
Abstract
Background
Terminal deletion of chromosome 10p is a rare chromosomal abnormality. We report a neonatal case with a large deletion of 10p15.3p13 diagnosed early because of severe clinical manifestations.
Case presentation
Our patient presented with specific facial features, hypoparathyroidism, sen sorineural deafness, renal abnormalities, and developmental retardation, and carried a 12.6 Mb deletion in the 10p15.3 p13 region. The terminal 10p deletion involved in our patient is the second largest reported terminal deletion reported to date, and includes the ZMYND11 and GATA3 genes and a partial critical region of the DiGeorge syndrome 2 gene (DGS2).
Conclusion
On the basis of a literature review, this terminal 10p deletion in the present case is responsible for a specific contiguous gene syndrome. This rare case may help the understanding of the genotype–phenotype spectrum of terminal deletion of chromosome 10p.
Similar content being viewed by others


Background
Terminal deletion of chromosome 10p is a rare chromosomal disorder. The haploinsufficiency in the distal region of 10p15.3 is responsible for 10p15.3 microdeletion syndrome (OMIM 608,668), characterized by specific facial features, neuropsychiatric retardation, and autism [1]. Additionally, hypoparathyroidism, deafness, and renal abnormalities (HDR syndrome; OMIM 146,255) occurs in patients with a haploinsufficiency of 10p14 in the distal region [2, 3], while deletions in a critical region of the DiGeorge syndrome 2 (DGS2; OMIM 601,362), located at the proximal region of 10p14-p13, are associated with congenital heart defects, thymus hypoplasia, and T cell defects [1].
Here, we report a Chinese infant showing specific facial features, congenital hypoparathyroidism, sensorineural hearing loss, absence of the right kidney, a sacrococcygeal mass, a right frontal cyst, and psychomotor retardation. The patient has a 12.6 Mb deletion in chromosome10p15.3p13. We summarized the clinical characteristics and laboratory data, in addition, we compared present case with previously published cases with terminal 10p deletions. Our data indicate that 10p15.3 microdeletion syndrome and HDR syndrome are associated with the partial monosomy 10p.
Case presentation
A male infant was born by cesarean delivery at 36 weeks secondary to fetal heart deceleration. His birth weight was 2300 g (10th–50th centile), and his length and head circumference were within the normal ranges. He was pale, weak, and apneic at birth, and required positive pressure ventilation in the delivery room. His Apgar scores were 7, 8, and 8 at 1, 5, and 10 min, respectively. He was transferred to the neonatal department for respiratory support shortly after birth. There was no family history of developmental delay or inherited disorders, and his parents were non-consanguineous.
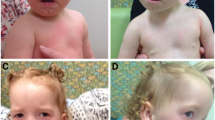
Clinical examination revealed an abnormal appearance (Fig. 1) including a prominent forehead, broad nasal bridge, small, low-set ears, micrognathia, and a sacrococcygeal mass. His anterior fontanelle was 3.5 × 3.0 cm, and cranial and sagittal sutures were approximately 1.0 cm. He also showed bilateral cryptorchidism. No deformity was found in the bones of his limbs, neck, chest, or abdomen.

The patient has a wide ocular distance, low-set ears, and an abnormal sacrococcygeal mass
He required continuous positive airway pressure for 36 h and was treated with ampicillin for 48 h. On the third day after birth, he was diagnosed with hypocalcemia, vitamin D deficiency, and hypomagnesemia, and was treated with intravenous calcium and magnesium and oral vitamin D. However, his blood calcium levels remained low, fluctuating between 1.32 and 1.45 mmol/l (normal: 2.11–2.52 mmol/l). On the fifth day, he was treated with intravenous calcium supplementation combined with oral calcitriol (0.1 µg/kg/d), after which serum calcium levels normalized. On the seventh day, calcium and calcitriol were administered orally. Two weeks after birth, serum 25-hydroxyvitamin D increased to normal levels. At the time of discharge at 16 days old, he failed a neonatal hearing test (otoacoustic emission and brainstem auditory response).
