
Abstract
Uncontrolled cell division is the hallmark of cancers. Full understanding of cell cycle regulation would contribute to promising cancer therapies. In particular, cyclin-dependent kinases 4/6 (CDK4/6), which are pivotal drivers of cell proliferation by combination with cyclin D, draw more and more attention. Subsequently, extensive studies were carried out to explore drugs inhibiting CDK4/6 and assess the efficacy and safety of these drugs in cancer, especially breast cancer. Due to the insuperable adverse events and the less activity observed in vivo, the drug development of the initial pan-CDK inhibitor flavopiridol was consequently discontinued, and then highly specific inhibitors were extensively researched and developed, including palbociclib (PD0332991), ribociclib (LEE011), and abemaciclib (LY2835219). Food and Drug Administration has approved palbociclib and ribociclib for the treatment of hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced or metastatic breast cancer, and recent clinical trial data suggest that palbociclib significantly improved clinical outcome when combined with letrozole or fulvestrant. Besides, the favorable effects of abemaciclib on prolonging survival of breast cancer patients have also been observed in clinical trials both for single-agent and combination strategy. In this review, we outline the preclinical and clinical advancement of these three orally bioavailable and highly selective CDK4/6 inhibitors in breast cancer.
Similar content being viewed by others


Background
Breast cancer is the most common female tumor type and accounts for the leading cancer mortality in women worldwide [1]. In spite of the great achievement in diagnosis and treatment, breast cancer remains a significant global burden [1]. Sequencing of breast cancer genome and transcriptome has identified breast cancer as a malignant disease with vast heterogeneity which is categorized into five distinct molecular subtypes including luminal A, luminal B, human epidermal growth factor receptor 2 (HER2)-enriched, basal-like, and claudin-low [2]. Among these, luminal-type accounts for the most part of breast cancer and is characterized with the typical expression of estrogen receptor (ER) and/or progesterone receptor (PR), which can be effectively targeted with hormone therapy. However, some patients have intrinsic resistance or acquired tolerance to hormone or endocrine therapy, which hampers the survival prolongation of these patients. Basal-like breast cancer, which is characterized with comparatively aggressive phenotype and the absent status of ER, PR and HER2, still lacks efficient treatment strategy. Thus, novel effective therapies are urgently required for breast cancer population.
Disordered cell cycle regulation is induced by complex mechanisms including the functional imbalance of oncogene and anti-oncogene, and contributes to uncontrolled cell proliferation resulting in cancer formation [3,4,5,6,7,8]. The past decades have witnessed the great progress in developing novel effective therapies [9,10,11,12], especially through diverting tumor cells from a proliferation phenotype towards a non-division state. Among the emerging therapies, cyclin-dependent kinase 4/6 (CDK4/6) inhibitors are the most attractive findings. CDK4/6 coordinates the cell cycle progression by reversible combination with cyclin D [13], and the bipartite complex of these elements phosphorylates pivotal tumor suppressors and transcription factors, contributing to cell cycle progression [14,15,16]. The essential roles of CDK4/6 in cell cycle regulation make them effective targets for cancer therapeutic intervention, especially in breast cancer [17,18,19]. The orally highly selective inhibitors of CDK4/6 are currently under active investigation, including palbociclib (PD0332991), ribociclib (LEE011), and abemaciclib (LY2835219). Among these, palbociclib and ribociclib remarkably prolonged the progression-free survival (PFS) in combination with letrozole for patients with ER-positive/HER2-negative advanced breast cancer, and have gained accelerated approval from Food and Drug Administration (FDA) as initial endocrine-based therapy for these patients.
In this review, we will first elaborate the anti-tumor mechanisms of CDK4/6 inhibitors, and then trace the preclinical and clinical evidence of these three highly specific inhibitors. At last, we will discuss the possible future directions in this field.
CDKs in cell cycle regulation
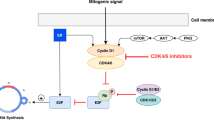
Cancer derives from uncontrolled cell division which results from the dysregulation of cell cycle progression including four stages of G1 (Gap phase 1), S phase (DNA synthesis), G2 (Gap phase 2), and M phase (mitosis) (Fig. 1). Cell cycle is monitored by a wide range of pathways including the retinoblastoma (RB)-E2F signaling [20]. RB, a well-known tumor suppressor, plays switching roles in cell cycle [20]. E2F is an evolutionarily conserved family of transcription factors, which functions in cell cycle control and contributes to tumor development [21]. The combination of RB and E2F makes E2F transcription modules in a suppressed state through inducing the recruitment of chromatin remodeling proteins, histone modifiers, and repressive chromatin marks, resulting in cell cycle block [22] (Fig. 1). The CDKs-RB axis is essential to cell cycle entry. CDK4/6 in combination with cyclin D, phosphorylates and inactivates RB [23], and then releases E2F, resulting in the recruitment of transcriptional activators, the alter transcription of genes involved during cell cycle process and subsequent G1-S block [22] (Fig. 1). Furthermore, the active combination of CDK4/6 and cyclin D is also involved in the phosphorylation of the cell proliferation-specific transcription factor forkhead box M1 (FOXM1), inducing the expression of genes which drive cell division and suppress cellular senescence in a FOXM1-dependent manner [24] (Fig. 1). However, the kinase activity of CDK4/6 is suppressed by p16INK4A [25,26,27] (Fig. 1), and cyclin D is regulated by a complex network such as ER/PR/androgen receptor (AR), nuclear factor kB (NF-kB), mitogen activated protein kinases (MAPKs), signal transducers and activators of transcription (STATs), Wnt/β-catenin and phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR [24] (Fig. 1).

Regulation and function of CDK4/6 in cell cycle progression. Active complex of CDK4/6 and cyclin D phosphorylates and inactivates RB protein and then releases transcription factor E2F, triggering the up-regulation of E2F-responsive gene which promotes cell proliferation with cell cycle G1/S transition. The combination of CDK4/6 and cyclin D can also phosphorylates transcription factor FOXM1, resulting in the FOXM1-dependent expression of gene which protects cancer cells from cell cycle block. The kinase activity of CDK4/6 is suppressed by p16INK4A and pharmacologic CDK4/6 inhibitors including palbociclib, ribociclib and abemaciclib. Cyclin D is regulated by multiple pathways such as ER/PR/AR, NF-kB, MAPKs, STATs, Wnt/β-catenin, and PI3K/AKT/mTOR. Besides, CDK2/cyclin E also participates in the RB phosphorylation. CDK2/cyclin A complex increases in stages S, G2, and M, while CDK1/Cyclin A/B complex mediates the transition from G2 to M stage
In addition, the active combination of CDK2 and cyclin E also participates in the phosphorylation of RB (Fig. 1). Besides, CDK2 is accumulating and active in stages S, G2, and M in combination with cyclin E and cyclin A, respectively, while CDK1/cyclin A/B complex mediates the transition from G2 to M stage [22] (Fig. 1).
