
Abstract
As a family of hormones with pleiotropic effects, natriuretic peptide (NP) system includes atrial NP (ANP), B-type NP (BNP), C-type NP (CNP), dendroaspis NP and urodilatin, with NP receptor-A (guanylate cyclase-A), NP receptor-B (guanylate cyclase-B) and NP receptor-C (clearance receptor). These peptides are genetically distinct, but structurally and functionally related for regulating circulatory homeostasis in vertebrates. In humans, ANP and BNP are encoded by NP precursor A (NPPA) and NPPB genes on chromosome 1, whereas CNP is encoded by NPPC on chromosome 2. NPs are synthesized and secreted through certain mechanisms by cardiomyocytes, fibroblasts, endotheliocytes, immune cells (neutrophils, T-cells and macrophages) and immature cells (embryonic stem cells, muscle satellite cells and cardiac precursor cells). They are mainly produced by cardiovascular, brain and renal tissues in response to wall stretch and other causes. NPs provide natriuresis, diuresis, vasodilation, antiproliferation, antihypertrophy, antifibrosis and other cardiometabolic protection. NPs represent body’s own antihypertensive system, and provide compensatory protection to counterbalance vasoconstrictor-mitogenic-sodium retaining hormones, released by renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS). NPs play central roles in regulation of heart failure (HF), and are inactivated through not only NP receptor-C, but also neutral endopeptidase (NEP), dipeptidyl peptidase-4 and insulin degrading enzyme. Both BNP and N-terminal proBNP are useful biomarkers to not only make the diagnosis and assess the severity of HF, but also guide the therapy and predict the prognosis in patients with HF. Current NP-augmenting strategies include the synthesis of NPs or agonists to increase NP bioactivity and inhibition of NEP to reduce NP breakdown. Nesiritide has been established as an available therapy, and angiotensin receptor blocker NEP inhibitor (ARNI, LCZ696) has obtained extremely encouraging results with decreased morbidity and mortality. Novel pharmacological approaches based on NPs may promote a therapeutic shift from suppressing the RAAS and SNS to re-balancing neuroendocrine dysregulation in patients with HF. The current review discussed the synthesis, secretion, function and metabolism of NPs, and their diagnostic, therapeutic and prognostic values in HF.
Similar content being viewed by others


Background
As a family of hormones with pleiotropic effects, natriuretic peptide (NP) system includes atrial NP (ANP), B-type NP (BNP, also called brain NP), C-type NP (CNP), dendroaspis NP (DNP) and urodilatin, with three receptors: NP receptor-A [guanylate cyclase (GC)-A or NPR-A], NP receptor-B (GC-B or NPR-B) and NP receptor-C (clearance receptor or NPR-C) [1]. These peptides are genetically distinct, but structurally and functionally related for regulating circulatory homeostasis in vertebrates, and each of them has a 17-amino acid (aa) cyclic structure constructed with an disulfide bond [2]. In humans, ANP and BNP are encoded by NP precursor A (NPPA) and NPPB genes on chromosome 1, whereas CNP is encoded by NPPC on chromosome 2 [3]. NPs are synthesized and secreted through certain mechanisms by cardiomyocytes, fibroblasts, endotheliocytes, immune cells (neutrophils, T-cells and macrophages) and immature cells, such as embryonic stem cells, muscle satellite cells and cardiac precursor cells (CPCs) [4]. They are mainly produced by cardiovascular, brain and renal tissues in response to wall stretch and other causes. NPs provide natriuresis, diuresis, vasodilation, antiproliferation, antihypertrophy, antifibrosis and other cardiometabolic protection [5, 6]. More importantly, NPs represent body’s own antihypertensive system, and provide compensatory protection to counterbalance vasoconstrictor-mitogenic-sodium retaining hormones, released by renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS) [7]. NPs are inactivated through not only NPR-C, but also neutral endopeptidase (NEP), dipeptidyl peptidase-4 (DPP-4) and insulin degrading enzyme (IDE). There is urinary excretion of NPs as well [3]. The current review discussed the synthesis, secretion, function and metabolism of NPs, and their diagnostic, therapeutic and prognostic values in heart failure (HF).
