
Abstract
Background
Uniparental disomy (UPD) is a rare genetic condition leading to potential disease risks. Maternal UPD of chromosome 6 upd(6)mat is exceptionally rare, with limited cases reported. This study reported two new cases of upd(6)mat and reviewed the literature of previous cases.
Case presentation
Both cases exhibited intrauterine growth restriction (IUGR), and genetic analysis confirmed upd(6)mat in each case. The literature review identified a total of 19 cases. IUGR and preterm labor were the most common two symptoms observed, and additional anomalies and genetic variations were also reported in some cases.
Conclusion
upd(6)mat is potentially associatied with IUGR, but the precise genotype–phenotype relationship remains unclear. The cases with upd(6)mat may present clinical features due to imprinting disorders.
Similar content being viewed by others


Introduction
Uniparental disomy (UPD) is a situation where both homologous chromosomes are inherited from a single parent [1]. The mechanisms leading to UPD include trisomy rescue, monosomy rescue, mitotic crossing over, and chromosomal rearrangements [2]. UPD manifests in two forms: isodisomy, characterized by two identical copies of the chromosome inherited from one parent, and heterodisomy, where both homologous chromosomes originate from one parent. UPD can cause diseases by increasing the risk of autosomal recessive diseases or imprinted disorders [3]. Imprinted regions, regulated by epigenetic mechanisms such as DNA methylation, encompass genes that express in parent-of-origin specific patterns. The occurrence of UPD can result in the overexpression or silencing of imprinted genes.
Specific phenotypes of UPD have been established in maternal UPD for chromosomes 7, 11, 14, 15, and 20 [2] and paternal UPD for chromosomes 6, 11, 14, 15, and 20. On the other hand, it is still disputable for some chromosomes whether UPD has phenotypic effects attributable to imprinted disorders. Segmental UPD refers to a situation where only a part of a chromosome is inherited from one parent, while the rest is biparental. Segmental UPD can be either heterodisomic or isodisomic. Mixed hetero- and isodisomy combines heterodisomy and isodisomy within the same chromosome [4]. Maternal UPD of chromosome 6 is scarce, with less than 20 cases reported to date. The clinical presentation of maternal UPD6 is variable. Intrauterine growth restriction (IUGR) and preterm labor were the most common presentation observed. No clear phenotype-genotype correlation has been established until now. It is also worth noting that the presence of (hidden) mosaicism for trisomy 6 poses a potential risk, and its impact on the phenotype should be considered in the analysis of UPD6 cases [5].
This study presents two new prenatal cases of maternal UPD6, both of which exhibited IUGR. The aim of this study was to contribute new cases to the existing literature on this rare genetic condition. Furthermore, we conducted a comprehensive review of previous literature to summarize the cases of maternal UPD6.
Case presentation
Case 1
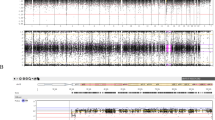
The mother was 33 years old, gravida 3 para 1, and the father was 34 years old. The couple was non-consanguineous and belonged to the Chinese Han population. The mother had a Hepatitis B Virus (HBV) infection. She gave birth to a healthy girl and had one abortion due to non-medical reasons. Fetal growth, as assessed during nuchal translucency (NT) screening at 12 + 6 gestational weeks (gw), was consistent with the expected measurements (Crown-Rump Length (CRL): 66.8 mm), and the NT was within a normal range (1.5 mm). The non-invasive prenatal testing (NIPT) plus test at 17 gw showed a low risk of chromosome aneuploidy. At 23 gw, ultrasound screening revealed signs of intrauterine growth retardation (IUGR), indicated by Biparietal Diameter (BPD): 5.37 cm (− 0.88SD), Head Circumference (HC): 20.18 cm (− 1.606SD), Abdominal Circumference (AC): 17.15 cm (− 1.606SD), Femur Length (FL): 3.77 cm (− 0.868SD), and an estimated fetal weight of 477 g (6.6%). At 30 gw, amniocentesis was performed for genetic testing and fetal magnetic resonance imaging (MRI) was performed for fetal structural examination. The FISH (Fluorescence in situ hybridizatio)analysis of the fetus did not indicate 13, 18, 21 and sex chromosome aneuploidy abnormalities. We utilized chromosome analysis by medium coverage whole genome sequencing (CMA-seq), a method optimized for reliable the absence of heterozygosity (AOH) detection, revealing the AOH of 26.1 Mb at 6p25.3p22.2 (200000-26300000) and 36.8 Mb at 6q15q22.31 (89300000-126100000) on chromosome 6, based on the hg19 human genome reference. Clinical exome-sequencing (CES) identified maternal UPD of at least 167.63 Mb (2959513-170592945) on chromosome 6. The heterodisomy UPD regions included 57.34 Mb at chr6: 30887972-88224164 and 41.08 Mb at chr6: 129511373-170592945. The isodisomy UPD region included 37.27 Mb at chr6: 91266350-123539904 (Fig. 1), all based on the hg19 reference. No pathogenic copy number variants (CNVs) or single nucleotide variants (SNVs) were detected in the fetus. Fetal MRI confirmed the ultrasound findings and did not reveal any additional malformations. IUGR was persistent at 33 gw, and ultrasound showed BPD 7.85 cm (− 2.45SD), HC 28.02 cm (− 2.67SD), AC 25.02 cm (− 3.14SD), FL 5.66 cm (− 2.08SD), and estimated fetal weight 1425 g (0.2%). Consequently, the parents elected to terminate the pregnancy due to severe IUGR. Post-mortem and examination of the placenta were declined.

