophobic EphA2 receptor ligand-binding channel, generating a salt bridge involving Arg103 [60], an essential amino acid in ephrin A1 recognition [32]. LCA was shown to competitively and reversibly inhibit EphA2-ephrin A1 binding (Ki = 49 μM) without reducing EphA2’s kinase activity [59]. Further functional assays revealed that LCA inhibits EphA2 autophosphorylation and blocks ephrin A1-related prostate cancer cell cytotoxicity.
The specificity of LAC in antagonizing Eph receptor has been demonstrated, with no detected effects on other RTKs, including EGFR, vascular endothelial growth factor receptor (VEGFR), insulin-like growth factor 1 receptor (IGF-1R), and the insulin receptor. However, LCA is also considered to interact with EphA and EphB receptors, indicating an interaction with the highly conserved region of Eph receptor family members [59]. Thus, LCA has been used as a prototype for designing or identifying other PPIs. Amino acid conjugates of LCA were shown to effectively disrupt EphA2 binding to ephrin A1 and to suppress EphA2 phosphorylation in intact cells, thereby blunting malignancy. UniPR126 (N-(3a-hydroxy-5b-cholan-24-oyl)-L-tryptophan), a novel antagonist derived from LCA, inhibits EphA2 phosphorylation and angiogenesis in cultured cells, in the low micromolar range [61]. UniPR126 was shown to disrupt the EphA2-ephrin A1 complex and to inhibit EphA2 phosphorylation in prostate cancer cells at a level 6-fold higher (pIC50 = 4.89). UniPR129 (N-(3a-hydroxy-5b-cholan-24-oyl)-Lb-homotryptophan, the L-homo-Trp conjugate of LCA, another newly developed PPI based on the in silico model of the EphA2-UniPR126 complex, also disrupts EphA2-ephrin A1 interaction (IC50 = 945 nM; Ki = 370 nM) [62]. In agreement, UniPR129 was shown to inhibit ephrin A1-Fc-associated prostate cancer cell cytotoxicity and angiogenesis in vitro. In addition, both UniPR129 and UniPR126 reduce polygon formation, but UniPR129 (IC50 = 5.2 μM) was 4-fold more potent than UniPR126 (IC50 = 20.5 μM). IC50 values in inhibiting ephrin A1-related EphA2 phosphorylation were 5 and 12 μM, respectively, for UniPR129 and UniPR126. Furthermore, UniPR126 showed cytotoxicity in HUVECs, increasing lactic dehydrogenase (LDH) release, unlike UniPR129. Comparing efficacy for prostate cancer cell retraction, UniPR129 and UniPR126 had similar strengths, and were much more potent compared with LCA. Likewise, a series of L-Trp derivatives of LCA have been synthesized, and a compound (defined as compound 20) was identified as the most potent antagonist disrupting EphA2 binding to ephrin A1 [60]. This compound blocking EphA2 phosphorylation (IC50 = 12 μM) was 4–5 times more efficient compared with LCA (IC50 = 50 μM) in inhibiting prostate cancer cells. Treatment with compound 20 significantly reduced the percentage of retracted cells stimulated by ephrin A1-Fc. In addition, UniPR1331 (N-(3b-hydroxy-D5-cholen-24-oyl)-L tryptophan) was identified as the first orally bioavailable small molecule antagonizing the Eph-ephrin system [103]. UniPR1331 was obtained by conjugating L-tryptophan with the parent compound 3β-hydroxy-D5-cholenic acid, which serves as bioisostere analogues of LCA. The activity of UniPR1331 in blunting EphA2 binding to ephrin A1 (pIC50 = 5.45) was ten times increased compared with that of the parent 3β-hydroxy-D5-cholenic acid (pIC50 = 4.40), and barely stronger than LCA (pIC50 = 4.25). Administration of UniPR1331 was shown to inhibit GBM growth and to extend the time to progression in a subcutaneous xenograft model through inhibition of angiogenesis [63, 104]. Cholanic acid ((5b)-cholan-24-oic acid) is another molecule competitively inhibiting EphA2 binding to ephrin A1 with increased potency compared with LCA [64]. Cholanic acid has a specific and reversible interaction with EphA2’s ligand-binding domain, blocking EphA2 phosphorylation and prostate cancer cell cytotoxicity. In contrast to LCA (promiscuous binding), cholanic acid is more selective for EphA receptors. Cholanic acid inhibits Eph receptor phosphorylation at non-cytotoxic levels. It inhibits EphA2 activation by ephrins (IC50 = 12 μM) more effectively compared with LCA (IC50 = 46 μM) [64]. In addition, cholanic acid suppresses EphA2 phosphorylation via direct binding to the EphA2 kinase domain rather than inhibiting EphA2 kinase activity.
Besides LCA and its analogues, small molecules that interfere with the EphA2-ephrin A1 system comprise the following: (i) the FXR agonist GW4064 [65], a stilbene carboxylic acid, dose-dependently disrupts the EphA2-ephrin A1 complex (IC50 = 23 μM), inhibits EphA2 phosphorylation (IC50 = 31 μM) and blocks EphA2 activation in prostate cancer cells; (ii) the disalicylic acid-furanyl derivative 76D10 (5,5′-(5,5′-((1E,4E)-3-oxopenta-1,4-diene-1,5-diyl)bis(furan-5,2-diyl))bis(2-hydroxybenzoic acid) inhibits ephrin interaction with EphA2, reducing EphA2 phosphorylation stimulated by ephrin-A1 Fc and inhibiting EphA2-mediated cell retraction in prostate cancer cells [66].
Inhibiting kinase activity of EphA2
The successful development of specific RTK inhibitors has prompted subsequent efforts for identifying comparable targets. Unlike other anticancer approaches, targeted therapies are relatively less toxic. Multiple small molecule EphA2 inhibitors interacting with the intracellular kinase domain have been described.
Dasatinib (BMS-354825) represents an oral kinase inhibitor simultaneously targeting breakpoint cluster region-Abelson (BCR-ABL), c-KIT, platelet-derived growth factor receptor (PDGFR), and SFKs [105, 106]. Its anticancer features have been demonstrated in early- and late-phase clinical studies of chronic myelogenous leukemia (CML). A variety of studies have demonstrated that dasatinib directly reduces EphA2 phosphorylation and kinase activity [67, 68, 107]. However, the promiscuous targeting profile of dasatinib makes data interpretation ambiguous. Dasatinib has also been recently used as a lead structure for developing EPHA2-inhibitors with ameliorated targeting profiles. The novel EphA2 inhibitor candidate 4a based on dasatinib was shown to feature an ameliorated selectivity profile while maintaining potent inhibitory effects against EphA2 as well as cytotoxic properties in glioblastoma cells [69].