Synthesis and secretion
Synthesis and secretion of ANP
ANP is mainly produced and stored in atrial granule, and normal ventricle actually produces little ANP [8]. Failing ventricle secretes ANP in patients with HF, and becomes a main part of plasma ANP [9]. NPPA gene has the following exons: exon 1 [5’-untranslated region (5’-UTR, a 25-aa signal peptide) and 16 aa of proANP sequence], exon 2 (most of proANP sequence) and exon 3 [terminal tyrosine and 3’-untranslated region (3’-UTR)] (Fig. 1). Proximal 5’-flanking region (5’-FR) of NPPA gene can regulate its spatio-temporal expression [10]. Mechanical stretch of cardiomyocytes, exercise, hypoxia, cold, angiotensin, endothelin, vasopressin, catecholamine or glucocorticoid induces transcription factor GATA to bind promoters, suggesting an active involvement of neurohormonal system in the regulation of NP synthesis. ANP mRNA is translated to 151-aa preproANP, and 126-aa proANP is produced and stored after removing 25-aa signal peptide. ProANP is cleaved upon secretion by transmembrane serine endoprotease (corin) into the active carboxy-terminal (C-terminal) 28-aa α-ANP with relatively short half-life, which can bind to receptor and have biologic function, and the inactive 98-aa amino-terminal (N-terminal) proANP (NT-proANP) more stable with relatively long half-life [11]. ANP secretion is caused by atrial and ventricular wall stretch due to transmural pressure or volume overload, and also affected by age, sex, heart rate and renal function [2]. ANP is distributed into coronary sinus, and then to various target organs. Meanwhile, human β-ANP, an antiparallel dimer of α-ANP, is present in failing heart, and has elevated levels in patients with severe HF [12]. Additionally, urodilatin is a 32-aa NP of renal origin with local function in regulating renal sodium and water excretion through interacting with NPR-A. It is produced as N-terminal 4-aa-extended form of α-ANP like γ-ANP after the cleavage of proANP by an unknown protease in renal distal tubules [13].

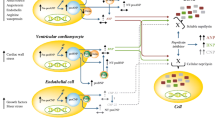
Synthesis, metabolism and function of natriuretic peptides. Abbreviations: ANP: atrial natriuretic peptide; BNP: B-type natriuretic peptide; cGMP: cyclic guanosine monophosphate; CNP: C-type natriuretic peptide; DPP-4: dipeptidyl peptidase-4; GC: guanylate cyclase; GTP: guanosine triphosphate; IDE: insulin degrading enzyme; NEP: neutral endopeptidase; NPR: natriuretic peptide receptor; NT-proANP: N-terminal proANP; NT-proBNP: N-terminal proBNP; NT-proCNP: N-terminal proCNP; PDE: phosphodiesterase; PKG: protein kinase; RAAS: renin-angiotensin-aldosterone system; SNS: sympathetic nervous system
Synthesis and secretion of BNP
Firstly discovered in porcine brain, BNP is stored in atrial granule with ANP, but not in granule in ventricle [14]. BNP is mainly secreted by normal atrium [15]. However, as a hallmark for maladaptive remodeling of left ventricle (LV), BNP is mainly secreted in ventricle when LV function is insufficient and cardiac wall is stretched due to transmural pressure or volume overload [16]. BNP has more obviously elevated levels than ANP in patients with HF [17]. NPPB gene has the following exons: exon 1 (5’-UTR, a 26-aa signal peptide, and 15 aa of proBNP sequence), exon 2 (most of proBNP sequence) and exon 3 (terminal tyrosine and 3’-UTR) (Fig. 1). Various causes, such as tissue hypoxia, transmural pressure or volume overload, induce the transcription of NPPB gene in endoplasmic reticulum to produce 134-aa preproBNP [18]. Pro-inflammatory cell factors, including interleukin-1β, interleukin-6 and tumor necrosis factor-α, induce BNP synthesis in cardiomyocytes, suggesting an active involvement of immune system in the regulation of NP synthesis [19]. Repeated AUUUA in 3’-UTR of BNP mRNA make it easier to be degraded than ANP mRNA. After removing 26-aa signal peptide, 108-aa proBNP is produced and then cleaved upon secretion by furin (or corin) into the active BNP1-32 (or BNP4-32), and the inactive 76-aa N-terminal proBNP (NT-proBNP) [20].