CDK4/6 inhibitors
In consideration of the pivotal role of CDK4/6 in cell cycle progression, amounting studies have been conducted to suppress cancer cell proliferation through targeting the CDK4/6 signaling for effective cancer therapies during the past decades. Several CDK inhibitors have been explored for potential tumor treatment and assessed for pharmacokinetics, efficacy, and safety in many clinical trials.
Flavopiridol, also named as alvocidib developed by Sanofi-Aventis, is the most extensively investigated one among the first generation pan-CDK inhibitors, showing inhibitory effects on CDK1, CDK2, CDK4, CDK6, CDK7, and CDK9 [28,29,30], with half-maximal inhibitory concentration (IC50) values ranging from 20 to 170 nM (Fig. 2). In addition to cell cycle inhibition, flavopiridol-associated distal cellular effects also include apoptosis, transcriptional suppression, autophagy and endoplasmic reticulum stress [31,32,33], leading to several unacceptable high rates of dose-limiting toxicities, including neutropenia, hyperglycemia, cardiac, and pulmonary dysfunction [34]. The development of this non-selective compound was discontinued due to the low specificity for CDKs and narrow therapeutic window [17].

Chemical structures of CDK4/6 inhibitors. The chemical structures of the pan-CDK inhibitor (a) flavopiridol and the highly selective inhibitors including (b) palbociclib (PD0332991), (c) ribociclib (LEE011) and (d) abemaciclib (LY2835219) are shown. The reported half-maximal inhibitory concentration (IC50) values of these inhibitors are shown
Subsequently, inhibitors highly selective for CDK4/6 were explored and developed, primarily through chemical screening and optimization via adding pyrido [2,3-d] pyrimidin-7-one compounds with a 2-amino pyridine side chain at the C2 position [35]. Chief among this new waves are palbociclib, ribociclib, and abemaciclib, all of which are orally administrated inhibitors and exhibit little or no suppression of other CDK activities at clinically achievable doses. Of these selective inhibitors, palbociclib is the most extensively investigated and has been evaluated in vitro and in vivo till recent times [36, 37]. It potently induces G1-S cell cycle block through blocking the phosphorylation of RB and related proteins and then down-regulating S-phase cyclins and mitotic regulatory genes as well as suppressing nucleotide biosynthesis and DNA replication [38, 39]. But its biological function has been limited because the anti-tumor effects of palbciclib is dependent on the presence of an active RB protein [36, 37]. Scientists at Eli Lilly and Novartis have also developed the parallel drugs ribociclib and abemaciclib, respectively [40,41,42,43,44].
The highly selective inhibitors are analogous to the pan-CDK inhibitor flavopiridol in structure but different in chemical function. The chemical structures of these four CDK4/6 inhibitors described above are showed in Fig. 2.
Pre-clinical evidence
In vitro an in vivo study
Palbociclib
Palbociclib halters the phosphorylation of RB protein and subsequently down-regulates E2F-targeted gene, which accounts for cell cycle arrest [11, 45]. Furthermore, palbociclib is also implicated to inactivate the transcription factor FOXM1 and its transcription targets which induce cell proliferation [45,46,47,48]. In addition to the well-known anti-proliferation effects, palbociclib also demonstrates dramatic promotion of epithelial-mesenchymal transition and tumor cell invasion [49]. Besides, this compound can also sensitize cancer cells to other regimes when in combination, such as chemotherapy [50] and ionizing radiation [51]. The potent anti-tumor effects of palbociclib have been observed in several tumor types, including T cell acute lymphoblastic leukemia (T-ALL) [45], hepatocellular carcinoma [39], neuroblastoma [40], renal cell carcinoma [52], myeloma [53], mantle cell lymphoma [54], pancreatic ductal adenocarcinoma [49], esophageal adenocarcinoma [55], medulloblastoma [51], melanoma [56], non-small cell lung carcinoma [57], and particularly in breast cancer [50, 58, 59].
Palbociclib was most extensively investigated in breast cancer. As a monotherapy, this compound potently blocks the cell cycle progression through inhibiting the hyperphosphorylation of RB protein in sensitive breast cancer cells [50]. Besides, when combined with tamoxifen or trastuzumab, it displayed obvious synergistic effects in ER+ cells and HER2-amplified cells, respectively [50]. Furthermore, this drug could re-sensitize the MCF7 cell line with acquired tamoxifen-resistance to this estrogen modulator [50]. Triple-negative breast cancer (TNBC), the most malignant molecular type and lack of well-established markers, appeared to be partially responsive to cytotoxic chemotherapy [58]. McClendon AK reported that combination therapy of palbociclib and doxorubicin displayed additive cytostatic effects in RB-proficient TNBC cells [58]. For combination therapy, palbociclib contributed to profound G1 block through targeting RB signaling, while doxorubicin led to cell accumulation in G2/M stage due to DNA damage response [58]. However, palbociclibin combination with doxorubicin ultimately resulted in recurrent subpopulation, which might be explained that palbociclib could antagonize the cell death induced by doxorubicin [58]. However, this issue might be resolved by an intermittent dosing schedule.
The efficacy of palbociclib have been evaluated in multiple animal models bearing different kinds of caner, including liver cancer [39], glioblastoma multiforme [60], pancreatic neuroendocrine tumors [61], gliomas [62, 63], colon carcinoma [36], especially breast cancer [36, 45, 64]. This compound led to growth arrest of xenograft tumors and prolonged survival of treated animals. Mice models with ERBB2-overexpressing breast carcinoma showed that CDK4/6 was pivotal to the maintenance of the disease [45]. Palbociclib administration delayed the progression of transplanted tumor through stimulating cancer cell senescence via strongly suppressing the phosphorylation of RB protein and subsequently down-regulating E2F-targeted genes, but did not have any effects on the apoptosis [45].