ALW-II-41-27, a type II small molecule inhibitor targeting the ATP-binding region of the kinase domain as well as an allosteric site following the “DFG” motif in EphA2, has been shown to bind to and potently inhibit EphA2 kinase activity [70, 108]. Treatment with ALW-II-41-27 inhibits EphA2 kinase activity in NSCLC cells (IC50 = 11 nM) and suppresses cell survival and proliferation, while inducing cell apoptosis in vitro [70]. In the NSCLC xenograft model, oral treatment with ALW-II-41-27 revealed a relatively poor pharmacokinetic profile and low oral bioavailability. Mice treated with ALW-II-41-27 intraperitoneally showed significantly inhibited tumor growth. In addition, an in vivo study confirmed ALW-II-41-27 specificity for EphA2 among Eph receptors, although significant interactions were detected with multiple intracellular kinases such as Abelson (ABL), p38 MAPK, and many steroid receptor coactivator (SRC)-family kinases. ALW-II-41-27 was also shown to decrease both survival and proliferation in cultured erlotinib-resistant lung cancer cells, inhibiting tumor growth in mouse xenografts [25]. Furthermore, no statistical difference in body weight was detected, and no significant histopathologic differences were found in the heart, liver, or kidney tissue in mice treated with ALW-II-41-27 [70].
EphA2 as drug delivery target
Peptides and antibodies selectively binding cancer cells followed by internalization provide a powerful vehicle to guide therapeutic delivery to specific cell types and to determine the format of peptide/antibody-drug conjugates or antibody-directed nanotherapeutics. These conjugates or targeted systems could deliver toxic compounds selectively to the tumors while sparing noncancerous tissues. For example, EphA2 could selectively deliver therapeutics to EphA2-overexpressing cancers while simultaneously regulating EphA2-signaling related events.
Peptide/antibody-drug conjugates
Ephrin A1-Fc has been utilized in EphA2 targeted therapy upon conjugation with PE38QQR, a derivative of Pseudomonas aeruginosa endotoxin A, to generate ephrin A1–PE38QQR conjugated cytotoxin [71]. Pseudomonas endotoxin A is a bacterial toxin with high cytotoxicity in eukaryotic cells; it can be genetically modified by replacing the natural eukaryotic cell receptor binding domain by a tumor-specific target antibody or ligand [109, 110]. It was shown that EphA2 protein levels are significantly decreased following ephrin A1–PE38QQR administration in glioblastoma cells [71]. Unsurprisingly, ephrin A1–PE38QQR was cytotoxic to glioblastoma, breast cancer, and prostate cancer cells that overexpress EphA2.
1C1 is a fully human monoclonal antibody with selective binding to EphA2, but no other Eph receptor family member [72]. After cell binding in prostate cancer cells, 1C1 rapidly induces tyrosine phosphorylation, internalization, and degradation of EphA2. However, 1C1 does not demonstrate direct cytotoxicity and antitumor effects, but allows highly toxic chemotherapeutics to be directly and specifically delivered to EphA2-expressing tumor cells. The EphA2 immunoconjugate MEDI-547 (1C1-mcMMAF) was generated by conjugating 1C1 with the chemotherapeutic drug monomethyl auristatin phenylalanine (MMAF) via the non-cleavable linker maleimidocaproyl (mc) [72]. MEDI-547 was shown to interact with EphA2 via the highly conserved extracellular domain with similar binding affinity observed for 1C1, with internalization in EphA2-expressing tumor cells and subsequent reduction of EphA2 protein levels. In vitro experiments revealed that MEDI-547 decreases viability and increases apoptosis in ovarian, endometrial carcinoma cells in an EphA2-specific fashion [73, 74]. Indeed, administration of MEDI-547 significantly reduced tumor growth with minimal adverse effects in mice and rats (evaluated by body weight lose). In addition, mice treated with MEDI-547 showed decreased rate of distant metastasis.
Antibody-directed nanotherapeutics
Off-target drug toxicity frequently causes treatment discontinuation, restricted dose escalation, and worse outcomes. Antibody-mediated tumor targeting and nanoparticle encapsulation decrease the toxic effects of anticancer agents, improving treatment efficacy. Trametinib (TMB) is a MAP/ERK kinase (MEK) inhibitor, but its off-target toxicities frequently prompt dose interruption as well as treatment discontinuation [111]. Thus, YTPL, an ephrin A1-mimicking peptide (YSA; amino acid sequence: YSAYPDSVPMMS) with high stability [112], has been anchored on TMB-loaded PEGylated nanoliposomes [75]. YTPL was shown to display elevated cell internalization in comparison with non-targeted nanoliposomes (TPL) due to receptor-associated uptake. Due to elevated EphA2 amounts in vemurafenib-resistant cells in comparison with parent cells, YTPL shows higher intracellular uptake in the former cells. In addition, TMB was confirmed to be released upon TPL internalization in tumor cells. Such a delivery approach markedly reduces the amounts of circulating free TMB, consequently minimizing undesirable effects. Likewise, MM-310 (EphA2-ILs-DTXp) are immunoliposomes encapsulating the readily hydrolysable docetaxel prodrug (DTXp) with conjugation to the high-affinity signal-chain variable fragment (scFv-3) targeting EphA2 [76, 113]. Administration of MM-310 was shown to remarkably enhance anticancer activity in multiple mouse tumor xenografts (from breast, prostate, gastric, and esophageal cancer cells), in comparison with the free docetaxel and non-targeted nanotherapeutic control groups [76]. Moreover, pharmacokinetic analysis revealed that the AUC of docetaxel was increased by 15-fold. Delivery via MM-310 resulted in slow and sustained release of DTXp, decreased circulatory amounts of active docetaxel, and significantly reduced hematologic toxicity in comparison with docetaxel. Administration of MM-310 maintained adequate drug levels in tumors. These findings suggest that MM-310 has improved pharmacokinetic features, with reduced plasma docetaxel and selective tumor exposure, resulting in ameliorated toxicity profile and augmented anticancer effects. Based on these findings, a phase 1 clinical trial was initiated for evaluating the effectiveness of MM-310 in many solid tumors (ClinicalTrials.gov: NCT03076372).
EphA2-based immunotherapy
Immunotherapy, which relies on enhancing the patient’s immune defenses to combat tumor cells, has become a game changer in cancer treatment. EphA2 overexpression on tumor cells could constitute a new antigen for tumor immunotherapy.