BNP-32 and proBNP-108
Not only proBNP and O-Glycosylated proBNP, but also BNP and NT-proBNP, are present in blood [14]. Current immunoassays for BNP-32 also recognize proBNP-108, and plasma BNP in HF rather includes proBNP-108 and O-Glycosylated proBNP-108 [21]. There is an elevated ProBNP-108/BNP-32 ratio in patients with HF [22]. ProBNP-108/BNP-32 ratio increases in response to ventricular overload rather than atrial overload in patients with HF [23]. Moreover, proBNP-108 has much less ability to induce the synthesis of cyclic guanosine monophosphate (cGMP) than BNP-32 in vascular smooth muscle and endothelial cells [24]. Despite high levels of plasma BNP, there is an attenuation of BNP bioactivity in patients with HF [25]. In mild to moderate HF, plasma cGMP levels increase in proportion to HF severity. However, plasma cGMP levels have an attenuated increase relative to disease state, and no longer correlate with BNP levels in severe HF [26]. Even relatively small increase in proBNP-108/BNP-32 ratio, if it lasts for a long time, can sufficiently reduce total potential of cGMP production and attenuate compensatory benefit of plasma BNP, thereby leading to HF progression.
O-Glycosylation and proBNP-108
O-Glycosylated proBNP-108 levels correlate with HF severity [24]. Within Golgi apparatus of ventricular myocytes, proBNP-108 is post-translationally glycosylated to various extent at seven sites of N-terminal region: threonine (Thr)36, serine (Ser)37, Ser44, Thr48, Ser53, Thr58 and Thr71 [27]. Within trans-Golgi network, O-Glycosylated proBNP-108 is cleaved into BNP-32 and NT-proBNP [23]. O-Glycosylation has potential effect on the secretion and processing of proBNP-108. Thr71 is near to cleavage site of proBNP-108, and O-Glycosylation at Thr71 inhibits processing of proBNP-108 in human embryonic kidney (HEK) 293 cells and human leukemia 1 cells [28, 29]. Mechanisms governing cardiac secretion and peripheral metabolism of proBNP-108 remain unclear in patients with severe HF [30]. Venous furin-like enzyme activity has been proposed to correlate with NTproBNP/proBNP-108 ratio, and cleave proBNP-108 into BNP-32 and NT-proBNP in peripheral blood of patients with acute HF [31]. However, it has also been reported that proBNP-108 has similar levels between peripheral vein and artery, and is unlikely to be metabolized in peripheral blood and tissue. These inconsistent results may be caused by the diversity between in vivo and in vitro settings, between glycosylated and non-glycosylated proBNP-108, and between clinical status of patients with HF [32].
New forms of BNP
Many new plasma forms of BNP with low molecular mass are present, but BNP-32 is nearly absent in patients with HF [33]. DPP-4 cleaves N-terminal serine-proline dipeptide from BNP1-32 in blood to produce BNP3-32 and BNP5-32, which are more rapidly degraded and have much less abilities to induce beneficial responses than BNP1-32 [34]. DPP-4 also cleaves N-terminal histidine-proline dipeptide from proBNP1-108 to produce proBNP3-108. Current immunoassays can not distinguish these forms in blood [35]. Patients using DPP-4 inhibitors have exhibited an increased risk of HF hospitalization in Saxagliptin Assessment of Vascular Outcomes Recorded in patients with diabetes mellitus-Thrombolysis in Myocardial Infarction (SAVOR-TIMI) trial [36]. However, other studies have not confirmed an association between DPP-4 inhibitors and HF hospitalization and shown adverse prognosis in patients with HF using DPP-4 inhibitors [37,38,39,40,41]. Previous meta-analyses can not rule out the concern that DPP-4 inhibitors have cardioprotective roles by affecting BNP1-32/BNP3-32 ratio [42,43,44,45].
