
Abstract
Background
Ring chromosome instability may influence a patient’s phenotype and challenge its interpretation.
Results
Here, we report a 4-year-old girl with a compound phenotype. Cytogenetic analysis revealed her karyotype to be 46,XX,r(22). aCGH identified a 180 kb 22q13.32 duplication, a de novo 2.024 Mb subtelomeric 22q13.32-q13.33 deletion, which is associated with Phelan-McDermid syndrome, and a maternal single gene 382-kb TUSC7 deletion of uncertain clinical significance located in the region of the 3q13.31 deletion syndrome. All chromosomal aberrations were confirmed by real-time PCR in lymphocytes and detected in skin fibroblasts. The deletions were also found in the buccal epithelium. According to FISH analysis, 8% and 24% of the patient’s lymphocytes and skin fibroblasts, respectively, had monosomy 22.
Conclusions
We believe that a combination of 22q13.32-q13.33 deletion and monosomy 22 in a portion of cells can better define the clinical phenotype of the patient. Importantly, the in vivo presence of monosomic cells indicates ring chromosome instability, which may favor karyotype correction that is significant for the development of chromosomal therapy protocols.
Similar content being viewed by others


Background
Terminal deletions at 22q13 are often associated with ring chromosome formation. To date, no more than a hundred patients with r(22) have been described, and their clinical phenotype is similar to those with a terminal 22q deletion [1,2,3,4,5,6,7]. Ring chromosomes are known to be unstable during mitotic divisions: the ring may change in size or may be lost, and dicentric and interlocking rings may appear [8, 9]. The loss of the ring chromosome followed by the amplification of the remaining normal homolog in induced pluripotent stem cells (iPSCs) formed the basis of in vitro karyotype correction and chromosomal therapy [10,11,12].
A combination of r(22) and cells with monosomy for chromosome 22 was described previously in one of two mentally retarded monozygotic twins with minor physical abnormalities [13]. In one twin, two of 50 metaphases had a 45,XX,-22 karyotype. Significantly, two cells of the 50 metaphases of the second twin had an apparently normal chromosome number, and one cell had 46 chromosomes with a dicentric ring. The remaining cells in both patients were 46,XX,r(22).
Heterozygous contiguous gene deletion at 22q13 or mutations in the SHANK3 gene (OMIM 606230), located within the minimum critical region, cause Phelan-McDermid syndrome (PHMDS, OMIM 606232). The frequent clinical findings are intrauterine and postnatal growth retardation, intellectual disability, speech delay, delayed motor development, microcephaly, large and misshapen ears, mild hypertelorism, strabismus, epicanthic folds, ptosis of the upper lips, bushy eyebrows and synophrys, a depressed and broad nasal bridge, short mandible, malocclusion and irregular position of the teeth, high palate, clinodactyly of the little fingers, partial syndactyly between the 2nd and 3rd toes, hypotonia, seizures and behavior problems.
Here, for the first time, we report a patient with a compound phenotype who exhibits ring chromosome 22, mosaic monosomy for chromosome 22 in lymphocytes and skin fibroblasts, and a microdeletion at 3q13.31 of maternal origin. Both microdeletions, del3q13.31 and del22q13.32-q13.33, were also detected by real-time PCR in the buccal epithelium.
The single-gene deletion of TUSC7 at 3q13.31 in the index patient is located in the region of the 3q13.31 deletion syndrome (OMIM 615433). Although the TUSC7 gene encodes the long non-coding RNA associated with various types of cancer [14], its expression has been shown to be closely related to the expression of the LSAMP gene [15], which is involved in neurodevelopmental impairments [16, 17].
Clinical report
The patient (Fig. 1), a 4-year-old girl, was referred to the clinical geneticist for the first time when she was one year and eight months old because of development delay, hyperexcitability, mood swings, and sleep disturbance. The girl did not walk alone, did not speak, and her head circumference did not increase. She is an only child of non-consanguineous healthy parents. Her father’s nephew is intellectually disabled and receives education at home.