Vaccines
Dendritic cell (DC)-based vaccines represent attractive anticancer tools, since DCs induce both tumor antigen-specific cytotoxic T lymphocytes (CTLs) and helper T cells [114]. Tumor antigen-derived peptide-DC vaccines could result in improved clinical outcome. Yamaguchi and collaborators [77] evaluated immunization with DCs pulsed with EphA2-derived peptides (Eph-DCs) in a mouse model of colorectal cancer, demonstrating the inhibition of relevant EphA2-positive mouse colon carcinoma MC38 cell-subcutaneous xenografts in comparison with the unpulsed DC and PBS groups. Interestingly, there was no significant difference in EphA2-negative melanoma BL6 cell-derived subcutaneous xenografts between the Eph-DC and unpulsed DC groups, suggesting vaccination with Eph-DCs provides specific antitumor effects against tumors positively express EphA2. In addition, both CD4+ and CD8+ CTLs, but not natural killer (NK) cells, were required for the anticancer effects detected upon immunization with Eph-DCs. The authors further demonstrated that treatment with Eph-DCs in mice results in higher tumor-specific CTL activity, in comparison with unpulsed DCs [78]. The MC38 and BL6 cell-derived intrahepatical xenografts in mice immunized with both Eph-DCs and unpulsed DCs were markedly inhibited in comparison with the PBS treatment group. Finally, Eph-DC immunizations were more effective against rechallenged tumor, consistent with their superior capacity to elicite EphA2-specific CTLs.
CAR-T
A strategy to treat cancer is chimeric antigen receptors (CARs) modified T (CAR-T) cell therapy, a cell-based tumor immunotherapeutic approach [115, 116]. CAR-T cells are T cells genetically engineered for producing a tumor targeting receptor, with normal T cells modified to recognize specific antigens for tumor cell targeting. The receptor combines a signaling domain of the T cell receptor (TcR) complex and an antigen-binding domain, e.g., an antibody’s scFv [117]. Therefore, independently of the native TcR, CAR-T cells recognize cancer cells through the CAR receptor. CAR-T cells that target CD19 show stark anticancer effects for chemo-refractory B cell-derived hematological cancers, which has resulted in FDA approval [118].
Several EphA2-specific T cells have been developed and evaluated in preclinical studies, recognizing EphA2-expressing tumors as assessed by interferon-γ (IFN-γ) and IL-2 synthesis and conferring cancer cell cytotoxicity. A second-generation EphA2-specific CAR was engineered on the basis of the humanized EphA2 monoclonal antibody 4H5, a CD28.ζ signaling domain, a CD28 transmembrane domain, and a CH2CH3 spacer. The final T cell populations comprised both CD4+ and CD8+ cells, all expressing EphA2-specific CARs [79]. These EphA2-specific T cells were demonstrated to identify and kill glioblastoma cells expressing EphA2. In addition, treatment with these EphA2-specific T cells suppressed EphA2-positive U373 glioma xenografts in severe combined immunodeficiency (SCID) mice and markedly increased animal survival in comparison with non-treated mice and those administered non-transduced T cells [79]. However, the CH2CH3 spacer might compromise the anticancer effects of CAR-T cells in vivo by promoting T cell sensitivity to immune cells expressing Fc receptors [119]. The EphA2-specific T cells were subsequently improved via CH2CH3 spacer replacement with an IgG1-derived short spacer, increasing the anti-glioma effects of CD28.ζ CAR T cells by 20-fold [80]. In addition to targeting gliomas, EphA2-specifc T cells have also been developed and evaluated in NSCLC [81] and esophageal squamous cell carcinoma (ESCC) [82]. Of interest, these tested EphA2-specifc T cells were shown to exhibit the ability to kill EphA2-positive tumor cells [81, 82]. In addition, administration of EphA2-specifc T cells results in inhibited lung cancer in vivo [81]. However, mice administered both EphA2-specifc and non-transduced T cells died within 7–8 weeks from non-tumor causes, which deserves further investigation.
EphA2-based combination therapeutics
Various modalities of combination therapy based on EphA2 targeting have been evaluated in preclinical studies (Table 2). For example, combined use of ALW-II-41-27 and WW437, a histone deacetylase inhibitor that could suppress phosphorylated EphA2 and EphA2 expression, results in remarkably increased effects on breast cancer cell growth and migration compared with either drug administered as monotherapy [87]. In another study, Martini et al. [27] demonstrated in cetuximab-resistant colorectal cancer cells a more pronounced EphA2 activation in comparison with sensitive ones. Joint administration of ALW-II-41-27 and cetuximab was shown to revert primary and acquired resistance to cetuximab, inhibit proliferation, and induce apoptosis in cultured cells. Likewise, significantly decreased growth of xenografts in vivo was found compared with the cetuximab alone group. Zhou et al. [86] showed in glioma cells that treatment with siRNA EphA2 exerts almost the same cell growth inhibitory effects as 3 chemotherapeutics, including cisplatin, etoposide, and minustine hydrochloride. Combining siRNA EphA2 and these anticancer agents markedly enhanced their effects. In addition, UniPR1331 was also shown to significantly increase the efficacy of bevacizumab, further reducing tumor growth in glioblastoma cells in in vivo mouse xenografts [63]. Also of interest, a study by Hasegawa et al. showed cisplatin alone does not suppress SNU-16 tumor growth at 10 mg/kg; however, combination with DS-8895a resulted in a therapeutic benefit in comparison with administration of the drug alone [56]. In a mouse breast cancer model, combination of MM-310 with anti-PD-1 (anti-mouse PD-1 antibody J43 and anti-PD-L1 antibody MPL3280) resulted in a 60% complete response rate, with durable responses that were resistant to re-challenge [76]. This combination resulted in a 93% TGI, which was greater than the effect observed with MM-310 and anti-PD-1 as monotherapies (81% and 54% TGI, respectively).
EphA2-based clinical development
Based on the preclinical studies mentioned above, several therapies have entered clinical trials, including dasatinib, MEDI-547, DS-8895a, BT5528, MM-310, EphA2-targeting DOPC-encapsulated siRNA, vaccine, and CAR-T cell immunotherapy. Dasatinib represents the only molecule already administered to humans in multiple clinical studies of cancer. However, dasatinib is frequently used as a BCR-ABL kinase inhibitor for treating malignant diseases. EphA2 is used as a biomarker for assessing patient response to dasatinib. Unfortunately, few of the remaining EphA2-target therapies have exhibited successful clinical outcomes. Drugs targeting EphA2 in clinical trials are shown in Table 3.