Synthesis and secretion of CNP
As the most abundant NP in brain, CNP is also synthesized in atrium, ventricle, kidney, chondrocyte, endothelium and blood cells. NPPC gene has the following exons: exon 1 (5’-UTR, a 23-aa signal peptide, and 7 aa of proCNP sequence), exon 2 (most of proCNP sequence) and exon 3 (3’-UTR) (Fig. 1). After removing 23-aa signal peptide from 126-aa preproCNP, 103-aa proCNP is produced and then cleaved upon secretion into 53-aa CNP by intracellular serine endoprotease (furin) [46]. CNP expression is up-regulated by shear stress [47]. Transforming growth factor-β stimulates CNP production and secretion in endotheliocytes [2]. CNP-53 is then cleaved into 22-aa CNP, and CNP-53 and CNP-22 are equipotent [48]. Although CNP-22 and CNP-53 have similar activity and function, CNP-53 predominates in heart, endothelium and brain, whereas CNP-22 predominates in cerebral spinal fluid and plasma [49].
Function
Receptors of NPs
NP system has significant autocrine, paracrine and endocrine function. As two of five transmembrane GC receptors in humans, NPR-A and NPR-B induce pathophysiologic functions of NP system (Fig. 1). NPR-A is activated by ANP and BNP, and NPR-B is activated by CNP [50]. NPRs are present in heart, brain, kidney, adrenal, liver, pancreas, vascular and gastrointestinal smooth muscle, adipocytes, chondrocytes, fibroblasts and platelets [3]. NPR-A is highly present in kidney, adrenal, lung, terminal ileum, aorta and adipose. NPR-B is highly present in fibroblasts [51]. They have an extracellular ligand-binding and membrane-spanning region, an intracellular particulate GC region and an intracellular cGMP-dependent protein kinase (PKG) region [52]. Activated NPRs catalyze conversion of guanosine triphosphate (GTP) to cGMP. As an intracellular second messenger, cGMP activates PKG and phosphodiesterase (PDE) to regulate various pathways including ion channels, protein phosphorylation, nuclear translocation and gene expression, all of which exert biologic effects [53, 54]. NPR-C is highly present in atrium, kidney, adrenal, lung, mesentery, placenta, cerebral cortex, cerebellum, aorta and vein [51]. NPR-C has a disulfide-bonding dimer homologous to extracellular region of NPR-A and NPR-B and intracellular 37 aa for potential signaling functions. NPR-C is a clearance receptor for ANP, BNP and CNP [55]. Although NPR-C has no GC activity, it induces pathophysiologic functions, such as affecting cell growth, through adenylate cyclase, inhibitory guanine nucleotide-regulatory protein (G-protein) and phospholipase C [56]. Modulation of NPR expression in target organ may be determinant for local bioavailability and regulation of NPs. Therefore, NP resistance may be caused by NPR-C upnregulation or NPR-A downregulation. In early stage of HF, NPs provide compensatory actions including not only natriuresis, diuresis and vasodilation, but also RAAS and SNS inhibition. In severe HF, NPs have attenuated effects despite high plasma NP levels assessed by current immunoassays. Several possible explanations include increased NP degradation, reduced NP bioactivity, increased secretion of inactive NP forms, increased cGMP degradation and reduced NPR-A activity due to receptor dephosphorylation and degradation [57,58,59]. However, limited information is available regarding bioactivity change in NPR and its clinical significance in HF.