UPD for chromosome 6 in two patients. Medium coverage whole genome sequencing showed AOH regions of chromosome 6 for Case 1 (A) and Case 2 (C). Clinical exome sequencing showed the UPD chromosome 6 had both isodisomy and heterodisomy for Case 1 (B) and Case 2 (D) The AOH scatter plots and the Z-score distribution of chromosome 6 were shown. Z-scores were calculated for each 25 kb window for each sample across the batch. Marked AOH regions when at least four consecutive windows had Z-scores below − 2 or above 2
Case 2
The mother was 32 years old, gravida 2 para 1, and the father was 33 years old. The couple was non-consanguineous, and both were Chinese Han populations. The mother presented with a decreased mean corpuscular volume (MCV), and thalassemia gene screening yielded the expected results. Her previous child was a healthy boy. Fetal ultrasound at 29 gw revealed the following measurements: BPD: 6.63 cm (− 2.668SD), HC: 24.81 cm (− 3.139SD), AC: 21.95 cm (− 2.842SD), FL: 5.13 cm (− 1.31SD), indicating IUGR. The ultrasound also detected a cystic mass in the lower right abdomen, raising suspicion of intestinal duplication. Amniocentesis and fetal MRI were performed on the same day. The FISH analysis of the fetus did not indicate 13, 18, 21 and sex chromosome aneuploidy abnormalities. CMA-seq identified three regions of AOH: 26.2 Mb at 6p21.32p11.1 (chr6:32700000-58900000), 28.4 Mb at 6q11.1q15 (62400000-90800000), and 29.8 Mb at 6q23.3q27 (138400000-168200000). Clinical Exome Sequencing (CES) revealed maternal UPD of at least 170.49 Mb (393359-170878793) at chromosome 6. The heterodisomy regions included 31.98 Mb at chr6:393359-32370927 and 91.27 Mb at chr6:91266350-138196066. The isodisomy UPD region included 16.57 Mb at chr6:33408632-88224164 and 14.96 Mb at chr6:152469188-167424293 (Fig. 1). No pathogenic copy number variants (CNVs) or single nucleotide variants (SNVs) were detected in the fetus. Fetal abdominal MRI findings were consistent with the ultrasound results. The pregnancy was terminated, and a post-mortem examination was declined.All participants provided informed consent prior to their inclusion in the study.
Literature review
The literature search was conducted using PubMed (https://pubmed.ncbi.nlm.nih.gov) with the search terms "Maternal uniparental disomy chromosome 6" or "Maternal UPD6". A total of 19 cases, including 2 cases from our study, were summarized from 15 articles [6,7,8,9,10,11,12,13,14,15,16,17,18,19]. The following information of each case was reviewed: sex, prenatal/postnatal phenotypes, placenta findings, size of UPD, type of UPD, karyotype, and other genetic findings (Table 1).
Prenatal conditions were mentioned in 16 cases, while the other 3 cases lacked description. Among the cases with available information, 13 cases (68.4%) presented with IUGR, the most common prenatal symptom. Ten cases (19.0%) combined with other anomalies, including oligohydramnios and malformations in the brain, heart, face, or genitalia. Preterm and low birth weight was reported in 8 cases (42.1%), and developmental delay were shown in 7 cases (36.8%). Postnatal phenotypes varied considerably due to the range of ages at the time of reporting, spanning from neonatal to 52 years old, as well as the specific diagnoses of associated diseases. Placental findings were not documented in the majority of cases. However, placental insufficiency was reported in 3 cases (15%), and mosaic trisomy 6 was observed in 2 cases (10.5%).