The abovementioned preclinical antitumor effects of DS-8895a on EphA2-overexpressing tumor cells advocate for its further clinical development. There are currently two phase I open-label studies determining the safety, tolerability, and pharmacokinetic features of DS-8895a in individuals with advanced-stage solid tumors (NCT02004717, NCT02252211). The NCT02004717 trial was a two-step, study with step 1 evaluating a dose escalation cohort (six dose levels from 0.1 to 20 mg/kg) in patients with advanced solid tumors and step 2 assessing dose expansion in individuals with EphA2-positive esophageal and gastric cancers. The maximum tolerated dose was not reached in step 1 and the planned highest dose (20 mg/kg) was used in step 2. A total of 37 cases (22 and 15 in steps 1 and 2, respectively) were included, but all discontinued the study for overt disease progression (20 and 13 in steps 1 and 2, respectively) or adverse events (AEs, the remaining cases) [120]. Similarly, the NCT02004717 trial enrolled 9 patients, who did not complete the study. There were 55.56% patients (5/9) with progressive disease and 22.22% (2/9) with serious adverse events, including cancer pain and spinal cord compression.
The safety, pharmacokinetic features, and anticancer effects of MEDI-547 were evaluated in a phase 1, open-label trial with included dose-escalation and dose-expansion cohorts [121]. Cases underwent a 1-h intravenous infusion of MEDI-547 (0.08 mg/kg) at 3-week intervals. This trial enrolled 6 patients but all discontinued the therapy due to treatment-associated bleeding (n = 3) and coagulation (n = 2) events, and the planned dose escalation was not pursued. Clinical responses comprised disease progression (n = 5, 83.3%) and stable disease (n = 1, 16.7%). The safety profile of MEDI-547 prevents its further clinical development for advanced-stage solid tumors. However, the causes of the detected AEs remain unclear.
Perspective and conclusion
The crucial roles in tumor biology have defined EphA2 as a promising therapeutic target. Due to intensive investigation and remarkable advances in understanding some of the mechanisms associated with EphA2 effects, multiple potential targets have been described. The advantages of the EphA2 as the target of tumor therapy include (i) possibility of targetable by cellular, molecular, and pharmaceutical approaches; (ii) possibility of developing anticancer immune-therapy; and (iii) possibility of combination with conventional therapeutics to improve efficacy.
However, to date, the precise mechanisms of EphA2, especially the effects of EphA2:ephrin reverse signaling, are largely unknown, and the outcomes of its regulation cannot be predicted with confidence. Indeed, EphA2-ephrin A1 signaling is extremely complex, with both interacting cells receiving interdependent signals from the identical signaling complex that is frequently associated with other Eph receptors and receptor tyrosine kinases, as well as additional signaling pathways. Meanwhile, EphA2 responses following stimulation could result from cell/tissue-specific kinase-dependent or kinase-independent signaling pathways. However, most of the preclinical work presented in this review tend to simplify the EphA2-ephrin A1 system. EphA2 inhibition or targeting confers marked benefits in experimental studies. However, these findings could not be translated into clinical use. For the currently developed EphA2-based targets, existing challenges include the following: (i) downregulation of EphA2 by siRNA affects both the forward and reverse signaling, together with compensatory stimulation of other Eph receptors and oncogenic signalings, which potentially regulate the biological behavior of a cell; (ii) compound that act on the extracellular ligand-binding domain of EphA2 block both reverse and forward signaling, and show poor physicochemical characteristics; (iii) the highly conserved cytoplasmic domain, particularly the kinase domain, among different Eph kinases, could lead to non-specific inhibition of other Eph family members and unwanted toxicities by the use of small molecule inhibitors to antagonize EphA2’s enzymatic activity; (iv) although antibody-drug conjugates are highly specific and stable, and possess antibody-like pharmacokinetic features, the non-negligible technological limitations impact drug activity and/or safety; (v) the significant proportion of non-responsive cases and treatment-related toxicities remain obstacles to successful immunotherapy treatment.
Sustained and systematic efforts in the future may involve an in-depth understanding of EphA2-ephrin A1 signaling and a precise elucidation of the crosstalk with other oncogenic pathways. The performed clinical studies and novel biological findings provide clues for developing next-generation EphA2-targeting therapies: (i) focus should be placed on enhancing efficacy and selectivity while preventing off-target secondary effects; (ii) combination with other therapies may be helpful. Despite multiple challenges due to the complex biological properties of the EphA2-ephrin A1 system, exciting possibilities still exist for novel treatment approaches based on these molecules.
Availability of data and materials
Not applicable.
Abbreviations
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- AEs:
-
Adverse events
- AONs:
-
Antisense oligonucleotides
- BCR-ABL:
-
Breakpoint cluster region-Abelson
- CARs:
-
Chimeric antigen receptors
- CAR-T:
-
(CARs) modified T cell
- CML:
-
Chronic myelogenous leukemia
- CTLs:
-
Cytotoxic T lymphocytes
- DC:
-
Dendritic cell
- DFS:
-
Disease-free survival
- DOPC:
-
1,2-dioleoyl-sn-glycero-3-phosphatidylcholine
- DTXp:
-
Docetaxel prodrug
- ECM:
-
Extracellular matrix
- eck:
-
Epithelial cell kinase
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
- Ephrin:
-
Eph receptor-interacting
- ER:
-
Estrogen receptor
- ERK:
-
Extracellular regulated protein kinases
- ESCC:
-
Esophageal squamous cell carcinoma
- FAK:
-
Focal adhesion kinase
- FcγRs:
-
Fcγ receptors
- FXR:
-
Farnesoid X receptor
- GBM:
-
Glioblastoma multiforme
- GPBAR1:
-
G protein-coupled bile acid receptor 1
- HUVECs:
-
Human umbilical vein endothelial cells
- IFN-γ:
-
Interferon-γ
- IGF-1R:
-
Insulin-like growth factor I receptor
- IgG:
-
Immunoglobulin g
- LCA:
-
Lithocholic acid
- LDH:
-
Lactic dehydrogenase
- LMW-PTP:
-
Low-molecular-weight protein phosphatase
- MMAF:
-
Monomethyl auristatin phenylalanine
- MEK:
-
MAP/ERK kinase
- NK:
-
Natural killer
- NSCLC:
-
Non-small cell lung cancer
- MAPK:
-
Mitogen-activated protein kinase
- mEA1:
-
Monomeric ephrin A1
- OS:
-
Overall survival
- PBS:
-
Phosphate buffer saline
- PDGFR:
-
Platelet-derived growth factor receptor
- Raf:
-
Rapidly accelerated fibrosarcoma
- Ras:
-
Rat sarcoma
- PPI:
-
Protein-protein interaction
- RNAi:
-
RNA interference
- RTKs:
-
Receptor tyrosine kinases
- RTKs:
-
Receptor tyrosine kinases
- SCID:
-
Severe combined immunodeficiency
- sgRNAs:
-
Single guide RNAs
- siRNAs:
-
Short interfering RNAs
- SRC:
-
Steroid receptor coactivator
- TcR:
-
T cell receptor
- TKIs:
-
Tyrosine kinase inhibitors
- TMB:
-
Trametinib
- UV:
-
Ultravioletray
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Ireton RC, Chen J. EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Curr Cancer Drug Targets. 2005;5(3):149–57.