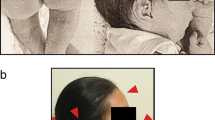
The patient at 4 years of age (note plagiocephaly, high anterior hairline, broad forehead, epicanthus, widely spaced eyes, upslanted palpebral fissure, upper eyelid fullness, straight eyebrows, prominent ears, wide and depressed nasal bridge, bulbous nose, smooth and short philtrum, and thin vermillion of the upper lip)
The patient was born at the 34th week of gestation via Cesarean section. Her birth weight was 1422 g (< 3rd centile); birth length was 48 cm (25th centile); head circumference was 25 cm (< 3rd centile); and chest circumference was 24 cm (< 3rd centile). Her Apgar score was 6. She was able to sit at the age of 8 months and walk independently at the age of 2 years.
The patient had neonatal hypotonia and hyporeflexia. Anticonvulsant therapy was prescribed at the age of 2 months because of seizures. At the age of 3 months, the seizures stopped, and the drugs were discontinued. After birth, the girl was placed on artificial feeding and had trouble gaining weight.
Currently, the patient is regularly observed by a neurologist, with diagnoses of cerebral palsy, atonic-astatic form, psychomotor and speech development delay, and by a psychiatrist due to mental disorder and decreased intelligence to the level of pronounced debility, psychomotor disinhibition syndrome, and unformed control functions for physiological sentiments.
At the age of 4 years, the girl’s weight was 14.5 kg (25th centile); height was 108 cm (95th centile); and head circumference was 45.5 cm (<2th centile). She exhibited plagiocephaly, flat occiput, high anterior hairline, broad forehead, micrognathia, epicanthus, widely spaced eyes, upslanted palpebral fissure, upper eyelid fullness, straight eyebrows, prominent ears, wide and depressed nasal bridge, bulbous nose, smooth and short philtrum, thin vermillion of the upper lip, widely spaced teeth, high palate, clinodactyly (radial, F5, bilateral), proximally placed thumb (bilateral), thickening of the distal phalanx of the thumb (bilateral), pes planus, sandal gap, cutaneous syndactyly of the toes (T2-3, bilateral), short toes (T3-5, bilateral), dysplastic nails of the feet, asthenic body, wide umbilical ring, and a sacral dimple. The patient demonstrated aggression, signs of autism, attention deficit hyperactivity disorder (ADHD), and enuresis.
When the girl was 6 months old, echoencephalography showed ventriculomegaly. At the age of 1 year and 6 months, magnetic resonance imaging of the brain revealed a Dandy-Walker variant: hypoplasia of the cerebellar tonsils. Echocardiography revealed small heart anomalies, including an open oval window with a minimal left-right discharge, an enlarged coronary sinus, and a diagonal false chord of the left ventricle.
Methods
The patient was subject to aCGH analysis due to her compound phenotype and r(22), as determined by conventional cytogenetic analysis of her blood lymphocytes. The aCGH findings were confirmed by real-time PCR in three different tissues: lymphocytes, skin fibroblasts, and buccal epithelium. FISH analysis was used to confirm the ring form of chromosome 22 and to determine the level of cells with monosomy 22 among lymphocytes and skin fibroblasts. SNP analyses identified the maternal origin of the ring chromosome. All techniques were performed by using equipment of the Center "Medical Genomics" of Research Institute of Medical Genetics, Tomsk NRMC.
Materials
For molecular genetic analyses, peripheral blood was collected in tubes containing EDTA. A primary culture of the patient’s skin fibroblasts was obtained from two full-thickness skin biopsies. A buccal smear was collected with a cotton swab, which was transported in a dry tube for further DNA extraction.
Cytogenetic analyses
Conventional cytogenetic analysis was performed based on GTG-banded metaphases from peripheral blood lymphocytes from the patient at a 400-band resolution.
Fibroblast culture
The biopsies of fibroblasts were washed twice with Hank’s solution containing antibiotics and antifungal agents and then treated with 0.2% collagenase in culture medium for 3 h at 37°C. The suspension was subsequently cultured in AminoMax culture medium. A confluent monolayer formed in one day.
Array-based Comparative Genomic Hybridization (aCGH) analyses
aCGH was performed using the SurePrint G3 Human CGH Microarray Kit (8×60K) (Agilent Technologies, Santa Clara, CA USA), according to the manufacturer’s recommendations. Labeling and hybridization of patient and reference DNA (#5190-3796, Human Reference DNA, Agilent Technologies, Santa Clara, CA USA) were performed using enzymatic labeling and hybridization protocols (v. 7.5 Agilent Technologies, Santa Clara, CA USA). Array images were acquired with an Agilent SureScan Microarray Scanner (Agilent Technologies, Santa Clara, CA USA). Data analysis was performed using Cytogenomics Software (v. 3.0.6.6) (Agilent Technologies), the publicly available Database of Genomic Variants (DGV), and the Database of Chromosomal Imbalance and Phenotype in Humans employing Ensembl Resources (DECIPHER). Annotations of the genes located within the region of genomic imbalance were retrieved from the NCBI Gene Database, OMIM, and the literature.