Ten cases (52.6%) were identified as isolated upd(6)mat without other genetic findings. Six cases (36.8%) had autosomal recessive (AR) diseases due to isodisomy maternal UPD6 involving CYP21A2, MOCS1, CUL7, and TULP1. One case (5.3%) had an X-linked disease due to a heterozygous variant in Wiskott-Aldrich Syndrome Protein (WASP). Three cases (15.8%), including one case with AR disease, exhibited mosaic chromosomal aneuploidies involving chromosome 6 or chromosome X. The size of maternal UPD6 mainly affected whole chromosome 6 (42.1%).
Discussion
UPD has an overall prevalence of 1 in 2000 births, in which the prevalence of disease-associated UPD is approximately 1 in 3500–5000 [20, 21]. upd(6)pat is associated with neonatal transient diabetes. Historically, upd(6)mat was considered a rare occurrence, with only 19 reported cases worldwide since 1996. Among the total 19 cases, 13 reported IUGR, 3 cases did not present IUGR, and 3 cases whose prenatal condition were not mentioned. One case (Case 9) reported low birth weight, which indicates IUGR, while the other 2 cases (Case 11 and 17) were both preterm. Thus, the proportion of IUGR in upd(6)mat cases might be higher than previously assumed. Recent research by Mackay et al. [22] has emphasized the previously underestimated significance of upd(6)mat in imprinting diagnostics, especially concerning Silver-Russell syndrome (SRS). The widely used commercial diagnostic kit for SRS has been instrumental in the global diagnosis of children with suspected SRS. This has led to the discovery of numerous upd(6)mat cases in major diagnostic labs, often attributing growth restriction to this UPD. Their study highlighted seven such cases detected across nine diagnostic labs, suggesting that upd(6)mat is emerging as a more frequent related diagnosis, even surpassing upd(20)mat, challenging previous notions of its rarity.
In China, IUGR is one of the prevalent reasons for delayed termination of pregnancy. upd(6)mat, as a rare genetic anomaly, presents unique challenges in the Chinese context. The intersection of medical complexities and cultural factors may significantly influence parental attitudes and decisions toward fetal anomalies, highlighting the need for a comprehensive approach that considers both medical and cultural impacts on family decision-making.
When isodisomy occurs, the entire chromosome shows an absence of heterozygosity, significantly increasing the risk of autosomal recessive (AR) diseases. According to the exome-based sequencing results, there was no overlap of isodisomy regions in our two cases. Furthermore, clinical exome sequencing for genes with definite corresponding diseases collected from the Online Mendelian Inheritance in Man (OMIM) database revealed no suspicious variants that could account for IUGR. These results substantially diminished the possibility that AR diseases accounted for IUGR in these two cases. In our study, we provided NIPT results for only one case, indicating a low possibility of mosaic trisomy 6 in the placenta. However, we acknowledge that a normal NIPT result cannot definitively exclude the presence of low-level mosaicism in the placenta. Furthermore, while karyotyping of placenta can offer insights into the presence of mosaic trisomy 6, we did not conduct this analysis in our study. This omission limits our understanding of the potential contribution of an underlying mosaic trisomy 6 cell line in the placenta to the observed growth restriction.