Function of ANP
ANP boosts natriuresis and diuresis. Na+ reabsorption in inner medullary collecting ducts depends mainly on the apical amiloride-sensitive Na+ channel (cyclic nucleotidegated ion channel) and basolateral Na+-K+-adenosine triphosphatase (Na+-K+-ATPase). Apical amiloride-sensitive Na+ channel allows passive Na+ entry from renal tubular lumen. Basolateral Na+-K+-adenosine triphosphatase helps Na+-K+-2Cl- cotransporter actively pump Na+ out of epithelial cell into peritubular space and eventually bloodstream. ANP inhibits apical Na+ channel and basolateral Na+-K+-ATPase activity, resulting in decreased Na+ reabsorption from inner medullary collecting ducts and increased Na+ excretion from urine known as natriuresis [53]. ANP induces cGMP to inhibit Na+ channel by a mechanism of activating PKG independent of phosphorylation [2]. ANP inhibits basolateral Na+-K+-ATPase through PKG-induced phosphorylation in a cGMP-dependent manner [53]. ANP inhibits RAAS with reduced angiotensin II-induced sodium and water transport in renal proximal tubules [60]. ANP suppresses renin secretion from juxtaglomerular (granular) cells in a cGMP-dependent manner without changing intracellular Ca2+. ANP also suppresses aldosterone synthesis in adrenal glomerulosa (adrenocorticotropic hormone-induced, angiotensin-II-induced and basic aldosterone), which enhances natriuretic effect and reduces extracellular volume [61]. ANP/NPR-A activity induces natriuresis and diuresis, but appears to be downregulated in HF with RAAS activation [62]. Although conflicting studies exist, ANP may suppress angiotensin-II-induced secretion of vasopressin from posterior pituitary without blood-brain barrier, and inhibit V2 receptor-mediated effect of vasopressin on water reabsorption in collecting ducts [63,64,65]. ANP increases glomerular filtration rate (GFR) through its direct vasodilative effect on afferent arterioles. ANP also reverses norepinephrine-induced vasoconstriction of afferent arterioles [66]. Vasodilative effect of ANP on vascular smooth muscle may involve following mechanisms: reducing Ca2+ influx, enhancing Ca2+ extrusion, and inhibiting Ca2+ release from sarcoplasmic reticulum [67]. ANP directly relaxes contractile intraglomerular mesangial cells and expands glomerular capillary surface area available for filtration. ANP also inhibits angiotensin-II-induced constriction of mesangial cells [67]. There have been controversies on vasoconstrictive effect of ANP on efferent arterioles: some have observed no change in diameter of efferent arterioles, whereas others have reported the dilatation of afferent arterioles and constriction of efferent arterioles [68].
Lowering blood pressure
ANP induces hypovolemia and decreases blood pressure (BP). ANP lowers BP with increased permeability of capillaries and fluid efflux from blood [69]. ANP decreases cardiac load by shifting intravascular fluid into interstitial space. ANP stimulates Ca2+/calmodulin-dependent endothelial nitric oxide (NO) synthase in aorta, ventricle and kidney to produce more NO for relaxing vascular smooth muscle cells by binding to either NPR-A or NPR-C, causing a decrease in BP levels [70]. ANP decreases vascular resistance also by inhibiting RAAS [71]. Thus, ANP/NPR-A activity reduces basic BP levels through its combined effects on vascular relaxation and intravascular volume. Moreover, ANP relaxes air passages and blood vessels in lung [72]. ANP and BNP have elevated levels due to wall stretch of right ventricle, and inhibit pulmonary hypertension caused by chronic hypoxia [73].
Counteracting sympathetic activity
ANP not only modulates baroreflex mechanism, but also inhibits sympathetic activity and enhances vagal afferent. SNS is inhibited in peripheral vessels, perhaps by lowering activation threshold of baroreceptors, by decreasing catecholamine release from nerve endings, and by reducing sympathetic outflow [74]. It lowers activation threshold of vagal afferent, reflex tachycardia and vascular constriction, and causes a sustained decrease in mean arterial pressure [75]. ANP decreases sympathetic outflow by modulating ganglionic neurotransmission rather than increasing discharge from cardiac mechanoreceptors with inhibitory vagal afferent [76].