Ribociclib
Amounting in vitro studies have been carried out to investigate the function of ribociclib in cancers including leukemia [65], neuroblastoma [40, 66, 67], neuroendocrine tumors [68], liposarcoma [69], particularly breast cancer [70]. Combination of ribociclib and 3-phosphoinositide dependent protein kinase 1 (PDK1) inhibitor GSK2334470 potently suppressed the proliferation and increased apoptosis in ER-positive breast cancer cell lines [70].
The efficacy of ribociclib was also assessed in animal models bearing multiple cancer types including breast cancer [42, 70, 71], neuroblastoma [40, 66], and liposarcoma [69]. In consideration that the PI3K/Akt/mTOR signaling participates in the regulation of cyclin D and this pathway is activated in most breast tumors, the combination of ribociclib and PI3K/mTOR inhibitor alpelisib (BYL719) was investigated in breast carcinoma [20]. These two agents synergistically impaired breast cancer cell proliferation and tumor growth in mouse models in comparison with alpelisib incubation alone, and this combination was also effective in breast cancer models with alpelisib-resistance [42]. Similar phenomenon was also noted by Jansen VM that ribociclib in combination with alpelisib potently suppressed tumor progression of MCF7 mice xenografts [70]. In addition, there are also studies assessing the role of ribociclib in HER2-positive breast cancer. The results of the research by Goel S indicated that the complex of cyclin D1 and CDK4 played important roles in the resistance of HER2-positive breast cancer cells to the anti-HER2 therapy. In this context, the inhibition of CDK4/6 resensitized acquired-resistant xenograft mice models to HER2-targeted therapies and suppressed tumor recurrence in vivo [71].
Abemaciclib
Abemaciclib, the third parallel CDK4/6 inhibitor, has been assessed in several tumors up to now, including breast cancer [72], melanoma [73], and bladder cancer [74]. ABCB1 and ABCG2, which are important ATP-binding cassette (ABC) transporters, contribute to multidrug resistance in tumor chemotherapy through transporting anti-tumor drugs to the outside of cancer cells [72]. Abemaciclib treatment significantly sensitized ABCB1 or ABCG2 over-expressing cancer cells to respective chemotherapeutic drugs through impairing the “porter” roles of ABCB1 and ABCG2 in these transporters-amplified carcinoma cells, which implicated that abemaciclib could reverse the ABCB1 or ABCG2-induced multidrug resistance to some extent [72].
The efficacy of ribociclib was also assessed in animal models bearing multiple cancer types including breast cancer [75], melanoma [73], glioblastoma [76],and head and neck squamous cell carcinoma [77]. A representative ER-positive/HER2-negative human breast cancer T47D xenograft model displayed the obvious anti-tumor effects of single-agent treatment of abemaciclib [75].
Therapy resistance
Palbociclib
The incoming resistance to targeted therapies is a major limitation to treatment efficacy [78]. According to the study of human breast cancer cell lines representing different molecular subtypes, ER-positive subtype (9/10) was the most responsible to the proliferation inhibition of palbociclib, followed by HER2-enriched type (10/16) [50].
Full understanding of the CDK4/6-RB-E2F pathway is pivotal to guiding the utilization of palbociclib treatment. Cancer cells that were intrinsically lacking of RB or harbored inactivation of this tumor suppressor were implicated to fail to effectively response to palbociclib treatment [36, 60, 79], probably due to the absence of the target for palbociclib in the RB-deficient tumor cell lines [38]. Nevertheless, this concept is not universal. The results from Dean JL reflected that knockdown of RB could facilitate only a partial resistance to CDK4/6 inhibition-induced cell cycle arrest, while overexpression of E2F was capable of leading to complete passby of CDK4/6 inhibition irrespective of RB status and palbociclib exposure [38]. This phenomenon might be explained by the elevated levels of p107 and p16INK4A in breast cancer. It was observed that p107 accumulated and augmented in RB-deficient environment and was associated with the moderate suppression of E2F-regulated proteins with the treatment of palbociclib, compensating for the RB loss in multiple breast cancer cell lines [38] and hepatocellular carcinoma models [39]. Besides, CDK4/6 inhibitor p16INK4A and CDKN2A gene were also implicated to play profound roles in drug resistance [56]. RB-deficient tumors tend to demonstrate extremely high expression of p16INK4A [80]. P16INK4A-enriched breast cancer models displayed unresponsive status to palbociclib treatment because CDK4/6 had already been largely suppressed by the endogenous p16INK4A [38]. P16INK4A level plus RB status could be utilized together to predict the response of breast cancer patients to palbociclib therapy [79, 81]. Besides, the deletion or inactivation of CDKN2A gene also predicted sensitivity to palbociclib treatment [56].
Ribociclib
ER-positive breast cancer cell line was the well-established population which was most sensitive to CDK4/6 inhibitors. However, drug-resistant subgroup emerged after chronic ribociclib treatment with no cell cycle G1 block and the upregulation of pCDK2, cyclin A, cyclin D1, and cyclin E in comparison with the parental cells [70]. PDK1 was identified to be capable of sensitizing ER-positive MCF7 cells [70]. The exposure of PDK1 inhibitor GSK2334470 could eliminate the resistance of ribociclib-tolerance breast cancer cells to this compound, with remarkable reduction of pRB, pCDK2, cyclin A, cyclin D1, cyclin E, pS6, and pRSK2 [70]. These results indicated that PDK1 might be involved in the acquired resistance of ER-positive breast cancer to ribociclib treatment.
Rader J reported that most of the studied human neuroblastoma-derived cell lines were sensitive to the proliferation inhibition induced by ribociclib and the other models were completely resistant [40]. Furthermore, it was found that MYCN-enriched cell lines and sensitive cells mostly overlapped, while MYCN-nonamplified cells and resistant cells partially overlapped, indicating that MYCN level was positively associated with the sensitivity of neuroblastoma cells to ribociclib treatment [40].
In spite of the potent anti-proliferaion effects of short-term ribociclib exposure, chronic continuous treatment of ribociclibre-established cell cycle progression with the recovery of RB hyperphosphorylation at sites S780 and S807/811 as well as the up-regulation of cyclins D1, D2, and D3, implicating a compensatory retroaction promoting cell cycle progression [69].
Clinical evidence
Pharmacokinetics
Given the promising results from in vitro and in vivo studies, these three highly selective CDK4/6 inhibitors were further investigated for pharmacokinetics, efficacy, and safety in clinical trials. Reported clinical trials on palbociclib, ribociclib, and abemaciclib in breast cancer are listed in Table 1.