Hirai H, Maru Y, Hagiwara K, Nishida J, Takaku F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science. 1987;238(4834):1717–20.
Dodelet VC, Pasquale EB. Eph receptors and ephrin ligands: embryogenesis to tumorigenesis. Oncogene. 2000;19(49):5614–9.
Heroult M, Schaffner F, Augustin HG. Eph receptor and ephrin ligand-mediated interactions during angiogenesis and tumor progression. Exp Cell Res. 2006;312(5):642–50.
Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3(7):475–86.
Ieguchi K, Maru Y. Roles of EphA1/A2 and ephrin-A1 in cancer. Cancer Sci. 2019;110(3):841–8.
Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133(1):38–52.
Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harb Perspect Biol. 2013;5(9).
Tandon M, Vemula SV, Mittal SK. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin Ther Targets. 2011;15(1):31–51.
Biao-xue R, Xi-guang C, Shuan-ying Y, Wei L, Zong-juan M. EphA2-dependent molecular targeting therapy for malignant tumors. Curr Cancer Drug Targets. 2011;11(9):1082–97.
Zhou Y, Sakurai H. Emerging and diverse functions of the EphA2 noncanonical pathway in cancer progression. Biol Pharm Bull. 2017;40(10):1616–24.
Lindberg RA, Hunter T. cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Mol Cell Biol. 1990;10(12):6316–24.
Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T, Goldfarb M, Yancopoulos GD. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science. 1994;266(5186):816–9.
Ruiz JC, Robertson EJ. The expression of the receptor-protein tyrosine kinase gene, eck, is highly restricted during early mouse development. Mech Dev. 1994;46(2):87–100.
Unified nomenclature for Eph family receptors and their ligands, the ephrins. Eph Nomenclature Committee. Cell. 1997;90(3):403–4.
Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. 2008;6(12):1795–806.
Holzman LB, Marks RM, Dixit VM. A novel immediate-early response gene of endothelium is induced by cytokines and encodes a secreted protein. Mol Cell Biol. 1990;10(11):5830–8.
Bartley TD, Hunt RW, Welcher AA, Boyle WJ, Parker VP, Lindberg RA, Lu HS, Colombero AM, Elliott RL, Guthrie BA, et al. B61 is a ligand for the ECK receptor protein-tyrosine kinase. Nature. 1994;368(6471):558–60.
Murai KK, Pasquale EB. Eph’ective signaling: forward, reverse and crosstalk. J Cell Sci. 2003;116(Pt 14):2823–32.
Himanen JP, Rajashankar KR, Lackmann M, Cowan CA, Henkemeyer M, Nikolov DB. Crystal structure of an Eph receptor-ephrin complex. Nature. 2001;414(6866):933–8.
Miao H, Wang B. EphA receptor signaling--complexity and emerging themes. Semin Cell Dev Biol. 2012;23(1):16–25.
Zantek ND, Azimi M, Fedor-Chaiken M, Wang B, Brackenbury R, Kinch MS. E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ. 1999;10(9):629–38.
Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16(1):9–20.
Kurose H, Ueda K, Kondo R, Ogasawara S, Kusano H, Sanada S, Naito Y, Nakiri M, Nishihara K, Kakuma T, et al. Elevated expression of EPHA2 is associated with poor prognosis after radical prostatectomy in prostate cancer. Anticancer Res. 2019;39(11):6249–57.
Amato KR, Wang S, Tan L, Hastings AK, Song W, Lovly CM, Meador CB, Ye F, Lu P, Balko JM, et al. EPHA2 blockade overcomes acquired resistance to EGFR kinase inhibitors in lung cancer. Cancer Res. 2016;76(2):305–18.
Miyazaki T, Kato H, Fukuchi M, Nakajima M, Kuwano H. EphA2 overexpression correlates with poor prognosis in esophageal squamous cell carcinoma. Int J Cancer. 2003;103(5):657–63.
Martini G, Cardone C, Vitiello PP, Belli V, Napolitano S, Troiani T, Ciardiello D, Della Corte CM, Morgillo F, Matrone N, et al. EPHA2 is a predictive biomarker of resistance and a potential therapeutic target for improving antiepidermal growth factor receptor therapy in colorectal cancer. Mol Cancer Ther. 2019;18(4):845–55.
Wu D, Suo Z, Kristensen GB, Li S, Troen G, Holm R, Nesland JM. Prognostic value of EphA2 and EphrinA-1 in squamous cell cervical carcinoma. Gynecol Oncol. 2004;94(2):312–9.
Lin YG, Han LY, Kamat AA, Merritt WM, Landen CN, Deavers MT, Fletcher MS, Urbauer DL, Kinch MS, Sood AK. EphA2 overexpression is associated with angiogenesis in ovarian cancer. Cancer. 2007;109(2):332–40.
Youngblood VM, Kim LC, Edwards DN, Hwang Y, Santapuram PR, Stirdivant SM, Lu P, Ye F, Brantley-Sieders DM, Chen J. The Ephrin-A1/EPHA2 signaling axis regulates glutamine metabolism in HER2-positive breast cancer. Cancer Res. 2016;76(7):1825–36.
Mo J, Zhao X, Dong X, Liu T, Zhao N, Zhang D, Wang W, Zhang Y, Sun B. Effect of EphA2 knockdown on melanoma metastasis depends on intrinsic ephrinA1 level. Cell Oncol (Dordr). 2020.
Kinch MS, Moore MB, Harpole DH Jr. Predictive value of the EphA2 receptor tyrosine kinase in lung cancer recurrence and survival. Clin Cancer Res. 2003;9(2):613–8.
Garcia-Monclus S, Lopez-Alemany R, Almacellas-Rabaiget O, Herrero-Martin D, Huertas-Martinez J, Lagares-Tena L, Alba-Pavon P, Hontecillas-Prieto L, Mora J, de Alava E, et al. EphA2 receptor is a key player in the metastatic onset of Ewing sarcoma. Int J Cancer. 2018;143(5):1188–201.
Kinch MS, Carles-Kinch K. Overexpression and functional alterations of the EphA2 tyrosine kinase in cancer. Clin Exp Metastasis. 2003;20(1):59–68.
Faoro L, Singleton PA, Cervantes GM, Lennon FE, Choong NW, Kanteti R, Ferguson BD, Husain AN, Tretiakova MS, Ramnath N, et al. EphA2 mutation in lung squamous cell carcinoma promotes increased cell survival, cell invasion, focal adhesions, and mammalian target of rapamycin activation. J Biol Chem. 2010;285(24):18575–85.
Shentu XC, Zhao SJ, Zhang L, Miao Q. A novel p.R890C mutation in EPHA2 gene associated with progressive childhood posterior cataract in a Chinese family. Int J Ophthalmol. 2013;6(1):34–8.