Laboratory examination included an electrolyte test on the third day that revealed the following: serum calcium: 1.18 mmol/l (normal 2.11–2.52 mmol/l), magnesium: 0.50 mmol/L (normal: 0.53–1.11 mmol/l), and phosphorus: 2.81 mmol/l (normal: 0.85–1.51 mmol/l). Serum parathyroid hormone levels on the fifth day were 2.8 pmol/l (normal 1.6–6.9 pmol/l), and 25-hydroxyvitamin D levels were 14.90 ng/ml (normal > 25 ng/ml). Brain magnetic resonance imaging (MRI; 1.5 T) performed on the 14th day revealed a malformation cyst in the left frontal lobe (Fig. 2a). Lumbosacral MRI was normal (Fig. 2b). Ultrasound of the scrotum revealed bilateral cryptorchidism, and ultrasound of the urinary system showed absence of the right kidney. At the age of 3 months, he failed a short-sound auditory brainstem response test, confirming binaural sensorineural hearing loss.

Brain MRI showing a left frontal lobe cyst
During follow-up, hearing loss and psychomotor retardation were noted. He was unable to laugh or control his head at the age of 5 months. On the basis of this observation, a motor evaluation was performed, resulting in an Alberta Infant Motor Scale score of 16 points (20th centile). Long-term follow-up visits showed that serum calcium and vitamin D levels were maintained within normal ranges. He is currently 6 months old (corrected gestational age: 5 months), and still unable to turn over or sit without help. His weight is 6.30 kg (3rd ~ 10th centile), his body length is 66.5 cm (10th ~ 50th centile), and his head circumference is 41.0 cm (3rd ~ 10th centile). He is scheduled for continued physical therapy and routine follow-up by pediatrician.
Genetic testing
This research was approved by the Ethics Committee of the First Affiliated Hospital of Fujian Medical University, and the patient’s parents provided their written informed consent. Whole blood was used to extract genomic DNA for genetic testing using the TIANamp blood genomic DNA kit (Tiangen, Beijing, China). Genome-wide copy number variation (CNV) was detected by DNA sequencing using the Illumina HiSeq series sequencer (Illumina, San Diego, CA) and analyzed by the cloud platform system using the human population CNV database, OMIM database, and PubMed database. CNV analysis revealed a pathogenic deletion about 12.71 Mb in size in chr10p15.3p13 (chr10: 105,001–12,815,001) (Fig. 3). The deletion involved the ATP5C1, IL2RA, DHTKD1, KLF6, AKR1C2, AKR1C4, ZMYND11, and GATA3 genes (Table 1). The genetic tests of the parents were normal (Fig. 4).

Distribution of chromosome 10 copy numbers in the proband

Size of the deletion in patients with partial monosomy 10p
Discussion and Conclusions
We describe herein a case with terminal 10p deletions presenting with specific facial features, hypoparathyroidism, sensorineural deafness, renal abnormalities, and developmental retardation. Hypoparathyroidism accompanied with dysmorphic features in this newborn were the reasons for performing chromosome analysis. We used CNV detection to analyze the whole genome chromosomal copy number of this patient. The results showed that our patient presented with a terminal deletion in the 10p15.3p13 region, involving ZMYND11, GATA3, and a critical region of DGS2.
Chromosome 10p terminal deletions have been described in more than 50 cases, since the first observation by Elliott et al. [12]. in 1970. GATA3 deletion is associated with hypoparathyroidism, sensorineural hearing loss, and nephrotic syndrome, while heterozygous loss of ZMYND11 is responsible for specific facial features and mental retardation. Additionally, the partial deletion of key regions of DGS2 may cause DiGeorge anomaly, also known as DiGeorge syndrome 2 (DGS2), and characterized by congenital heart malformation, thymus dysplasia, and T cell defects [13].