Palboliclib is slowly absorbed and eliminated in cancer patients after orally administrated [82,83,84]. The phase I clinical study in Japanese patients with solid tumors was conducted to assess the pharmacokinetics of palbociclib in patients with solid tumors [83]. Its half-life was 23–26 h and there were no drug to drug interactions between letrozole and palbociclib in this study [83]. Besides, 125 mg once daily over 3 weeks on followed by 1 week off schedule was the maximum tolerated dose (MTD) and was recommended for both monotherapy and combination strategy in ER-positive/HER2-negative advanced breast cancer [83]. Another two phase I studies both enrolling different RB-positive solid tumor patients indicated that palbociclib was slowly absorbed with median time from oral dose to maximum plasma concentration (T max) 4.2 or 5.5 h and slowly eliminated with mean half-life 26.7 or 25.9 h, respectively [82, 84].
According to results of the phase II clinical trial conducted by Infante JR, MTD and recommended dose for expansion (RDE) of ribociclib were 900 and 600 mg daily of 3 weeks on and 1 week off over a 28-day schedule, respectively, based on the assessment on the safety and efficacy of the dose-escalation schedules [85]. Upon oral administration, ribociclib was absorbed with median T max varying from 1 to 5 h [85]. The half-life of ribociclib was approximately 36 h [86] and the average effective half-life was approximately 32 h at the dose of 600 mg daily of 3/1 schedule [85]. During 17 days following oral dosing, plasma concentrations rapidly increased about two- to threefolds because of accumulation [85]. The level of LEQ803, the main active metabolite of ribociclib, was positively linked to the dose of the parent drug ribociclib [85].
Abemaciclib is absorbed slowly ranging from 4 to 6 h from oral dose to maximum plasma concentration [75]. Abemaciclib was widely eliminated and distributed, and the average terminal elimination half-life varied from 17.4 to 38.1 h without significant dose-dependent clearance [75]. The mean top of plasma concentration of patients with 150 and 200 mg twice daily treatment reached 249 and 298 ng/mL, respectively [75]. Also, the cerebrospinal fluid concentration of abemaciclib ranged from 2.2 to 14.7 nmol/L, which was beyond the dissociation constant of CDK4/cyclin D1 combination and was close to the unbound plasma concentrations [75].
Single-agent strategies
According to the phase II clinical study on palbociclib monotherapy enrolling RB-positive advanced breast cancer patients including 31 patients with hormone receptor (HR)-positive/HER2-negative disease, 2 patients with HR-positive/HER2-positive disease, and 4 patients with HR-negative/HER2-negative disease, clinical benefit (CB) was noted in 7 individuals overall, all of which were HR-positive patients after the treatment of palbociclib at the recommonded dose 125 mg daily on the 3/1 schedule [87]. The median PFS of the HR-positive group versus HR-negative population was 4.5 and 1.5 months (P = 0.03), indicating ER-positive breast tumors were more responsive to palbociclib treatment than ER-negative disease [87]. Furthermore, the degree of previous endocrine therapy impacted the efficacy of palbociclib in breast cancer [87]. HR-positive patients who had received more than two lines of anti-hormone regimens enjoyed 3 months longer median PFS than patients who had received less than two lines of these regimens after palbociclib treatment [87]. However, prior therapy of cytotoxic drugs did not significantly affect the median PFS on palbociclib treatment [87].
A phase I dose-escalation clinical study on ribociclib for single-agent therapy, enrolling 132 Rb-positive solid tumors including 20 breast cancer cases, demonstrated that one breast cancer patient with positive status of CCND1 and ER achieved partial responses (PR) at the dose of 600 mg daily during continuous ribociclib treatment [85].
A phase I study was conducted by Patnaik A to assess the pharmacokinetic profile, efficacy, and safety of abemaciclib in cancer patients. In this study, a total of 225 patients were enrolled including breast cancer patients [75]. The efficacy of abemaciclib monotherapy was investigated in 47 breast cancer patients including the following three subtypes: HR-positive/HER2-positive (N = 11), HR-positive/HER2-negative (N = 25), and HR-negative (N = 9) [75]. The overall level of complete response (CR) plus PR plus stable disease (SD) was much higher in HR-positive population than HR-negative subgroup (80 versus 33%) [75]. About 31% achieved PR and 50% achieved SD among 36 HR-positive patients, while none had PR and 33% had SD in nine HR-negative individuals [75]. Furthermore, abemaciclib treatment improved median PFS to greater extent in HR-positive breast cancer patients (8.8 months) than in HR-negative patients (1.1 months) [75]. However, the HER2 status did not make significant difference in the effects of abemaciclib on PFS of HR-positive breast cancer population (7.2 versus 8.8 months) [75]. These data indicated that abemaciclib was highly effective in HR-positive breast cancer for single-agent therapy. In order to further investigate the efficacy and safety of abemaciclib monotherapy, a phase II study was conducted, which included 132 female patients bearing HR-positive/HER2-negative advanced or metastatic breast cancer with disease progression during both hormone therapy and 1 or 2 lines of chemotherapy [86]. Patients received abemaciclib treatment at the dose of 200 mg twice daily continuously. Of patients evaluable for response, the clinical benefit rate including CR, PR, and SD reached 42.4%, and the median PFS was 6 months [86].
Combination strategies
Palbociclib
The phase II trial by Finn RS, enrolled 165 postmenopausal patients with advanced ER-positive/HER2-negative breast cancer who had not received any treatment for this malignant disease [88]. Patients were randomly assigned into two groups receiving continuous oral aromatase inhibitor letrozole 2.5 mg daily alone (N = 81) or letrozole 2.5 mg daily in combination with palbociclib 125 mg daily (N = 84) for 3 weeks on followed by 1 week off over 28-day schedule [88]. Median PFS of these patients was assessed, showing that palbociclib plus letrozole group enjoyed about 10 months longer to progression (20.2 months) than the letrozole group (10.2 months) (hazard ratio (HR) 0.488; 95% confidence interval (CI), 0.319–0.748; one-sided P = 0.0004) [88].
In order to further confirm and extend efficacy and safety data for palbociclib plus letrozole from the phase II study, Finn RS conducted a double-blind phase 3 study enrolling 666 postmenopausal patients with ER-positive/HER2-negative breast cancer who had not received any treatment for this deadly disease [89]. A total of 444 patients were randomly assigned to receive palbociclib plus letrozole and the other patients received matching placebo plus letrozole [89]. The primary endpoint PFS was evaluated and the results indicated that palbociclib in combination with letrozole dramatically prolonged PFS (24.8 months) in comparison with letrozole monotherapy (14.5 months) (HR 0.58; 95% CI, 0.46 to 0.72; P < 0.001) [89].