Considering the genetic test results and NIPT results, we highly suspected that upd(6)mat was responsible for IUGR in these two cases. It is important to note that while in many known imprinting disorders, paternal UPD is often associated with overgrowth and maternal UPD with growth retardation, there are exceptions. Specifically, paternal UPD 6 has been observed to result in IUGR/postnatal growth retardation(PNGR). For instance, Temple syndrome associated with UPD(14)mat [23], Russell-Silver syndrome associated with UPD(7)mat or UPD(11)mat [24, 25], and Mulchandani-Bhoj-Conlin syndrome caused by UPD(20)mat [24,25,26], all result in prenatal and postnatal growth retardation. Conversely, UPD(14)pat, associated with Kagami-Ogata syndrome, is a severe overgrowth syndrome characterized by fetal macrosomia, polyhydramnios, and abdominal wall defects. Paternal UPD11 is also associated with fetal overgrowth, identified as Beckwith-Wiedemann syndrome (BWS) [2]. Among the upd(6)mat cases in the literature, most of the UPDs encompass the entire chromosome 6 or involve the known imprinting region 6q24.2. Cases 15 and 16 received methylation-specific single-nucleotide primer extension (MS-SNuPE) and methylation-specific multiplex ligation-dependent probe amplification (MLPA) for PLAGL1 and IGF2R differentially methylated regions (DMRs) in 6q24. The results from these cases showed hypermethylation in these regions compared with normal controls, indicating an imprinting defect. In the broader context of chr6, beyond PLAGL1, there are other imprinted genes. Some of these, as detailed by Court et al. [27], are germline-imprinted, while others, as described by Hanna et al. [28], are placental. While the phenotype of upd(6)mat remains somewhat unclear, the association of upd(6)mat with certain imprinting defects, such as the hypermethylation of PLAGL1 TSS-DMR, is well-established. As sequencing technology advances in its application for disease diagnosis and prenatal diagnosis, more cases of upd(6)mat will likely be identified, further solidifying the genotype–phenotype correlation. For future upd(6)mat cases, systemic methylation tests such as methylation sequencing are recommended to uncover the full spectrum of imprinting defects caused by upd(6)mat.
Contrary to previous assumptions, upd(6)mat is not as rare as once believed. It has gained recognition in imprinting diagnostics and is even considered a potentially related disorder to SRS. The widely-used commercial diagnostic kit for Silver-Russell syndrome (SRS) facilitates the global diagnosis of children with suspected SRS. Many diagnostic labs have identified cases of upd(6)mat, often linking this upd to growth restriction. A recent study by Mackay et al. highlighted seven such cases detected across nine diagnostic labs, suggesting that upd(6)mat is a more frequent related diagnosis than previously thought, even surpassing upd(20)mat.
In conclusion, upd(6)mat mainly manifests as IUGR and preterm labor. It is hypothesized that upd(6)mat may contribute to IUGR through imprinting defects. Nevertheless, based on the available information from existing cases, the precise association between upd(6)mat and IUGR, as well as other symptoms, cannot be definitively determined, necessitating further research with a larger sample size. It is also crucial to consider the limitations of NIPT results, especially when evaluating the potential of low-level mosaicism in the placenta.
References
Engel E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet. 1980;6(2):137–43.
Del Gaudio D, Shinawi M, Astbury C, Tayeh MK, Deak KL, Raca G, et al. Diagnostic testing for uniparental disomy: a points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2020;22(7):1133–41.
Benn P. Uniparental disomy: origin, frequency, and clinical significance. Prenat Diagn. 2021;41(5):564–72.
Kotzot D. Complex and segmental uniparental disomy (UPD): review and lessons from rare chromosomal complements. J Med Genet. 2001;38(8):497–507.
Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet. 2010;19(7):1263–75.
van den Berg-Loonen EM, Savelkoul P, van Hooff H, van Eede P, Riesewijk A, Geraedts J. Uniparental maternal disomy 6 in a renal transplant patient. Hum Immunol. 1996;45(1):46–51.
Spiro RP, Christian SL, Ledbetter DH, New MI, Wilson RC, Roizen N, et al. Intrauterine growth retardation associated with maternal uniparental disomy for chromosome 6 unmasked by congenital adrenal hyperplasia. Pediatr Res. 1999;46(5):510–3.
Parker EA, Hovanes K, Germak J, Porter F, Merke DP. Maternal 21-hydroxylase deficiency and uniparental isodisomy of chromosome 6 and X results in a child with 21-hydroxylase deficiency and Klinefelter syndrome. Am J Med Genet A. 2006;140(20):2236–40.
Cockwell AE, Baker SJ, Connarty M, Moore IE, Crolla JA. Mosaic trisomy 6 and maternal uniparental disomy 6 in a 23-week gestation fetus with atrioventricular septal defect. Am J Med Genet A. 2006;140(6):624–7.
Haag M, Beischel L, Rokeach J. First prenatal detection of maternal uniparental disomy (UPD) of chromosome 6 and “rescue” of trisomy 6. Am Soc Hum Genet. 2007;66:2428.
Gumus H, Ghesquiere S, Per H, Kondolot M, Ichida K, Poyrazoglu G, et al. Maternal uniparental isodisomy is responsible for serious molybdenum cofactor deficiency. Dev Med Child Neurol. 2010;52(9):868–72.