Inhibiting cardiac hypertrophy
Both prolonged exposure to systemic hypertension and lacking the inhibition of heart growth lead to cardiac hypertrophy [77, 78]. Moreover, ANP has direct effects on heart, and inhibits cardiac hypertrophy and fibrosis [79]. ANP induces cardiomyocyte apoptosis and inhibits fibroblast growth [3]. ANP suppresses fibroblast migration and proliferation by counteracting angiotensin-II, aldosterone and endothelin-1, as well as inflammatory reaction including pro-inflammatory factor and macrophage infiltration [80, 81]. ANP inhibits stress-activated protein kinase and extracellular-regulated kinase-2 induced by platelet-derived growth factor [82]. ANP exhibits antimitogenic and antineoplastic properties by reducing cell adhesion and inflammatory reaction through p38 mitogen-activated protein kinase [83]. ANP attenuates the growth of cardiomyocytes and fibroblasts by inhibiting norepinephrine-induced Ca2+ influx in a cGMP-mediated manner [84]. Meanwhile, ANP and BNP reduce systemic and pulmonary BP, and inhibit cardiac hypertrophy in HF [85]. ANP and CNP exert direct effects on vessels by reducing adhesion molecules of endotheliocytes (P-selectin and monocyte chemotactic protein-1) and inflammatory infiltration on atheromatous plaques [86].
Regulating energy homeostasis
ANP stimulates exercise-induced lipolysis through cGMP and PKG in primates with an increased NPR-A/NPR-C ratio [87]. ANP affects the conversion from white to brown fat through mitochondrial uncoupling protein-1 and p38 mitogen-activated protein kinase [88]. BNP may also have hypoglycemic effect and regulate energy homeostasis [89]. ANP and BNP stimulate oxidative ability of skeletal muscle and lipolytic action in subcutaneous adipose [90]. ANP and BNP induce hormone-sensitive lipase of adipocytes in a cGMP-mediated manner [91].
Function of BNP
In addition to the well documented natriuresis, diuresis and vasodilation, BNP also has direct effects on heart [14]. BNP may provide compensatory protection, such as inhibiting myocardial apoptosis and necrosis and reducing cardiac hypertrophy and fibrosis [92, 93]. BNP may also modulate immune and inflammatory reaction to cardiac injury. BNP depletes monocytes, B lymphocytes and natural killer cells in peripheral blood [94]. BNP regulates the chemotaxis of monocytes and production of inflammatory molecules by macrophages [95]. BNP may promote cardiac neutrophil infiltration and metalloprotease-9 expression after myocardial infarction (MI), and also have direct effects on matrix remodeling and wound healing [96].
Affecting cardiac embryogenesis
BNP plays significant roles in cardiac embryogenesis. There are high BNP levels in embryonic heart during the midgestation, and peaks of BNP secretion correlate with cardiac development [97]. Plasma BNP levels in humans are high at birth, progressively declining thereafter, to stabilize at around ten years of age to the levels found in adults [98]. ANP and BNP may regulate cardiomyocyte differentiation and proliferation in the developing embryo [99]. Embryonic stem cells express high levels of BNP, which are crucial for their proliferation and differentiation [100]. BNP may also be involved in the process of angiogenesis following skeletal muscle ischemia [101]. BNP secretion by vascular satellite cells has been found to activate the regeneration of adjacent endothelium in a paracrine manner. Moreover, BNP has been addressed in cardiac regeneration by evaluating the relationships between cardiac precursor cells (CPCs) and BNP in neonatal and adult mice. Firstly, all forms of proBNP are more abundant in neonatal heart than in adult heart [102]. Secondly, CPCs express NPR-A and NPR-B, supporting that CPCs can respond to BNP [102]. NPR-A contributes to self-renewal and maintenance of CPC pluripotency, whereas NPR-B is involved in CPC proliferation [103]. ANP, BNP and CNP stimulate CPC proliferation and differentiation into new cardiomyocytes by NPR-B binding, cGMP increase and PKG activation [104]. Thirdly, exogenous BNP has increased proliferating CPCs and new cardiomyocytes, which are associated with improved cardiac function and remodeling after MI [102]. Finally, CPCs stain positive for BNP, suggesting that CPCs can also synthesize and secrete BNP in an autocrine manner to regulate their proliferation and differentiation into new cardiomyocytes. Thus, BNP and CPCs may be useful therapies for HF and MI [102].