In addition to the combination of palbociclib and letrozole, the efficacy of palbociclib plus fulvestrant was also investigated in breast cancer patients. Turner NC carried out a phase III trial including a total of 521 patients with HR-positive/HER2-negative advanced breast cancer that progressed in the process of prior hormone therapy [90]. Patients were randomly grouped into two cohorts. Patients in cohort 1 (N = 347) recieved palbociclib (125 mg daily orally for 3/1 schedule) plus fulvestrant (500 mg intramuscularly every 3 weeks for the first three injections and then every 4 weeks), and cohort 2 population received matching placebo plus fulvestrant [90]. Results implicated that there was a clinical meaningful and statistically significant improvement in PFS in patients receiving palbciclib plus fulvestrant (9.2 months) in comparison with the placebo group (3.8 months) (HR 0.42; 95% CI, 0.32 to 0.56; P < 0.001) [90].
Ribociclib
A phase 3 trial carried out by Hortobagyi GN, enrolled a total of 668 female postmenopausal patients with HR-positive/HER2-negative recurrent or metastatic breast cancer who had not receive systemic therapy for this advanced disease previously [91]. Among these patients, half were assigned to orally administer ribociclib (600 mg daily for 3 week on and 1 week off schedule) plus letrozole (2.5 mg daily), and the others were grouped to receive the treatment of matching placebo plus letrozole [91]. PFS was the primary endpoint of the study and overall response rate was one of the second endpoints [91]. The HR on PFS for ribociclib group versus placebo group was 0.56 (95% CI 0.43–0.72; P < 0.001). After 18 months, the PFS rate of ribociclib group was 63% (95% CI 54.6–70.3) and the placebo was 42.2% (95% CI 34.8–49.5) [91]. The overall response rates of ribociclib group and placebo group were 52.7 and 37.1%, respectively (P < 0.001) [91].
In consideration of the synergistic anti-tumor effects of ribociclib in combination with alpelisib in vitro and in vivo, this combination was also assessed in clinical trials. The phase Ib/2 study was conducted by Bardia A to investigate the safety and efficacy of triple combination of ribociclib plus exemestane and everolimus, enrolling 70 postmenopausal ER-positive/HER2-negative advanced breast cancer patients with letrozole- or anastrozole-resistance [86]. The recommended dose for phase II studies (RP2D) was established at 300 mg daily over 3/1 schedule plus 2.5 mg daily continously plus 25 mg daily continuously for ribociclib, everolimus and exemestane, respectively. Of 55 patients evaluable for response, 1 patient achieved CR, 5 patients achieved PR, and 26 individuals achieved SD [86].
Another phase Ib/2 trial assessed the triple combination of ribociclib plus letrozole and alpelisib in ER-positive/HER2-negative breast cancer patients. In this study, patients were grouped in three cohorts: cohort 1 (N = 41) for ribociclib plus letrozole, cohort 2 (N = 21) for alpelisib plus letrozole, and cohort (N = 36) for ribociclib plus alpelisib [86]. RP2D was established at 300 mg daily over 3 weeks on followed by 1 week off schedule plus 200 mg daily continuously plus 25 mg daily continuously for ribociclib, alpelisib, and letrozole, respectively [86]. Among 27 patients evaluable for response, PR was observed in 6 patients including 2 in confirmed PR and 4 in unconfirmed PR, and SD was also noted in 6 patients [86].
Abemaciclib
The phase I study carried out by Patnaik A which was described previously in this review, not only assessed the efficacy of abemaciclib monotherapy but also investigated the combination of abemaciclib and the antiestrogen agent fulvestrant [75]. In this study, 19 HR-positive breast cancer patients were enrolled in this cohort. Among this population, four patients (21%) achieved PR. The clinical benefit rate was 63%, which was similar to that of the single-agent strategy [75]. A phase Ib study showed that the disease control rate of CR, PR and SD was 67% in 36 breast cancer patients with the treatment of abemaciclib plus letrozole or abemaciclib plus anastrozole, and 75% of 16 patients with abemaciclib plus tamoxifen [86].
Safety profile
The management of drug-related adverse events is a pivotal aspect of treatment. Reported clinical adverse events caused by palbociclib include neutropenia, leucopenia, fatigue, pulmonary embolism, back pain, and diarrhea. Among these, neutropeniais the primary toxicity of palbociclib [82,83,84, 87, 88, 92]. Previous study conducted by Flaherty KT enrolled 41 patients with distinct RB-positive solid tumors including melanoma, breast and other types, demonstrated that neutropenia is the only dose-limiting event and the most common non-hematologic adverse effects included fatigue, nausea, and diarrhea [84]. According to the phase 2 study by Finn RS, grades 3–4 neutropenia was noted in about half of advanced breast cancer patients treated with palbociclib plus letrozole, while in only 1% of patients treated with letrozole alone [88]. For leucopenia and fatigue, it was 19% versus none and four (4%) versus one (1%), respectively [88]. Furthermore, serious adverse events such as back pain, pulmonary embolism and diarrhea occurred in 2, 4, and 2% of palbociclib plus letrozole group, respectively [88]. But, febrile neutropenia or neutropenia-related infections were not observed among these patients during this study [88]. Also, according to the phase 3 study of 521 women with ER-positive/HER2-negative advanced or metastatic breast cancer, the adverse events were most commonly observed in palbociclib plus fulvetrant group in comparison with the placebo plus fulcestrant group, including neutropenia (62.0 versus 0.6%), leukopenia (25.2 versus 0.6%), anemia (2.6 versus 1.7%), thrombocytopenia (2.3 versus 0%), and fatigue (2.0 versus 1.2%) [90].
The safety of ribociclib was also assessed in clinical trials. According to the phase 3 clinical trial by Hortobagyi GN, common grade 3 or 4 adverse events were neutropenia (59.3% in the ribociclib group and 0.9% in the placebo group) and leukopenia (21.0 versus 0.6%) [91]. Infante JR reported that neutropenia and thrombocytopenia were the most common dose limiting toxicities (DLT) according to the MTD determination on seventy patients after cycle 1 treatment [85]. The most common hematologic adverse events were treatment-related neutropenia, leukopenia, thrombocytopenia and anemia, and the most common non-hematologic treatment-related adverse events were fatigue, nausea, and vomiting for all grades [85]. Approximately 9% of patients treated at 600 mg daily of 3 weeks on followed by 1 week off schedule experienced treatment-related asymptomatic QTcF prolongation, but grade 3/4 asymptomatic QTcF prolongation only occurred at the dose of more than 900 mg daily [85]. It was reversible and parallel with the maximal plasma concentration kinetics [85].