Confirmation of copy number variation using quantitative real-time PCR
Target sequences within and outside the deleted chromosomal regions and specific amplification primers for quantitative real-time PCR assays were selected using Primer 3 software (Additional file 1). The presence of 3q13.31 microdeletion, 22q13.32 microduplication and 22q13.32-q13.33 microdeletion was tested using genomic DNA from peripheral blood lymphocytes from the patient, her parents, and maternal mother (maternal father was not available for the analysis) as well as in cultured skin fibroblasts and a buccal smear from the patient and using the AriaMX Real-Time PCR System (Agilent Technologies, Santa Clara, CA USA). Control genomic DNA was obtained from the peripheral blood lymphocytes, skin fibroblasts and a buccal smear of a healthy donor. The control gene was HEXB, which encodes the beta subunit of hexosaminidase and is located at 5q13 (Additional file 1).
Fluorescent In Situ Hybridization (FISH)
FISH was performed using PCR-based probes for the centromeres of chromosomes 14 and 22 and the TBC1D22A gene located close to del22q13.32-q13.33 in lymphocytes and cultured skin fibroblasts from the proband following the standard protocol. E. coli clones carrying plasmids with inserts of centromere-specific alpha-satellite DNA sequences as well as the BAC clone RP11-569D9 were kindly provided by Professor M. Rocchi (Resources for Molecular Cytogenetics, Institute of Genetics, Bari, Italy). The probe for the TBC1D22A gene was generated using a long-range PCR kit (BioLabMix, Novosibirsk, Russia) (Additional file 1). Probes 14/22 and TBC1D22A were labeled with TAMRA-dUTP and Fluorescein-dUTP (BioSan, Russia), respectively.
The results of the aCGH and FISH analyses are described further according to the International System for Human Cytogenomic Nomenclature (ISCN 2016).
SNP analyses
Nine SNPs (rs11541025, rs2272837, rs8238, rs11703226, rs6010218, rs28637964, rs11547734, rs28379706, rs5771206) located at 22q13.32-q13.33 were selected to determine the parental origin of chromosome 22 with deletion. SNP investigation was performed via SNaPshot analysis.
Results
Metaphase analysis of the G-banded chromosomes from peripheral blood lymphocytes showed a karyotype 46,XX,r(22) [11] (Fig. 2a). FISH analysis detected 8% of monosomic cells (Table 1). aCGH using an Agilent 60K microarray revealed del3q13.31, dup22q13.32, and del22q13.32-q13.33 of 382 kb, 180 kb, and 2.024 Mb, in size, respectively: arr[hg19] 3q13.31(116233164_116615500)×1,22q13.32(48886812_49059015)×3, 22q13.32-q13.33(49115584_51178264)×1 (Fig. 3a). The distal breakpoint of dup22q13.32 is located within intron 2 of the FAM19A5 gene, i.e., exons 1-2 are duplicated, and the proximal breakpoint of del22q13.32-q13.33 is located within intron 3 of the FAM19A5 gene, i.e., exon 4 is deleted. The 3q13.31 microdeletion involved the single TUSC7 gene. The microdeletion was confirmed via quantitative real-time PCR analysis and was shown to be inherited from the intellectually normal mother and grandmother (Fig. 3c). TUSC7 is known to be regulated together with the neighboring LSAMP [15], which is associated with neuropsychiatric disorders in patients with 3q13.31 deletion syndrome (OMIM615433). We also investigated the number of copies of the latter gene, which appeared to be normal (Fig. 3c). Similar microdeletions and reciprocal microduplications were present in DGV in one and two reports, respectively, and in DECIPHER (nos. 264508 and 299972). However, the gene located within this region in DECIPHER is LSAMP, with the coordinates - chr3:115521235-117716095 (hg19) vs. chr3:115521210-116164385 (hg19) in DGV. The deletion in the index patient (chr3:116233164-116615500 (hg19)) is within the DECIPHER region.