Function of CNP
All of ANP, BNP and CNP provide cardiorenal protection, although CNP has the most antifibrotic and least renal effects [13]. CNP is a vasodilator mainly secreted from endothelial cells in response to vascular injury. Patients with HF have minimally increased CNP levels, and HF severity is significantly relevant to CNP levels [105]. Although CNP dose not predominantly behave as a cardiac hormone, it has cardiovascular actions as well, such as re-endothelialization, hyperpolarization, antithrombosis and antifibrosis [106, 107]. CNP inhibits proliferation and migration of coronary artery smooth muscle cells mediated by oxidized low-density lipoprotein in a cGMP-dependent manner [108]. CNP inhibits platelet aggregation and thrombosis formation by suppressing plasminogen activator inhibitor-1, perhaps through NPR-C [109]. As an endothelium-derived hyperpolarizing factor, CNP may regulate several vasodilative factors, including prostacyclin and NO [110]. CNP inhibits cardiac hypertrophy and fibrosis in autocrine and paracrine manners within myocardium [111]. CNP has antifibrotic effect by regulating PKG-derived phosphorylation of Smad3 and transforming growth factor-β-derived nuclear translocation [112]. CNP may have compensatory actions in HF in a cAMP-dependent manner, which are incompletely understood. CNP-dependent NPR-B activity is about half of ANP-dependent NPR-A activity in normal ventricle. ANP-dependent NPR-A activity is unaltered or reduced, and CNP-dependent NPR-B activity is mildly or significantly elevated in failing ventricle [113, 114]. Failing ventricle has increased fibroblasts, and NPR-B is highly present in cardiac fibroblasts [115]. Additionally, CNP inhibits pulmonary hypertension and fibrosis in a similar manner in HF [116]. Fibroblast growth factor receptor 3 (FGFR3) is an important regulator of bone formation. Its gene gain-of-function mutations activate mitogen-activated protein kinase pathway and result in achondroplasia. CNP acts as a key regulator of longitudinal bone growth by downregulating mitogen-activated protein kinase pathway [117].
Metabolism
Mechanisms of metabolism
Main mechanisms are in the following: 1) NPR-C-derived and clathrin-mediated endocytosis, lysosomal ligand hydrolysis and ligand-free receptor recycling; and 2) NEP (neprilysin), a zinc-dependent exoenzyme with broad substrates [53]. ANP is degraded effectively in most organs, and more effectively in some than others. Although 30%–50% of ANP has been shown to be degraded in kidney, liver or lower limbs rather than lung, subsequent studies have reported that 19%-24% of ANP is degraded in lung (lung > liver > kidney) [118, 119]. Degradation of BNP and CNP have been discussed previously [120]. BNP binds to NPR-C 7% as tightly as ANP, and BNP has long half-life due to less degradation by NPR-C [121]. With a part of aa sequence similar to NPs, osteocrin (also known as musclin) is an important decoy ligand for NPR-C, and acts to increase plasma levels of NPs [122].
NEP and IDE
NEP is a dominant enzyme for NP degradation. In heart, NEP is expressed on membrane of cardiomyocytes, fibroblasts, vascular smooth muscle and endothelial cells [123]. Although expressed in many epithelial tissues, NEP levels are particularly high at the luminal side of renal proximal tubules [124]. Initial attack of breaking ring and inactivating peptide occurs between cysteine and phenylalanine (Fig. 1) [125]. Cleavage sites in ring structure are crucial for degradation [48]. ANP and CNP have differences of zero or one aa between species, and are similarly degraded by NEP. BNP differs obviously between species, and is species-specifically degraded by NEP [126]. BNP is a worse substrate than ANP and CNP, and human NEP cleaves BNP at three sites [127]. Most BNP is degraded by NEP in rat renal membrane, but NEP dose not degrade all BNP in human renal membrane [128]. IDE, a zinc-dependent protease with broad substrates, degrades not only insulin, but also ANP [129, 130].
Roles of NPR-C
Relative effects of NPR-C and NEP degradation on NP levels are still controversial and unclear [131]. Under normal condition, NPR-C-blocking peptides affect physiologic roles of ANP mildly more than or equally to NEP inhibitors, and ANP has maximal roles with both NPR-C-blocking peptides and NEP inhibitors [132]. Under pathologic condition, NEP inhibitors become important due to elevated NP levels and possible NPR-C saturation [133].