We reviewed the previous literatures on terminal deletion of chromosome 10, and compared the clinical phenotypes and genotypes of chromosome 10 terminal deletion (see Table 2). In order to avoid the overlap of clinical phenotypes caused by different genovariation, reports with other variations or unclear variations were not included in the summary. Only the proband information was included in the cases with family disease. A total of 18 cases of chromosome 10 terminal deletion were included. In all the cases reviewed, more than half of the patients had specific facial anomalies, including a prominent forehead, broad nasal bridge, low-set ears, and micrognathia. Most of patients showed a wide range of psychomotor retardation, including hypotonia, seizures or autism. More than two-thirds of patients had HDR syndrome. Among them, there were 12 patients with hypoparathyroidism, 10 patients with sensorineural hearing loss, 9 patients with renal or urinary system abnormalities, and 6 patients with the HDR triad. Approximately one-third of the patients had congenital heart disease, which was the major cause of death within the first months of life. (patient17, 18 and patient 19). Urinary tract anomalies were found in approximately one-third of the patients. Frontal lobe cyst, dilatation of cerebral ventricles, and hypoplasia of brainstem were presented in some patients. However, a sacrococcygeal mass, which was observed in our patient, has not been described in any of the other published patients. By comparing the clinical features of our patients with the previously reported patients with pure 10p terminal deletion, we found that the size of the 10p terminal deletion is the most important influence on the clinical phenotype of our patients.
The deletion of the 10p15p13 terminal fragment, identified in our patient, is closely related to 10p15.3 microdeletion syndrome which is associated with neurodevelopmental disorders and characterized by developmental retardation, intellectual disability, craniofacial deformity, behavioral abnormalities, hypotonia, and seizures [14]. Mutations in ZMYND11 are reported to be causative for 10p15.3 microdeletion syndrome [15]. Coe et al. [16] created an expanded CNV morbidity map from 29,085 children with developmental delay in comparison with 19,584 healthy controls, and pinpointed the haploinsufficiency of ZMYND11 as being associated with subtle facial dysmorphism, mild intellectual disability, and neuropsychiatric behavioral features. Moskowitz et al. [17] identified ZMYND11 heterozygous variants in patients with specific facial features, autism spectrum disorders, mental retardation, aggression, and complex neuropsychiatric characteristics, supporting the association of ZMYND11 with 10p15.3 deletion syndrome. Moreover, Tumiene et al. [18] compared the clinical phenotypes of 14 patients with 10p15.3 deletions with the phenotypes of eight patients with loss-of-function ZMYND11 mutations, then further confirmed that ZMYND11 was the critical gene for the 10p15.3 microdeletion clinical phenotype. Our patient presenting with the entire deletion of ZMYND11 gene, has specific facial features that are consistent with 10p15.3 microdeletion clinical phenotype. Although still young, he has already developed psychomotor retardation. Therefore, we should continue to follow up his future neuropsychiatric development.
Our patient carries a GATA3 deletion in the 10p15.3p13 region, accompanied by hypoparathyroidism, sensorineural deafness, and renal dysplasia, and was diagnosed with HDR syndrome, also known as Barakat syndrome.The main clinical features of HDR syndrome are the triad of hypoparathyroidism (H), sensorineural hearing loss (d) and renal dysplasia (R). The other clinical features of HDR syndrome are variable, including hypomagnesemia and vitamin D deficiency, insulin-dependent diabetes mellitus, congenital heart disease, specific facial features, cerebral infarction, severe cognitive impairment and autism [19]. In 2000, Van Esch et al. [20] found that GATA3 was essential for embryonic development of the parathyroids, auditory system, and kidneys, and also revealed that GATA3 haploinsufficiency causes human HDR syndrome. To date, several types of GATA3 mutations related to HDR syndrome have been reported in nearly 124 families (177 patients) worldwide. These include about 40% frameshift deletions or insertions, 23% missense mutations, 14% nonsense mutations, 6% splice site mutations, 1% in-frame deletions or insertions, 15% complete gene deletions, and 1% complete gene duplications [21]. Vidavalur et al. [22] reported a male premature infant who was small for gestational age, with micrognathia and facial malformation, combined with hypoparathyroidism and bilateral sensorineural hearing loss. CNV examination revealed the absence of chromosome 10p (10p15.3p14), including GATA3 and other genes. This further confirmed that GATA3 is a critical gene for HDR syndrome. Our patient presents with a typical triad of hypoparathyroidism, sensorineural hearing loss, and renal dysplasia, and we suggest that his GATA3 deletion is the main cause of the disease.