Cytogenetic analysis. a Conventional cytogenetics analysis: ring chromosome 22; b FISH analysis with centromere-specific probes for chromosomes 14/22 (red) and a PCR-based probe for TBC1D22A (green) in cultured lymphocytes from the proband. The arrow indicates ring chromosome 22. C, D - FISH analysis with centromere-specific probes for chromosomes 14/22 (red) and PCR-based probes for TBC1D22A (green) in cultured skin fibroblasts from the proband. Ring chromosome 22 is designated by a white arrow (c); blue and pink arrows designate 46,XX,r(22) and 45,XX,-22 cells, respectively (d)

Molecular genetic analysis. a An aCGH image of chromosomes 3 and 22 in the lymphocytes of the patient. Deletions are designated by arrows. b Confirmation of the deletion at 22q13.32-q13.33 by quantitative real-time PCR analysis. c Confirmation of the deletion at 3q13.31 by quantitative real-time PCR analysis. d Identification of the deletions at 3q13.31 and 22q13.32-q13.33 in the buccal epithelium by quantitative real-time PCR analysis. e Confirmation of the duplication at 22q13.32 by quantitative real-time PCR analysis
The 22q13.32-q13.33 microdeletion overlaps with the region of PHMDS and includes 43 RefSeq genes. The main candidate gene within this region is SHANK3 (606230) [18]. Real-time PCR confirmed the presence of the microdeletion with primers for the FAM19A5, SHANK3, and ACR genes (Fig. 3b). According to the SNP analysis, the 22q13.32-q13.33 microdeletion originated de novo on the maternal chromosome.
The ring chromosome 22 was confirmed via FISH using centromere-specific DNA probes for chromosomes 14 and 22 and for the TBC1D22A gene located in the intact part of chromosome 22 close to del22q13.32-q13.33 (Fig. 2b). In addition, 8% of lymphocytes were observed to exhibit monosomy for chromosome 22 (nuc ish(D14Z1/D22Z1×3)[41/479],(TBC1D22A×1)[41/479]).
Skin fibroblasts of the patient were investigated using the Agilent 60K microarray as well. The 22q13.32-q13.33 microdeletion (arr[hg19]22q13.32-q13.33(49084185_51043490)×1) was found and further confirmed via real-time PCR, while the 3q13.31 deletion and 22q13.32 duplication were not revealed by aCGH but were also determined by real-time PCR (Fig. 3e). aCGH demonstrated the shift of chromosomal profiles at 3q13.31 and 22q13.32 towards the deletion and duplication, respectively, which, however, did not reach the significance level due to the high variance of the fluorescence intensity over the chromosomes. FISH analysis showed that 24% of the fibroblasts initially had monosomy of chromosome 22 (Fig. 2c and d): nuc ish(D14Z1/D22Z1×3)[27/113], (TBC1D22A×1)[27/113].
The buccal epithelium was investigated via real-time PCR. Both 3q13.31 and 22q13.32-q13.33 deletions were confirmed (Fig. 3d).
Discussion
We present a patient with a compound phenotype and a combination of chromosomal abnormalities: del3q13.31 (single TUSC7 gene), dup22q13.32 (LOC284933 and exons 1-2 of FAM19A5 gene), ring chromosome 22 associated with del22q13.32-q13.33 (43 RefSeq genes), and mosaic chromosome 22 monosomy in 8% of lymphocytes and 24% of fibroblasts. Both microdeletions were present in lymphocytes, fibroblasts, and the buccal epithelium. The microduplication was present in lymphocytes and fibroblasts; the buccal epithelium was not investigated. The del3q13.31 was inherited from the apparently healthy mother and grandmother and is located within the region of the 3q13.31 deletion syndrome (OMIM 615433).
The only gene located within the deleted 3q13.31 region is TUSC7, also called LSAMP antisense RNA3. It is a putative suppressor gene in various tumors, including glioma [19]. Low TUSC7 expression is associated with significantly unfavorable survival and may be a risk factor for distant metastases [20]. Frequent deletions in the 3q13.31, including LSAMP and TUSC7, have also been identified in patients with osteosarcomas [14]. Although the copy number of the LSAMP gene is preserved in our patient, LSAMP and TUSC7 expression are known to be closely related [15]. The authors also showed that, although the patient with acute myeloid leukemia had a 250-kb deletion in 3q13.31, which included the TUSC7 gene but not LSAMP, the expression of both genes was increased by an unknown mechanism. LSAMP, implicated in the regulation of emotional and social behavior in mice [21] and associated with major depressive disorder and schizophrenia in humans [16, 17], is also one of the candidate genes for the 3q13.31 deletion syndrome.