Abemaciclib treatment represents a distinct toxicity profile. In contrast to palbociclib and ribociclib, the DLT of abemaciclib was fatigue and this agent produced relatively less neutropenia might be due to the higher specific selectivity of this agent for CDK4 than for CDK6 [86]. The clinical study of a total of 225 patients with multiple cancer types showed that abemaciclib treatment related adverse events of all grades included diarrhea, nausea, fatigue, vomiting, leukopenia, thrombocytopenia, neutropenia, anemia, anorexia, increased creatinine, and weight loss [75]. The most common adverse events caused by abemaciclib treatment included fatigue and the gastrointestinal, renal and hematopoietic systems [75]. Grade 3 fatigue was DLT and the MTD was 200 mg every 12 h [75]. At 200 mg twice daily, one out of seven patients experienced DLT of grade 3 fatigue, and 275 mg twice daily endowed two out of three patients with the DLT of grade 3 fatigue [75]. According to a phase 1 clinical trial of 12 cancer patients demonstrated that diarrhea was the most common treatment–emergent adverse event and it could be managed to have no effects on the continuation of abemaciclib treatment at the dose of 200 mg twice daily [93].
Conclusions
Taken together, oral highly selective CDK inhibitors, including palbociclib, ribociclib, and abemaciclib, represent an important therapeutic advancement in breast oncology. Apart from the clinical success of palbociclib and ribociclib, abemaciclib is in active investigation and the favorable effects of this agent on PFS of ER-positive/HER2-negative advanced breast cancer were observed in clinical trials. However, there are still some challenges in the optimization of CDK inhibitors in clinical practice. Firstly, there is still lack of predictive biomarkers to screen appropriate population who can benefit most from these agents. Selection of sensitive patients can improve the cost-effective ratio of these drugs. Although several studies have implied some potential candidates for sensitivity prediction such as the protein levels of RB and p16, further extensive clinical trials are urgently needed before applied as clinically useful biomarkers [94]. In consideration that liquid biopsy is a new technique for monitor tumor progression and treatment response [95,96,97], is it possible to identify potential biomarkers for predicting response to CDK inhibitors through analyzing circulating breast cancer cells or cell-free DNA? Secondly, aside from the already investigated combination strategies, whether CDK4/6 inhibitors in combination with other therapeutic regimens including chemotherapy, radiotherapy and immunecheckpoint inhibitors are also more effective than monotherapy is a problem, which is imperatively needed to be solved. For instance, there is some doubt whether palbociclib antagonizes the anti-tumor effects of cytotoxic chemotherapy and radiotherapy which function through killing cancer cells in cell cylce. Thirdly, triple-negative breast cancer, which is characterized with comparatively aggressive phenotype and the absent status of ER, PR, and HER2, is still lack of efficient treatment strategy. Previous study by Asghar U indicated that a subset of triple-negative breast cancer cells with expression of AR and the loss of cyclin E1 could be responsive to CDK4/6 inhibition [86]. Clinical trials are ongoing or in plans to address these questions.
Abbreviations
- ABC:
-
ATP-binding cassette
- AR:
-
Androgen receptor
- CB:
-
Clinical benefit
- CDK4/6:
-
Cyclin-dependent kinases 4/6
- CI:
-
Confidence interval
- CR:
-
Complete response
- DLT:
-
Dose-limiting toxicities
- ER:
-
Estrogen receptor
- FDA:
-
Food and Drug Administration
- FOXM1:
-
Forkhead box M1
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hormone receptor
- HR:
-
Hazard ratio
- MAPKs:
-
Mitogen-activated protein kinases
- MTD:
-
Maximum tolerated dose
- NF-kB:
-
Nuclear factor kB
- PDK1:
-
3-phosphoinositide-dependent protein kinase 1
- PFS:
-
Progression-free survival
- PI3K:
-
Phosphatidylinositol 3-kinase
- PR:
-
Progesterone receptor
- PR:
-
Partial responses
- RB:
-
Retinoblastoma
- RDE:
-
Recommended dose for expansion
- RP2D:
-
Recommended dose for phase II studies
- SD:
-
Stable disease
- STATs:
-
Signal transducers and activators of transcription
- T-ALL:
-
T cell acute lymphoblastic leukemia
- T max :
-
Time from oral dose to maximum plasma concentration
- TNBC:
-
Triple-negative breast cancer
Reference
Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nature reviews Cancer. 2009;9(3):153–66.
Xu H, Wu K, Tian Y, Liu Q, Han N, Yuan X, et al. CD44 correlates with clinicopathological characteristics and is upregulated by EGFR in breast cancer. Int JOncol. 2016;49(4):1343–50.
Xu HX, Wu KJ, Tian YJ, Liu Q, Han N, He XL, et al. Expression profile of SIX family members correlates with clinic-pathological features and prognosis of breast cancer: A systematic review and meta-analysis. Medicine. 2016;95(27), e4085.
Perurena N, Zandueta C, Martínez-Canarias S, Moreno H, Vicent S, Almeida AS, et al. EPCR promotes breast cancer progression by altering SPOCK1/testican 1-mediated 3D growth. J Hematol Oncol. 2017;10(1):23.
Knutson TP, Truong TH, Ma S, Brady NJ, Sullivan ME, Raj G, et al. Posttranslationally modified progesterone receptors direct ligand-specific expression of breastcancer stem cell-associated gene programs. J Hematol Oncol. 2017;10(1):89.
Di Lauro L, Pizzuti L, Barba M, Sergi D, Sperduti I, Mottolese M, et al. Role of gonadotropin-releasing hormone analogues in metastatic male breast cancer: results from a pooled analysis. J Hematol Oncol. 2015;8:53.
Zhang P, Tong Z, Tian F, Wang Y, Yang J, Li W, et al. Phase II trial of utidelone as monotherapy or in combination with capecitabine in heavily pretreated metastatic breast cancer patients. J Hematol Oncol. 2016;9(1):68.
Song DG, Ye Q, Poussin M, Chacon JA, Figini M, Powell Jr DJ. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. J Hematol Oncol. 2016;9(1):56.