Our patient was found to have partial monosomy for a proximal deletion of chromosome 10p14-p13, which is associated with DGS2. Typical DGS is caused by the microdeletion of chromosome 22q.12.2. However, in 1996, Daw first stated that haploinsufficiency of a gene or genes within chromosome 10p (the DGS2 locus) can cause DGS spectrum, defined as DSG2 (OMIM #601,362). Lichtner et al. [2] suggested that the DGS phenotype associated with a 10p deletion should be considered a contiguous gene syndrome owing to terminal deletions between D10S585 and D10S1720. Hemizygosity of the proximal region, designated DGCR2, can cause cardiac defects and T cell deficiency. In 2017, Kim et al. [23] reported a female infant (patient 3) with the largest deletion of the chromosome 10p, presenting with hypoparathyroidism, hearing loss, cardiac abnormalities, thymus hypoplasia, a double uterus, double cervix, and relatively small kidneys. Unexpectedly, our patient has so far only presented with a few clinical features of DGS2 (psychomotor retardation and cryptorchidism) and lacks other typical signs such as cardiac defects, a cleft palate, thymus dysplasia, and abnormal T cell levels. The mechanism for this is still unclear, but it may be related to the different effects of large fragment deletions on individuals and gene mutations. Some specific manifestations involving 10p13- p14 have yet to be determined as the patient is still very young; however, an adverse clinical course is to be expected.
In conclusion, the combined data from the present case and previous reported cases support that the terminal deletion of chromosome 10p can be considered as a contiguous gene syndrome, related to a variety of clinical characteristics including specific facial features, psychomotor retardation, hypoparathyroidism, sensorineural hearing loss, and renal dysplasia. In contrast to previously reported cases, our patient was diagnosed early in the neonatal period, which has been very important for early clinical intervention. For patients with similar clinical manifestations, we suggest that genetic analysis should be performed to identify the precise molecular defects to further elucidate the genotype–phenotype correlation of chromosome 10p deletions.
Availability of data and materials
All data generated in this study are included in this published article.
Abbreviations
- DGS:
-
DiGeorge syndrome
- MRI:
-
Magnetic resonance imaging
- CNV:
-
Copy number variation
- DNA:
-
Deoxyribonucleic acid
- OMIM:
-
Online Mendelian Inheritance in Man
- HDR:
-
Hypoparathyroidism, deafness and renal dysplasia
References
Schuffenhauer S, Lichtner P, Peykar-Derakhshandeh P, et al. Deletion mapping on chromosome 10p and definition of a critical region for the second DiGeorge syndrome locus (DGS2). Eur J Hum Genet. 1998;6(3):213–25.
Lichtner P, König R, Hasegawa T, Van Esch H, Meitinger T, Schuffenhauer S. An HDR (hypoparathyroidism, deafness, renal dysplasia) syndrome locus maps distal to the DiGeorge syndrome region on 10p13/14. J Med Genet. 2000;37(1):33–7.
Melis D, Genesio R, Boemio P, Del Giudice E, Cappuccio G, Mormile A, Ronga V, Conti A, Imperati F, Nitsch L, Andria G. Clinical description of a patient carrying the smallest reported deletion involving 10p14 region. Am J Med Genet A. 2012;158A(4):832–5.
Mari N. Terminal deletion of chromosome 10p13 as a cause of hypoparathyroidism in a neonate. Central Eur J Paed. 2018;14(1):88–91.
Gamba BF, Rosenberg C, Costa S. Cleft lip/ palate, short stature, and developmental delay in a boy with a 5.6-Mb interstitial deletion involving 10p15.3p14(Article). Molecul Syndromol. 2015;6(1):39–43.
Benetti E, Murer L, Bordug A, et al. 10p121 deletion: HDR phenotype without DGS2 features. Exp Mole Pathol. 2009;86(1):74–6.
Verri A, Maraschio P, Devriendt K, et al. Chromosome 10p deletion in a patient with hypoparathyroidism, severe mental retardation, autism and basal ganglia calcifications. Ann Genet. 2004;47(3):281–7.