Del22q13.32-q13.33 in our patient originated de novo on the maternal chromosome. This deletion is associated with the origin of the ring chromosome and includes part of the minimal critical region of PHMDS ([hg19] 22q13.33(51045516_51187844)) with the MAPK8IP2, ARSA, SHANK3, and ACR genes. PHMDS is a developmental disorder (OMIM 606232). The symptoms of the index proband commonly associated with PHMDS are listed in Table 2.
The most important contributing factor to PHMDS is SHANK3 haploinsufficiency [22]. Mutations in this gene have also been associated with schizophrenia [23]. Analysis of human neurons differentiated from the induced pluripotent cells of patients with PHMDS showed impaired synaptic transmission and increased input resistance [24].
The instability of r(22) in the index patient led to the loss of chromosome 22 in a portion of cells, resulting in monosomy for chromosome 22. Non-mosaic monosomy for chromosome 22 is incompatible with life. To date, only seven cases of monosomy 22 have been published [25,26,27,28,29,30,31], four of which were mosaic. The oldest described child was three years old. None of the patients had r(22), which could somehow, during the early stages of the development before its elimination from the cell, compensate for the phenotype. The symptoms of our proband are similar to those described in patients with mosaicism for monosomy 22 and are listed in Table 2. In addition, the index patient exhibits some symptoms that have not been described in any patient with either of the anomalies discussed above.
How can the clinical phenotype of our patient be interpreted? To our knowledge, two explanations may exist: phenotypic variability and a combined effect of del22q13.32-q13.33 and mosaic monosomy for chromosome 22. Phenotypic variability is typical for the manifestation of most CNV-associated syndromes. Variability can be associated with the variability of the breakpoints. In addition, the phenotype of a patient may also result from a combination of genetic and non-genetic modifiers.
The heterogeneous clinical phenotype in carriers of the ring chromosome 22 was first explained by mosaicism with monosomy 22 by Lejeune in 1968 [32]. Later, Stoll and Roth described a family with 46,XX/46,XX,r(22) and 46,XY/46,XY,r(22) karyotypes in three generations of individuals with both normal development and severe mental retardation. In these individuals, 40-50% of their lymphocytes had a ring chromosome 22, and a few cells were observed to carry two ring chromosomes. Skin biopsies were refused [33]. In 2014, a patient with a developmental delay and dysmorphic features was described, from whom 12% of lymphocytes and 40-48% of fibroblasts had r(22), and the remaining cells had a normal karyotype - 46,XX/46,XX,r(22). In some metaphase cells of a patient, the ring chromosome was duplicated or lost [23]. In our patient, the initial metaphase analysis revealed only cells with the 46,XX,r(22) karyotype. Using FISH, we performed a search for mosaicism of the ring chromosome 22 in interphase nuclei of peripheral blood lymphocytes and skin fibroblasts of the index patient. Approximately 8% of lymphocytes and 24% of fibroblasts had monosomy 22 in combination with 46,XX,r(22) cells. The discrepancy between the results of these two analyses can be due to the low level of monosomic cells among lymphocytes detected by FISH, which was not revealed by metaphase analysis, as well as to the potential inability of monosomic cells to proliferate in vitro.
To the best of our knowledge, only four live-born infants with mosaicism for monosomy of chromosome 22 have been described. The first one is a 2-year-old male child with moderate psychomotor retardation, generalized hypotonia, large ears, epicanthus, synophrys, and cutaneous syndactyly between all the fingers [28]. The patient’s karyotype was 45,XY,-22/46,XY [12/50], i.e., 24% of lymphocytes were monosomic. The second case was published by Verloes et al. [31], who observed a slightly dysmorphic and mentally defective three-year-old child with a 45,XY,-22/46,XY karyotype. Chromosome 22 was absent in 10.5% of lymphocytes and 8.3% of fibroblasts. The third case was a newborn with gastroschisis and absent cerebral diastolic flow. The baby died on its second day of life. The karyotype was 45,XY,-22/46,XY [3/50] [26]. The fourth child was an abnormal premature male infant with a 45,XY,-22/46,XY [15/100] karyotype [29]. He had a unique facial appearance, similar to those with DiGeorge syndrome (OMIM 188400), hypertonicity, limitation of extension at the major joints, and flexion contractures of all fingers. However, the direct comparison of the phenotypes of these patients with the symptoms of our proband seem to be incorrect because, in addition to monosomy for chromosome 22, she also has a deletion at 22q13.32-q13.33.