Lu J. Palbociclib: a first-in-class CDK4/CDK6 inhibitor for the treatment of hormone-receptor positive advanced breast cancer. J Hematol Oncol. 2015;8:98.
Yu S, Li A, Liu Q, Li T, Yuan X, Han X, et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol. 2017;10(1):78.
Udvardy A. The role of controlled proteolysis in cell-cycle regulation. Eur J Biochem. 1996;240(2):307–13.
Coudreuse D, Nurse P. Driving the cell cycle with a minimal CDK control network. Nature. 2010;468(7327):1074–9.
Hunt T, Nasmyth K, Novak B. The cell cycle. Philos Trans R Soc Lond B Biol Sci. 2011;366(1584):3494–7.
Harper JW, Adams PD. Cyclin-dependent kinases. Chem Rev. 2001;101(8):2511–26.
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–46.
Shah AN, Cristofanilli M. The Growing Role of CDK4/6 Inhibitors in Treating Hormone Receptor-Positive Advanced Breast Cancer. Curr Treat Options Oncol. 2017;18(1):6.
Sablin MP, Ricci F, Loirat D, Jobard A, Basse C, Romano E, et al. Cell cycle inhibitors in endocrine receptor positive breast cancer. Bull Cancer. 2017;104(2):114–22.
Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672–7.
Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9(11):785–97.
Johnson J, Thijssen B, McDermott U, Garnett M, Wessels LF, Bernards R. Targeting the RB-E2F pathway in breast cancer. Oncogene. 2016;35(37):4829–35.
Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016;45:129–38.
VanArsdale T, Boshoff C, Arndt KT, Abraham RT. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin Cancer Res. 2015;21(13):2905–10.
Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366(6456):704–7.
Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature. 1998;395(6699):237–43.
Jeffrey PD, Tong L, Pavletich NP. Structural basis of inhibition of CDK-cyclin complexes by INK4 inhibitors. Genes Dev. 2000;14(24):3115–25.
Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24(11):1770–83.
Sedlacek H, Czech J, Naik R, Kaur G, Worland P, Losiewicz M, et al. Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int J Oncol. 1996;9(6):1143–68.
Wu K, Wang C, D'Amico M, Lee RJ, Albanese C, Pestell RG, et al. Flavopiridol and trastuzumab synergistically inhibit proliferation of breast cancer cells: association with selective cooperative inhibition of cyclin D1-dependent kinase and Akt signaling pathways. Mol Cancer Ther. 2002;1(9):695–706.
Mahoney E, Byrd JC, Johnson AJ. Autophagy and ER stress play an essential role in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Autophagy. 2013;9(3):434–5.
Carlson BA, Dubay MM, Sausville EA, Brizuela L, Worland PJ. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res. 1996;56(13):2973–8.
Kelland LR. Flavopiridol, the first cyclin-dependent kinase inhibitor to enter the clinic: current status. Expert Opin Investig Drugs. 2000;9(12):2903–11.
Bose P, Simmons GL, Grant S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin Investig Drugs. 2013;22(6):723–38.
VanderWel SN, Harvey PJ, McNamara DJ, Repine JT, Keller PR, Quin 3rd J, et al. Pyrido[2,3-d]pyrimidin-7-ones as specific inhibitors of cyclin-dependent kinase 4. J Med Chem. 2005;48(7):2371–87.
Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–38.
Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48(7):2388–406.
Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29(28):4018–32.
Rivadeneira DB, Mayhew CN, Thangavel C, Sotillo E, Reed CA, Grana X, et al. Proliferative suppression by CDK4/6 inhibition: complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterology. 2010;138(5):1920–30.
Rader J, Russell MR, Hart LS, Nakazawa MS, Belcastro LT, Martinez D, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013;19(22):6173–82.
Dickson MA. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin Cancer Res. 2014;20(13):3379–83.
Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26(1):136–49.
Tate SC, Cai S, Ajamie RT, Burke T, Beckmann RP, Chan EM, et al. Semi-mechanistic pharmacokinetic/pharmacodynamic modeling of the antitumor activity of LY2835219, a new cyclin-dependent kinase 4/6 inhibitor, in mice bearing human tumor xenografts. Clin Cancer Res. 2014;20(14):3763–74.
Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. 2014;32(5):825–37.
Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22(4):438–51.
Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20(5):620–34.
Li SK, Smith DK, Leung WY, Cheung AM, Lam EW, Dimri GP, et al. FoxM1c counteracts oxidative stress-induced senescence and stimulates Bmi-1 expression. J Biol Chem. 2008;283(24):16545–53.
Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, et al. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009;28(19):2908–18.
Liu F, Korc M. Cdk4/6 inhibition induces epithelial-mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther. 2012;11(10):2138–48.
Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77.
Whiteway SL, Harris PS, Venkataraman S, Alimova I, Birks DK, Donson AM, et al. Inhibition of cyclin-dependent kinase 6 suppresses cell proliferation and enhances radiation sensitivity in medulloblastoma cells. J Neurooncol. 2013;111(2):113–21.
Logan JE, Mostofizadeh N, Desai AJ, VON Euw E, Conklin D, Konkankit V, et al. PD-0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity. Anticancer Res. 2013;33(8):2997–3004.
Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006;66(15):7661–7.
Marzec M, Kasprzycka M, Lai R, Gladden AB, Wlodarski P, Tomczak E, et al. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood. 2006;108(5):1744–50.
Ismail A, Bandla S, Reveiller M, Toia L, Zhou Z, Gooding WE, et al. Early G(1) cyclin-dependent kinases as prognostic markers and potential therapeutic targets in esophageal adenocarcinoma. Clin Cancer Res. 2011;17(13):4513–22.
Young RJ, Waldeck K, Martin C, Foo JH, Cameron DP, Kirby L, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014;27(4):590–600.
Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18(1):63–73.
McClendon AK, Dean JL, Rivadeneira DB, Yu JE, Reed CA, Gao E, et al. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle. 2012;11(14):2747–55.
Trape AP, Liu S, Cortes AC, Ueno NT, Gonzalez-Angulo AM. Effects of CDK4/6 Inhibition in Hormone Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer Cells with Acquired Resistance to Paclitaxel. J Cancer. 2016;7(8):947–56.
Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, Ozawa T, et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70(8):3228–38.
Tang LH, Contractor T, Clausen R, Klimstra DS, Du YC, Allen PJ, et al. Attenuation of the retinoblastoma pathway in pancreatic neuroendocrine tumors due to increased cdk4/cdk6. Clin Cancer Res. 2012;18(17):4612–20.