Zhai Y, Zhu S, Li J, Yao K. A novel human congenital cataract mutation in EPHA2 kinase domain (p.G668D) alters receptor stability and function. Invest Ophthalmol Vis Sci. 2019;60(14):4717–26.
Mudali SV, Fu B, Lakkur SS, Luo M, Embuscado EE, Iacobuzio-Donahue CA. Patterns of EphA2 protein expression in primary and metastatic pancreatic carcinoma and correlation with genetic status. Clin Exp Metastasis. 2006;23(7-8):357–65.
Dohn M, Jiang J, Chen X. Receptor tyrosine kinase EphA2 is regulated by p53-family proteins and induces apoptosis. Oncogene. 2001;20(45):6503–15.
Zantek ND, Walker-Daniels J, Stewart J, Hansen RK, Robinson D, Miao H, Wang B, Kung HJ, Bissell MJ, Kinch MS. MCF-10A-NeoST: a new cell system for studying cell-ECM and cell-cell interactions in breast cancer. Clin Cancer Res. 2001;7(11):3640–8.
Zelinski DP, Zantek ND, Walker-Daniels J, Peters MA, Taparowsky EJ, Kinch MS. Estrogen and Myc negatively regulate expression of the EphA2 tyrosine kinase. J Cell Biochem. 2002;85(4):714–20.
Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61(5):2301–6.
Sakamoto A, Kato K, Hasegawa T, Ikeda S. An agonistic antibody to EPHA2 exhibits antitumor effects on human melanoma cells. Anticancer Res. 2018;38(6):3273–82.
Bruckheimer EM, Fazenbaker CA, Gallagher S, Mulgrew K, Fuhrmann S, Coffman KT, Walsh W, Ready S, Cook K, Damschroder M, et al: Antibody-dependent cell-mediated cytotoxicity effector-enhanced EphA2 agonist monoclonal antibody demonstrates potent activity against human tumors. Neoplasia. 2009, 11(6):509-517, 502 p following 517.
Carles-Kinch K, Kilpatrick KE, Stewart JC, Kinch MS. Antibody targeting of the EphA2 tyrosine kinase inhibits malignant cell behavior. Cancer Res. 2002;62(10):2840–7.
Walker-Daniels J, Riese DJ 2nd, Kinch MS. c-Cbl-dependent EphA2 protein degradation is induced by ligand binding. Mol Cancer Res. 2002;1(1):79–87.
Kikawa KD, Vidale DR, Van Etten RL, Kinch MS. Regulation of the EphA2 kinase by the low molecular weight tyrosine phosphatase induces transformation. J Biol Chem. 2002;277(42):39274–9.
Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3(10):541–51.
Landen CN Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, Sood AK. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65(15):6910–8.
Wykosky J, Palma E, Gibo DM, Ringler S, Turner CP, Debinski W. Soluble monomeric EphrinA1 is released from tumor cells and is a functional ligand for the EphA2 receptor. Oncogene. 2008;27(58):7260–73.
Xu Q, Lin WC, Petit RS, Groves JT. EphA2 receptor activation by monomeric Ephrin-A1 on supported membranes. Biophys J. 2011;101(11):2731–9.
Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Ligation of EphA2 by Ephrin A1-Fc inhibits pancreatic adenocarcinoma cellular invasiveness. Biochem Biophys Res Commun. 2004;320(4):1096–102.
Nakamura R, Kataoka H, Sato N, Kanamori M, Ihara M, Igarashi H, Ravshanov S, Wang YJ, Li ZY, Shimamura T, et al. EPHA2/EFNA1 expression in human gastric cancer. Cancer Sci. 2005;96(1):42–7.
Coffman KT, Hu M, Carles-Kinch K, Tice D, Donacki N, Munyon K, Kifle G, Woods R, Langermann S, Kiener PA, Kinch MS. Differential EphA2 epitope display on normal versus malignant cells. Cancer Res. 2003;63(22):7907–12.
Goldgur Y, Susi P, Karelehto E, Sanmark H, Lamminmaki U, Oricchio E, Wendel HG, Nikolov DB, Himanen JP. Generation and characterization of a single-chain anti-EphA2 antibody. Growth Factors. 2014;32(6):214–22.
Hasegawa J, Sue M, Yamato M, Ichikawa J, Ishida S, Shibutani T, Kitamura M, Wada T, Agatsuma T. Novel anti-EPHA2 antibody, DS-8895a for cancer treatment. Cancer Biol Ther. 2016;17(11):1158–67.
Burvenich IJ, Parakh S, Gan HK, Lee FT, Guo N, Rigopoulos A, Lee ST, Gong S, O'Keefe GJ, Tochon-Danguy H, et al. Molecular imaging and quantitation of EphA2 expression in xenograft models with 89Zr-DS-8895a. J Nucl Med. 2016;57(6):974–80.
Dobrzanski P, Hunter K, Jones-Bolin S, Chang H, Robinson C, Pritchard S, Zhao H, Ruggeri B. Antiangiogenic and antitumor efficacy of EphA2 receptor antagonist. Cancer Res. 2004;64(3):910–9.
Giorgio C, Hassan Mohamed I, Flammini L, Barocelli E, Incerti M, Lodola A, Tognolini M. Lithocholic acid is an Eph-ephrin ligand interfering with Eph-kinase activation. PLoS One. 2011;6(3):e18128.
Incerti M, Tognolini M, Russo S, Pala D, Giorgio C, Hassan-Mohamed I, Noberini R, Pasquale EB, Vicini P, Piersanti S, et al. Amino acid conjugates of lithocholic acid as antagonists of the EphA2 receptor. J Med Chem. 2013;56(7):2936–47.
Giorgio C, Russo S, Incerti M, Bugatti A, Vacondio F, Barocelli E, Mor M, Pala D, Hassan-Mohamed I, Gioiello A, et al. Biochemical characterization of EphA2 antagonists with improved physico-chemical properties by cell-based assays and surface plasmon resonance analysis. Biochem Pharmacol. 2016;99:18–30.
Hassan-Mohamed I, Giorgio C, Incerti M, Russo S, Pala D, Pasquale EB, Zanotti I, Vicini P, Barocelli E, Rivara S, et al. UniPR129 is a competitive small molecule Eph-ephrin antagonist blocking in vitro angiogenesis at low micromolar concentrations. Br J Pharmacol. 2014;171(23):5195–208.
Festuccia C, Gravina GL, Giorgio C, Mancini A, Pellegrini C, Colapietro A, Delle Monache S, Maturo MG, Sferra R, Chiodelli P, et al. UniPR1331, a small molecule targeting Eph/ephrin interaction, prolongs survival in glioblastoma and potentiates the effect of antiangiogenic therapy in mice. Oncotarget. 2018;9(36):24347–63.