For the index patient, among 43 identified symptoms, 18 have previously been described in other patients with PHMDS and 15 in patients with mosaic monosomy 22, while 14 symptoms have never been observed in any of the mentioned cases (Table 2). These symptoms include skeletal abnormalities, facial dysmorphism, wide umbilical ring, and enuresis. Significantly, two important papers concerning the FAM19A5 gene, which is disrupted by the CNV breakpoints in the index patient, have been recently published. The first paper demonstrates that the gene is a crucial candidate for modulating osteoclast formation and bone disorders [34], i.e., we can assume that the skeletal abnormalities are due to FAM19A5 protein deficiency. In addition, the second paper is the first to show that FAM19A5 is highly expressed and secreted by adipocytes and adipose tissue protein [35]. Thus, its deficiency can probably be related to the low weight and asthenic body of the proband. Moreover, FAM19A5 is also expressed in the brain and acts as a modulator of immune response in nervous cells, which can make it responsible for neuropsychiatric processes [36].
Conclusions
In conclusion, we present the first patient with a compound phenotype and a combination of a 382-kb deletion at 3q13.31, encompassing the single TUSC7 gene, a 180-kb duplication at 22q13.32, a 2.024-Mb deletion at 22q11.32-q11.33, including part of the PHMDS critical region, and monosomy for chromosome 22 in 8% of lymphocytes and 24% of fibroblasts. The presence of deletions was detected by real-time PCR in the buccal epithelium. The clinical significance of the 3q13.31 deletion is unclear, though the TUSC7 gene is associated with various types of tumors. Special attention should thus be paid to the early prevention and diagnosis of cancer in a carrier. In addition, only the combination of the 22q13.32-q13.33 deletion and monosomy for chromosome 22 allowed us to explain most of the clinical features of the patient. Therefore, in cases with a compound phenotype, it is important to use a combination of different methods and sometimes to investigate more than one tissue. The presence of monosomic cells in patients with a ring chromosome indicates ring chromosome instability. A monosomic karyotype can be the intermediate step in the process of chromosomal defect correction, which can underlie the chromosomal therapy of genetic diseases.
Abbreviations
- aCGH:
-
Array comparative genomic hybridization
- CNV:
-
Copy number variation
- DECIPHER:
-
Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources
- iPSC:
-
Induced pluripotent stem cell
- OMIM:
-
Online Mendelian Inheritance in Man
- PCR:
-
Polymerase chain reaction
- PHMDS:
-
Phelan-McDermid syndrome
- SNP:
-
Single-nucleotide polymorphism
References
De Mas P, Chassaing N, Chaix Y, Vincent MC, Julia S, Bourrouillou G, et al. Molecular characterisation of a ring chromosome 22 in a patient with severe language delay: a contribution to the refinement of the subtelomeric 22q deletion syndrome. J Med Genet. 2002;39:e17.
Guilherme RS, Soares KC, Simioni M, Vieira TP, Gil-da-Silva-Lopes VL, Kim C, et al. Clinical, cytogenetic, and molecular characterization of six patients with ring chromosomes 22, including one with concomitant 22q11.2 deletion. Am J Med Genet A. 2014;164A:1659–65.
Ishmael HA, Cataldi D, Begleiter ML, Pasztor LM, Dasouki MJ, Butler MG. Five new subjects with ring chromosome 22. Clin Genet. 2003;63:410–4.
Jeffries AR, Curran S, Elmslie F, Sharma A, Wenger S, Hummel M, et al. Molecular and phenotypic characterization of ring chromosome 22. Am J Med Genet A. 2005;137:139–47.
MacLean JE, Teshima IE, Szatmari P, Nowaczyk MJ. Ring chromosome 22 and autism: report and review. Am J Med Genet. 2000;90:382–5.