Dasouki M, Jurecic V, Phillips JA, et al. DiGeorge anomaly and chromosome 10p deletions: One or two loci? Am Med Genet. 1997;73(1):72–5.
Shapira M, Borochowitz Z, Bar-El H, et al. Deletion of the short arm of chromosome 10 (10p13): report of a patient and review. Am J Med Genet. 1994;52(1):34–8.
Berger R, Larroche JC, Toubas PL. Deletion of the short arm of chromosome no 10. Acta Paediatr Scand. 1977;66(5):659–62.
Shokeir MH, Ray M, Hamerton JL, et al. Deletion of the short arm of chromosome No 10. J Med Genet. 1975;12(1):99–103.
Elliott D, Thomas GH, Condron CJ, et al. C-group chromosome abnormality (10p-) Occurrence in a child with multiple malformations. Am J Dis Child. 1970;119:72–3.
Anderlid BM, Schoumans J, Nordgren A, Malmgren H, Verri A, Blennow E, Eriksson M, Lindstrand A, Benetti E, Golovleva I. Molecular and clinical characterization of patients with overlapping 10p deletions. Am J Med Genet A. 2010;152A(5):1233–43.
DeScipio C, Conlin L, Rosenfeld J, et al. Subtelomeric deletion of chromosome 10p153: clinical findings and molecular cytogenetic characterization. Am J Med Genet A. 2012;158A(9):2152–2161ss.
Hong Wen, Yuanyuan Li, Haitao Li, et al. ZMYND11: an H3.3-specific reader of H3K36me3. Cell cycle (Georgetown, Tex), 2014, 13(14):2153–2154.
Coe BP, Witherspoon K, Rosenfeld JA, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46(10):1063–71.
Moskowitz AM, Belnap N, Siniard AL, et al. A de novo missense mutation in ZMYND11 is associated with global developmental delay, seizures, and hypotonia. Cold Spring Harb Mol Case Stud. 2016;2(5):a000851.
Tumiene B, Čiuladaitė Ž, Preikšaitienė E, et al. Phenotype comparison confirms ZMYND11 as a critical gene for 10p15.3 microdeletion syndrome(Article). J Appl Genet. 2017;58(4):467–74.
Barakat AJ, Raygada M, Rennert OM, et al. Barakat syndrome revisited. Am J Med Genet A. 2018;176(6):1341–8.
Van Esch H, Devriendt K. Human genome and diseases: review. Transcription factor GATA3 and the human HDR syndrome. Cell Mol Life Sci. 2011;58:1296–300.
Lemos MC, Thakker RV. Hypoparathyroidism, deafness, and renal dysplasia syndrome: 20 Years after the identification of the first GATA3 mutations. Hum Mutat. 2020;41(8):1341–50.
Vidavalur R, Devapatla S. A Unique Genomic Variant of HDR Syndrome in Newborn. Indian Pediatr. 2018;55(8):705–6.
Kim S, Kim Y, Jung J, et al. Clinical description of a neonate carrying the largest reported deletion involving the 10p15.3p13 region. Clin Case Rep. 2017;5(8):1369–75.
Acknowledgements
We are extremely grateful to the patient and his family for their participation and cooperation. We thank Sarah Williams, PhD, from Edanz Group (https://en-author-services.edanz.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
Q-YS obtained clinical information, collated literature data, and wrote the manuscript. P-LW, B-YL, and S-JC performed and coordinated the genetic analysis. JL helped draft and revise the manuscript. S-Q critically revised the final manuscript for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from the proband’s mother for genetic testing as part of standard care. A copy of the written consent is available for review by the Editor of this journal.
Consent for publication
Written informed consent was obtained from the proband’s mother for the publication of all personal information contained in this case report and accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shao, QY., Wu, PL., Lin, BY. et al. Clinical report of a neonate carrying a large deletion in the 10p15.3p13 region and review of the literature. Mol Cytogenet 14, 29 (2021). https://doi.org/10.1186/s13039-021-00546-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-021-00546-1