Tognolini M, Incerti M, Hassan-Mohamed I, Giorgio C, Russo S, Bruni R, Lelli B, Bracci L, Noberini R, Pasquale EB, et al. Structure-activity relationships and mechanism of action of Eph-ephrin antagonists: interaction of cholanic acid with the EphA2 receptor. ChemMedChem. 2012;7(6):1071–83.
Tognolini M, Incerti M, Pala D, Russo S, Castelli R, Hassan-Mohamed I, Giorgio C, Lodola A. Target hopping as a useful tool for the identification of novel EphA2 protein-protein antagonists. ChemMedChem. 2014;9(1):67–72.
Noberini R, De SK, Zhang Z, Wu B, Raveendra-Panickar D, Chen V, Vazquez J, Qin H, Song J, Cosford ND, et al. A disalicylic acid-furanyl derivative inhibits ephrin binding to a subset of Eph receptors. Chem Biol Drug Des. 2011;78(4):667–78.
Buettner R, Mesa T, Vultur A, Lee F, Jove R. Inhibition of Src family kinases with dasatinib blocks migration and invasion of human melanoma cells. Mol Cancer Res. 2008;6(11):1766–74.
Chang Q, Jorgensen C, Pawson T, Hedley DW. Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. Br J Cancer. 2008;99(7):1074–82.
Heinzlmeir S, Lohse J, Treiber T, Kudlinzki D, Linhard V, Gande SL, Sreeramulu S, Saxena K, Liu X, Wilhelm M, et al. Chemoproteomics-aided medicinal chemistry for the discovery of EPHA2 inhibitors. ChemMedChem. 2017;12(12):999–1011.
Amato KR, Wang S, Hastings AK, Youngblood VM, Santapuram PR, Chen H, Cates JM, Colvin DC, Ye F, Brantley-Sieders DM, et al. Genetic and pharmacologic inhibition of EPHA2 promotes apoptosis in NSCLC. J Clin Invest. 2014;124(5):2037–49.
Wykosky J, Gibo DM, Debinski W. A novel, potent, and specific ephrinA1-based cytotoxin against EphA2 receptor expressing tumor cells. Mol Cancer Ther. 2007;6(12 Pt 1):3208–18.
Jackson D, Gooya J, Mao S, Kinneer K, Xu L, Camara M, Fazenbaker C, Fleming R, Swamynathan S, Meyer D, et al. A human antibody-drug conjugate targeting EphA2 inhibits tumor growth in vivo. Cancer Res. 2008;68(22):9367–74.
Lee JW, Stone RL, Lee SJ, Nam EJ, Roh JW, Nick AM, Han HD, Shahzad MM, Kim HS, Mangala LS, et al. EphA2 targeted chemotherapy using an antibody drug conjugate in endometrial carcinoma. Clin Cancer Res. 2010;16(9):2562–70.
Lee JW, Han HD, Shahzad MM, Kim SW, Mangala LS, Nick AM, Lu C, Langley RR, Schmandt R, Kim HS, et al. EphA2 immunoconjugate as molecularly targeted chemotherapy for ovarian carcinoma. J Natl Cancer Inst. 2009;101(17):1193–205.
Fu Y, Rathod D, Abo-Ali EM, Dukhande VV, Patel K. EphA2-receptor targeted PEGylated nanoliposomes for the treatment of BRAF(V600E) mutated parent- and vemurafenib-resistant melanoma. Pharmaceutics. 2019;11(10).
Kamoun WS, Dugast AS, Suchy JJ, Grabow S, Fulton RB, Sampson JF, Luus L, Santiago M, Koshkaryev A, Sun G, et al. Synergy between EphA2-ILs-DTXp, a novel EphA2-targeted nanoliposomal taxane, and PD-1 inhibitors in preclinical tumor models. Mol Cancer Ther. 2020;19(1):270–81.
Yamaguchi S, Tatsumi T, Takehara T, Sakamori R, Uemura A, Mizushima T, Ohkawa K, Storkus WJ, Hayashi N. Immunotherapy of murine colon cancer using receptor tyrosine kinase EphA2-derived peptide-pulsed dendritic cell vaccines. Cancer. 2007;110(7):1469–77.
Yamaguchi S, Tatsumi T, Takehara T, Sasakawa A, Hikita H, Kohga K, Uemura A, Sakamori R, Ohkawa K, Hayashi N. Dendritic cell-based vaccines suppress metastatic liver tumor via activation of local innate and acquired immunity. Cancer Immunol Immunother. 2008;57(12):1861–9.
Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, Rainusso N, Wu MF, Liu H, Kew Y, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21(3):629–37.
Yi Z, Prinzing BL, Cao F, Gottschalk S, Krenciute G. Optimizing EphA2-CAR T cells for the adoptive immunotherapy of glioma. Mol Ther Methods Clin Dev. 2018;9:70–80.
Li N, Liu S, Sun M, Chen W, Xu X, Zeng Z, Tang Y, Dong Y, Chang AH, Zhao Q. Chimeric antigen receptor-modified T cells redirected to EphA2 for the immunotherapy of non-small cell lung cancer. Transl Oncol. 2018;11(1):11–7.
Shi H, Yu F, Mao Y, Ju Q, Wu Y, Bai W, Wang P, Xu R, Jiang M, Shi J. EphA2 chimeric antigen receptor-modified T cells for the immunotherapy of esophageal squamous cell carcinoma. J Thorac Dis. 2018;10(5):2779–88.
Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431(7006):371–8.
Kim HJ, Kim A, Miyata K, Kataoka K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv Drug Deliv Rev. 2016;104:61–77.
Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. EphA2: a determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene. 2004;23(7):1448–56.
Zhou Z, Yuan X, Li Z, Tu H, Li D, Qing J, Wang H, Zhang L. RNA interference targeting EphA2 inhibits proliferation, induces apoptosis, and cooperates with cytotoxic drugs in human glioma cells. Surg Neurol. 2008;70(6):562–8 discussion 568-569.
Zhang T, Li J, Ma X, Yang Y, Sun W, Jin W, Wang L, He Y, Yang F, Yi Z, et al. Inhibition of HDACs-EphA2 signaling axis with WW437 demonstrates promising preclinical antitumor activity in breast cancer. EBioMedicine. 2018;31:276–86.
Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011;11(1):59–67.
Wu SY, Lopez-Berestein G, Calin GA, Sood AK. RNAi therapies: drugging the undruggable. Sci Transl Med. 2014;6(240):240 ps247.
Wagner MJ, Mitra R, McArthur MJ, Baze W, Barnhart K, Wu SY, Rodriguez-Aguayo C, Zhang X, Coleman RL, Lopez-Berestein G, Sood AK. Preclinical mammalian safety studies of EPHARNA (DOPC nanoliposomal EphA2-targeted siRNA). Mol Cancer Ther. 2017;16(6):1114–23.