Schinzel A. Catalogue of unbalanced chromosome aberrations in man. 2. Berlin, Germany: Walter de Gruyter; 2001.7.
Kurtas N, Arrigoni F, Errichiello E, Zucca C, Maghini C, D'Angelo MG, Beri S, Giorda R, Bertuzzo S, Delledonne M, Xumerle L, Rossato M, Zuffardi O, Bonaglia MC. Chromothripsis and ring chromosome 22: a paradigm of genomic complexity in the Phelan-McDermid syndrome (22q13 deletion syndrome). J Med Genet. 2018;55:269-77.
Pristyazhnyuk IE, Menzorov AG. Ring chromosomes: from formation to clinical potential. Protoplasma. 2018;255:439-49.
Yip MY. Autosomal ring chromosomes in human genetic disorders. Transl Pediatr. 2015;4:164–74.
Kim T, Bershteyn M, Wynshaw-Boris A. Chromosome therapy. Correction of large chromosomal aberrations by inducing ring chromosomes in induced pluripotent stem cells (iPSCs). Nucleus. 2014;5:391–5.
Kim T, Plona K, Wynshaw-Boris A. A novel system for correcting large-scale chromosomal aberrations: Ring chromosome correction via reprogramming into induced pluripotent stem cell (iPSC). Chromosoma. 2017;126:457–63.
Plona K, Kim T, Halloran K, Wynshaw-Boris A. Chromosome therapy: potential strategies for the correction of severe chromosome aberrations. Am J Med Genet C Semin Med Genet. 2016;172:422–30.
Lindenbaum RH, Bobrow M, Barber L. Monozygotic twins with ring chromosome 22. J Med Genet. 1973;10:85–9.
Kresse SH, Ohnstad HO, Paulsen EB, Bjerkehagen B, Szuhai K, Serra M, et al. LSAMP, a novel candidate tumor suppressor gene in human osteosarcomas, identified by array comparative genomic hybridization. Genes Chromosomes Cancer. 2009;48:679–93.
Coccaro N, Zagaria A, Tota G, Anelli L, Orsini P, Casieri P, et al. Overexpression of the LSAMP and TUSC7 genes in acute myeloid leukemia following microdeletion/duplication of chromosome 3. Cancer Genet. 2015;208:517–22.
Innos J, Koido K, Philips MA, Vasar E. Limbic system associated membrane protein as a potential target for neuropsychiatric disorders. Front Pharmacol. 2013;4:32.
Koido K, Janno S, Traks T, Parksepp M, Ljubajev Ü, Veiksaar P, et al. Associations between polymorphisms of LSAMP gene and schizophrenia. Psychiatry Res. 2014;215:797–8.
Mitz AR, Philyaw TJ, Boccuto L, Shcheglovitov A, Sarasua SM, Kaufmann WE, Thurm A. Identification of 22q13 genes most likely to contribute to Phelan McDermid syndrome. Eur J Hum Genet. 2018;3:293–302.
Shang C, Guo Y, Hong Y, Xue YX. Long non-coding RNA TUSC7, a target of miR-23b, plays tumor-suppressing roles in human gliomas. Front Cell Neurosci. 2016;10:235.
Li N, Yang M, Shi K, Li W. Prognostic value of decreased long non-coding RNA TUSC7 expression in some solid tumors: A systematic review and meta-analysis. Oncotarget. 2017;8:59518–26.
Philips MA, Lilleväli K, Heinla I, Luuk H, Hundahl CA, Kongi K, et al. Lsamp is implicated in the regulation of emotional and social behavior by use of alternative promoters in the brain. Brain Struct Funct. 2015;220:1381–93.
Betancur C, Buxbaum JD. SHANK3 haploinsufficiency: A "common" but underdiagnosed highly penetrant monogeniccause of autism spectrum disorders. Mol Autism. 2013;4:17.
Guilmatre A, Huguet G, Delorme R, Bourgeron T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev Neurobiol. 2014;74:113–22.
Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–71.
DeCicco F, Steele MW, Pan S, Park SC. Monosomy of chromosome No. 22. A case report. J Pediatr. 1973;83:836–8.
Lewinsky RM, Johnson JM, Lao TT, Winsor EJ, Cohen H. Fetal gastroschisis associated with monosomy 22 mosaicism and absent cerebral diastolic flow. Prenat Diagn. 1990;10:605–8.