Sasaki K, Okamoto N, Kosaki K, Yorifuji T, Shimokawa O, Mishima H, et al. Maternal uniparental isodisomy and heterodisomy on chromosome 6 encompassing a CUL7 gene mutation causing 3M syndrome. Clin Genet. 2011;80(5):478–83.
Roosing S, van den Born LI, Hoyng CB, Thiadens AA, de Baere E, Collin RW, et al. Maternal uniparental isodisomy of chromosome 6 reveals a TULP1 mutation as a novel cause of cone dysfunction. Ophthalmology. 2013;120(6):1239–46.
Poke G, Doody M, Prado J, Gattas M. Segmental maternal UPD6 with prenatal growth restriction. Mol Syndromol. 2013;3(6):270–3.
Takimoto T, Takada H, Ishimura M, Kirino M, Hata K, Ohara O, et al. Wiskott–Aldrich syndrome in a girl caused by heterozygous WASP mutation and extremely skewed X-chromosome inactivation: a novel association with maternal uniparental isodisomy 6. Neonatology. 2015;107(3):185–90.
Lazier J, Martin N, Stavropoulos JD, Chitayat D. Maternal uniparental disomy for chromosome 6 in a patient with IUGR, ambiguous genitalia, and persistent mullerian structures. Am J Med Genet A. 2016;170(12):3227–30.
Leung WC, Lau WL, Lo TK, Lau TK, Lam YY, Kan A, et al. Two IUGR foetuses with maternal uniparental disomy of chromosome 6 or upd(6)mat. J Obstet Gynaecol. 2017;37(1):113–5.
Eggermann T, Oehl-Jaschkowitz B, Dicks S, Thomas W, Kanber D, Albrecht B, et al. The maternal uniparental disomy of chromosome 6 (upd(6)mat) “phenotype”: result of placental trisomy 6 mosaicism? Mol Genet Genomic Med. 2017;5(6):668–77.
Kerr ER, Stuhlmiller GM, Maha GC, Ladd MA, Mikhail FM, Koester RP, et al. Maternal uniparental isodisomy for chromosome 6 discovered by paternity testing: a case report. Mol Cytogenet. 2018;11:60.
Liehr T. Cytogenetic contribution to uniparental disomy (UPD). Mol Cytogenet. 2010;3:8.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences (0265-9247 (Print)).
Mackay D, Bliek J, Kagami M, Tenorio-Castano J, Pereda A, Brioude F, et al. First step towards a consensus strategy for multi-locus diagnostic testing of imprinting disorders. Clin Epigenet. 2022;14(1):143.
Ioannides Y, Lokulo-Sodipe K, Mackay DJ, Davies JH, Temple IK. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet. 2014;51(8):495–501.
Mulchandani S, Bhoj EJ, Luo M, Powell-Hamilton N, Jenny K, Gripp KW, et al. Maternal uniparental disomy of chromosome 20: a novel imprinting disorder of growth failure. Genet Med. 2016;18(4):309–15.
Eggermann T, Begemann M, Binder G, Spengler S. Silver–Russell syndrome: genetic basis and molecular genetic testing. Orphanet J Rare Dis. 2010;5:19.
Dixit A, Chandler KE, Lever M, Poole RL, Bullman H, Mughal MZ, et al. Pseudohypoparathyroidism type 1b due to paternal uniparental disomy of chromosome 20q. J Clin Endocrinol Metab. 2013;98(1):E103–8.
Court F, Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014;24(4):554–69.
Hanna CW, Penaherrera MS, Saadeh H, Andrews S, McFadden DE, Kelsey G, et al. Pervasive polymorphic imprinted methylation in the human placenta. Genome Res. 2016;26(6):756–67.
Acknowledgements
We thank the patients and their family members for their participation in this study.
Funding
No funding was received for this manuscript.
Author information
Authors and Affiliations
Contributions
YJ, VWZ, CD, LX reviewed the literature and wrote the main manuscript text; YXX and JJX Collected clinical information; FL performed the genetic counseling and contributed to manuscript drafting; YJ and FL were responsible for the revision of the manuscript for important intellectual content; all authors issued final approval for the version to be submitted. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiang, Y., Xiao, Y.X., Xiong, J.J. et al. Maternal uniparental disomy for chromosome 6 in 2 prenatal cases with IUGR: case report and literature review. Mol Cytogenet 17, 1 (2024). https://doi.org/10.1186/s13039-023-00670-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-023-00670-0