Aoki Y, Hashizume R, Ozawa T, Banerjee A, Prados M, James CD, et al. An experimental xenograft mouse model of diffuse pontine glioma designed for therapeutic testing. J Neurooncol. 2012;108(1):29–35.
Barton KL, Misuraca K, Cordero F, Dobrikova E, Min HD, Gromeier M, et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PloS One. 2013;8(10), e77639.
Roberts PJ, Bisi JE, Strum JC, Combest AJ, Darr DB, Usary JE, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104(6):476–87.
Tao YF, Wang NN, Xu LX, Li ZH, Li XL, Xu YY, et al. Molecular mechanism of G1 arrest and cellular senescence induced by LEE011, a novel CDK4/CDK6 inhibitor, in leukemia cells. Cancer Cell Int. 2017;17:35.
Wood AC, Krytska K, Ryles HT, Infarinato NR, Sano R, Hansel TD, et al. Dual ALK and CDK4/6 Inhibition Demonstrates Synergy against Neuroblastoma. Clin Cancer Res. 2016;doi: 10.1158/1078-0432.CCR-16-1114.
Hart LS, Rader J, Raman P, Batra V, Russell MR, Tsang M, et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. ClinCancer Res. 2016;doi: 10.1158/1078-0432.CCR-16-1131.
Aristizabal Prada ET, Noelting S, Spoettl G, Maurer J, Auernhammer CJ. The Novel Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) Alone and in Dual-Targeting Approaches Demonstrates Antitumoral Efficacy in Neuroendocrine Tumors in vitro. Neuroendocrinology. 2017;doi: 10.1159/000463386.
Zhang YX, Sicinska E, Czaplinski JT, Remillard SP, Moss S, Wang Y, et al. Antiproliferative effects of CDK4/6 inhibition in CDK4-amplified human liposarcoma in vitro and in vivo. Mol Cancer Ther. 2014;13(9):2184–93.
Jansen VM, Bhola NE, Bauer JA, Formisano L, Lee KM, Hutchinson KE, et al. Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Cancer Res. 2017;doi: 10.1158/0008-5472. [Epub ahead of print] .
Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell. 2016;29(3):255–69.
Wu T, Chen Z, To KK, Fang X, Wang F, Cheng B, et al. Effect of abemaciclib (LY2835219) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Biochem Pharmacol. 2017;124:29–42.
Yadav V, Burke TF, Huber L, Van Horn RD, Zhang Y, Buchanan SG, et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer Ther. 2014;13(10):2253–63.
Sathe A, Koshy N, Schmid SC, Thalgott M, Schwarzenbock SM, Krause BJ, et al. CDK4/6 Inhibition Controls Proliferation of Bladder Cancer and Transcription of RB1. J Urol. 2016;195(3):771–9.
Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016;6(7):740–53.
Raub TJ, Wishart GN, Kulanthaivel P, Staton BA, Ajamie RT, Sawada GA, et al. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab Dispos. 2015;43(9):1360–71.
Ku BM, Yi SY, Koh J, Bae YH, Sun JM, Lee SH, et al. The CDK4/6 inhibitor LY2835219 has potent activity in combination with mTOR inhibitor in head and neck squamous cell carcinoma. Oncotarget. 2016;7(12):14803–13.
Ellis LM, Hicklin DJ. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin Cancer Res. 2009;15(24):7471–8.
Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle. 2012;11(14):2756–61.
Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011;10(15):2497–503.
Konecny GE, Winterhoff B, Kolarova T, Qi J, Manivong K, Dering J, et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res. 2011;17(6):1591–602.
Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br JCancer. 2011;104(12):1862–8.
Tamura K, Mukai H, Naito Y, Yonemori K, Kodaira M, Tanabe Y, et al. Phase I study of palbociclib, a cyclin-dependent kinase 4/6 inhibitor, in Japanese patients. Cancer Sci. 2016;107(6):755–63.
Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012;18(2):568–76.
Infante JR, Cassier PA, Gerecitano JF, Witteveen PO, Chugh R, Ribrag V, et al. A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clin Cancer Res. 2016;22(23):5696–705.
Barroso-Sousa R, Shapiro GI, Tolaney SM. Clinical Development of the CDK4/6 Inhibitors Ribociclib and Abemaciclib in Breast Cancer. Breast Care. 2016;11(3):167–73.
DeMichele A, Clark AS, Tan KS, et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb + advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res. 2015;21(5):995–1001.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16(1):25–35.
Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med. 2016;375(20):1925–36.
Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2015;373(3):209–19.
Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med. 2016;375(18):1738–48.
Turner NC, Huang Bartlett C, Cristofanilli M. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2015;373(17):1672–73.
Fujiwara Y, Tamura K, Kondo S, Tanabe Y, Iwasa S, Shimomura A, et al. Phase 1 study of abemaciclib, an inhibitor of CDK 4 and 6, as a single agent for Japanese patients with advanced cancer. Cancer Chemother Pharmacol. 2016;78(2):281–8.
Polk A, Kolmos LL, Kumler L, Nielsen DL. Specific CDK4/6 inhibition in breast cancer: a systematic review of current clinical evidence. ESMO Open. 2017;1, e000093.
Leong SM, Tan KM, Chua HW, Tan D, Fareda D, Osmany S, et al. Sampling circulating tumor cells for clinical benefits: how frequent? J Hematol Oncol. 2015;8:75.
Sun W, Yuan X, Tian Y, Wu H, Xu H, Hu G, et al. Non-invasive approaches to monitor EGFR-TKI treatment in non-small-cell lung cancer. J Hematol Oncol. 2015;8:95.
Wang S, Su X, Bai H, Zhao J, Duan J, An T, et al. Identification of plasma microRNA profiles for primary resistance to EGFR-TKIs in advanced non-small cell lung cancer (NSCLC) patients with EGFR activating mutation. J Hematol Oncol. 2015;8:127.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) No. 81572608 and 81172422 (KW), and also supported in part by NIH grant R01CA132115 (RGP).
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Authors’ contributions
HX performed the selection of literature, drafted the manuscript, and prepared the figures. SY and XY collected the related references. SM revised the language. RP and KW carried out the design of this review and revised the manuscript. All authors contributed to this manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xu, H., Yu, S., Liu, Q. et al. Recent advances of highly selective CDK4/6 inhibitors in breast cancer. J Hematol Oncol 10, 97 (2017). https://doi.org/10.1186/s13045-017-0467-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-017-0467-2