Merino A, De Perdigo A, Nomballais F, Yvinec M, Lopes P. Digeorge syndrome with total monosomy 22 diagnosed prenatally. Prenat Diagn. 1995;15:189–92.
Moghe MS, Patel ZM, Peter JJ, Ambani LM. Monosomy 22 with mosaicism. J Med Genet. 1981;18:71–3.
Pinto-Escalante D, Ceballos-Quintal JM, Castillo-Zapata I, Canto-Herrera J. Full mosaic monosomy 22 in a child with DiGeorge syndrome facial appearance. Am J Med Genet. 1998;76:150–3.
Rosenthal IM, Bocian M, Krmpottik E. Multiple anomalies including thymic aplasia associated with monosomy-22. Pediatr Res. 1972;6:358.
Verloes A, Herens C, Lambotte C, Frederic J. Chromosome 22 mosaic monosomy (46,XY/45,XY,-22). Ann Genet. 1987;30:178–9.
Lejeune J. On the duplication of circular structures. Ann Genet. 1968;11:71–7.
Stoll C, Roth MP. Segregation of a 22 ring chromosome in three generations. Hum Genet. 1983;63:294–6.
Park MY, Kim HS, Lee M, Park B, Lee HY, Cho EB, et al. FAM19A5, a brain-specific chemokine, inhibits RANKL-induced osteoclast formation through formyl peptide receptor 2. Sci Rep. 2017;7:15575.
Wang Y, Chen D, Zhang Y, Wang P, Zheng C, Zhang S, et al. A Novel Adipokine, FAM19A5, Inhibits Postinjury Neointima Formation through Sphingosine-1-Phosphate Receptor 2. Circulation. 2018; https://doi.org/10.1161/CIRCULATIONAHA.117.032398.
Tom Tang Y, Emtage P, Funk WD, Hu T, Arterburn M, Park EE, Rupp F. TAFA: a novel secreted family with conserved cysteine residues and restricted expression in the brain. Genomics. 2004;83:727–34.
Tabet AC, Rolland T, Ducloy M, Lévy J, Buratti J, Mathieu A, et al. A framework to identify contributing genes in patients with Phelan-McDermid syndrome. NPJ Genom Med. 2017;2:32.
Acknowledgements
This study used data generated by the DECIPHER Consortium. A full list of the centers that helped to generate the data is available at http://decipher.sanger.ac.uk and can be obtained via e-mail from decipher@sanger.ac.uk. Funding for the DECIPHER project was provided by the Wellcome Trust.
We would like to thank the family of our patient for their assistance with the clinical evaluation.
Databases
Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources [http://decipher.sanger.ac.uk].
Database of Genomic Variants [http://projects.tcag.ca/variation].
NCBI Gene Database [http://www.ncbi.nlm.nih.gov/gene].
Online Mendelian Inheritance in Man [http://omim.org].
Primer3 (v. 0.4.0) [http://frodo.wi.mit.edu/primer3].
Funding
This study was supported by the Russian Science Foundation (project 16-15-10231).
Availability of data and materials
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Contributions
The patient was examined clinically by AMN, LPN, AAK, and EOB. Cytogenetic analysis was performed by OVP and Yu.S.Ya. Array CGH analysis was performed by NAS. Primer design and real-time PCR assays were performed by AAK, MEL, and RRS. FISH probes were created and the analysis was performed by ENT and SAV. Fibroblasts were obtained and cultured by TVN. The buccal epithelium was analyzed by ENT. SNP analysis was performed by NPB. The manuscript was written by AAK and INL. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the local Research Ethics Committee of the Research Institute of Medical Genetics, Tomsk NRMC. Written informed consent was obtained from the parents of the patient for her participation.
Consent for publication
Written informed consent was obtained from the parents of the patient for the publication of the clinical data and pictures.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Primers for real-time PCR and FISH-probe synthesis. (DOC 41 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kashevarova, A.A., Belyaeva, E.O., Nikonov, A.M. et al. Compound phenotype in a girl with r(22), concomitant microdeletion 22q13.32-q13.33 and mosaic monosomy 22. Mol Cytogenet 11, 26 (2018). https://doi.org/10.1186/s13039-018-0375-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-018-0375-3