Sukka-Ganesh B, Mohammed KA, Kaye F, Goldberg EP, Nasreen N. Ephrin-A1 inhibits NSCLC tumor growth via induction of Cdx-2 a tumor suppressor gene. BMC Cancer. 2012;12(309).
Guo H, Miao H, Gerber L, Singh J, Denning MF, Gilliam AC, Wang B. Disruption of EphA2 receptor tyrosine kinase leads to increased susceptibility to carcinogenesis in mouse skin. Cancer Res. 2006;66(14):7050–8.
Macrae M, Neve RM, Rodriguez-Viciana P, Haqq C, Yeh J, Chen C, Gray JW, McCormick F. A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell. 2005;8(2):111–8.
Hamaoka Y, Negishi M, Katoh H. Tyrosine kinase activity of EphA2 promotes its S897 phosphorylation and glioblastoma cell proliferation. Biochem Biophys Res Commun. 2018;499(4):920–6.
Cuyas E, Queralt B, Martin-Castillo B, Bosch-Barrera J, Menendez JA. EphA2 receptor activation with ephrin-A1 ligand restores cetuximab efficacy in NRAS-mutant colorectal cancer cells. Oncol Rep. 2017;38(1):263–70.
Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, Dhe-Paganon S. Architecture of Eph receptor clusters. Proc Natl Acad Sci U S A. 2010;107(24):10860–5.
Bufalo MC, Bordon-Graciani AP, Conti BJ, de Assis GM, Sforcin JM. The immunomodulatory effect of propolis on receptors expression, cytokine production and fungicidal activity of human monocytes. J Pharm Pharmacol. 2014;66(10):1497–504.
DiLillo DJ, Ravetch JV. Differential Fc-receptor engagement drives an anti-tumor vaccinal effect. Cell. 2015;161(5):1035–45.
DiLillo DJ, Ravetch JV. Fc-receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunol Res. 2015;3(7):704–13.
Mohamed IH, Giorgio C, Bruni R, Flammini L, Barocelli E, Rossi D, Domenichini G, Poli F, Tognolini M. Polyphenol rich botanicals used as food supplements interfere with EphA2-ephrinA1 system. Pharmacol Res. 2011;64(5):464–70.
Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res. 2012;53(9):1723–37.
Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7(8):678–93.
Castelli R, Tognolini M, Vacondio F, Incerti M, Pala D, Callegari D, Bertoni S, Giorgio C, Hassan-Mohamed I, Zanotti I, et al. Delta(5)-cholenoyl-amino acids as selective and orally available antagonists of the Eph-ephrin system. Eur J Med Chem. 2015;103:312–24.
Ferlenghi F, Castelli R, Scalvini L, Giorgio C, Corrado M, Tognolini M, Mor M, Lodola A, Vacondio F. Drug-gut microbiota metabolic interactions: the case of UniPR1331, selective antagonist of the Eph-ephrin system, in mice. J Pharm Biomed Anal. 2020;180:113067.
Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O'Brien S, Nicaise C, Bleickardt E, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–41.
Olivieri A, Manzione L. Dasatinib: a new step in molecular target therapy. Ann Oncol. 2007;18(Suppl 6):vi42–6.
Wang XD, Reeves K, Luo FR, Xu LA, Lee F, Clark E, Huang F. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: rationale for patient selection and efficacy monitoring. Genome Biol. 2007;8(11):R255.
Choi Y, Syeda F, Walker JR, Finerty PJ Jr, Cuerrier D, Wojciechowski A, Liu Q, Dhe-Paganon S, Gray NS. Discovery and structural analysis of Eph receptor tyrosine kinase inhibitors. Bioorg Med Chem Lett. 2009;19(15):4467–70.
Moolten FL, Cooperband SR. Selective destruction of target cells by diphtheria toxin conjugated to antibody directed against antigens on the cells. Science. 1970;169(3940):68–70.
Pastan I, Chaudhary V, FitzGerald DJ. Recombinant toxins as novel therapeutic agents. Annu Rev Biochem. 1992;61:331–54.
Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14.
Wang S, Placzek WJ, Stebbins JL, Mitra S, Noberini R, Koolpe M, Zhang Z, Dahl R, Pasquale EB, Pellecchia M. Novel targeted system to deliver chemotherapeutic drugs to EphA2-expressing cancer cells. J Med Chem. 2012;55(5):2427–36.
Geddie ML, Kohli N, Kirpotin DB, Razlog M, Jiao Y, Kornaga T, Rennard R, Xu L, Schoerberl B, Marks JD, et al. Improving the developability of an anti-EphA2 single-chain variable fragment for nanoparticle targeting. MAbs. 2017;9(1):58–67.
Hart DN. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood. 1997;90(9):3245–87.
Cheadle EJ, Gornall H, Baldan V, Hanson V, Hawkins RE, Gilham DE. CAR T cells: driving the road from the laboratory to the clinic. Immunol Rev. 2014;257(1):91–106.
Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017;168(4):724–40.
Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257(1):107–26.
Sheridan C. First approval in sight for Novartis’ CAR-T therapy after panel vote. Nat Biotechnol. 2017;35(8):691–3.
Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, Chang WC, Bretzlaff W, Starr R, Priceman S, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23(4):757–68.
Shitara K, Satoh T, Iwasa S, Yamaguchi K, Muro K, Komatsu Y, Nishina T, Esaki T, Hasegawa J, Kakurai Y, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the afucosylated, humanized anti-EPHA2 antibody DS-8895a: a first-in-human phase I dose escalation and dose expansion study in patients with advanced solid tumors. J Immunother Cancer. 2019;7(1):219.
Annunziata CM, Kohn EC, LoRusso P, Houston ND, Coleman RL, Buzoianu M, Robbie G, Lechleider R. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Invest New Drugs. 2013;31(1):77–84.
Acknowledgements
We thank Dr. Qianjin Liao and Wei Ou for the helpful discussion.
Funding
This study was supported by National Natural Science Foundation of China (Grant No. 81230053, 81874132, 81802947, 81472801, and 81672687); State Key Basic Research Program of China (2013CB910502); Changsha Science and Technology Board (kq1801110); Natural Science Foundation of Hunan Province (Grant No. 2019JJ50968); and Shenzhen Science and Technology Program of China (KQTD20170810160226082).
Author information
Authors and Affiliations
Contributions
All authors have contributed to the preparation of this manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xiao, T., Xiao, Y., Wang, W. et al. Targeting EphA2 in cancer. J Hematol Oncol 13, 114 (2020). https://doi.org/10.1186/s13045-020-00944-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00944-9