Genetic regulation
Genetic variants
Not only there are genetic variants in NPs and NPRs in humans, but also they have significant associations with cardiometabolic phenotypes [134]. NPPA gene has many variants in promoter, intronic, coding and 3’-UTR [134]. C-664G variant has been related to lower ANP levels, higher BP levels and more LV hypertrophy in Italian and Japanese populations [85]. Rs5063 variant has been related to lower BP levels in American and Chinese populations [135,136,137]. Rs5065 variant has been related to less hypertension and more MI [138,139,140]. However, lots of candidate genes have not been confirmed in big-sample population genetic studies and Genome Wide Association Studies. In a big-sample and genome-wide meta-analysis, rs5068 variant in 3’-UTR has been inversely regulated by micro-RNA (miR)-425 and related to higher ANP levels, lower BP levels and less LV hypertrophy [141]. Rs5068 variant has also been correlated with lower anthropometric indices, lower C-reactive protein, higher high density lipoprotein, as well as less susceptibility to HF [142,143,144]. In NPPB gene, rs198388 and 198389 variants have been related to lower BP levels, reduced LV remodeling, improved LV function and less diabetes mellitus [145, 146]. Variants in NPPC and NPR-B gene remain unclear [133]. Variants in NPR-C gene have been related to hypertension in Genome Wide Association Studies of Caucasian and Asian populations [147]. Variants in corin gene have been related to higher BP levels and more LV hypertrophy in African-American population [148].
Genetic manipulation
In order to determine the function of NPs and medication of NPRs, genetic manipulation has been widely applied through the knockout of NPRs in animal experiments. For example, ANP-dependent natriuresis and diuresis are mediated exclusively by NPR-A in mice because these effects are completely lost after NPR-A knockout [62]. Mice lacking ANP or NPR-A have an enlarged heart, whereas mice over-expressing ANP have a smaller heart [79]. Mice over-expressing ANP are resistant to hypoxia-induced hypertension, whereas mice lacking ANP exhibit increased pulmonary hypertension in response to chronic hypoxia [73]. Moreover, designer NPs have been engineered through genetic alteration of native NPs. Compared with native NPs, designer NPs have improved efficacy and safety [7].
Roles of micro-RNA
MiR-100 has been demonstrated to inhibit NPR-C expression in rat MI tissues and human LV cells, and increased miR-100 in HF may reflect a compensatory mechanism to prolong half-life of NPs [149]. MiR-425 inhibits ANP synthesis in human heart by interacting with 3’-UTR of NPPA gene, and antagonists of miR-425 may be a potential therapy for HF [144]. MiR-21 interacts with ANP in vascular smooth muscle cells through modulating downstream cGMP signaling [150]. MiR-30 is expressed in healthy heart, but suppressed in failing heart. MiR-30 inhibits GalNAc-transferase (GALNT) 1 and 2-mediated glycosylation of proBNP-108. MiR-30-GALNT pathway may be a novel therapeutic target for HF, and more researches are necessary in humans [32]. Although many miRs targeting NPRs have been suggested in silico analyses to modulate NP signaling pathways in HF, it is necessary to confirm their actual interactions in vivo experiments [151].
Epigenetic remodeling
Maladaptive remodeling of LV in HF is correlated with fetal-gene reactivation and epigenetic remodeling in promoters of NPPA and NPPB genes. Although there is nuclear export of histone deacetylase 4 (HDAC4), gene activation of NPPA and NPPB dose not require increased histone acetylation in promoters. In contrast, methylation of histone 3 lysine 9 (H3K9) and binding of heterochromatin protein 1 (HP1) in promoters of these genes are reduced by HDAC4, perhaps by forming a transcriptional repressor complex with histone methyltransferase (suppressor of variegation 3-9 homolog 1, SUV39H1). This complex is disrupted by Ca2+/calmodulin-dependent kinase II (CaMKII)δB-induced phosphorylation of HDAC4. Histone demethylase [Jumonji C (JmjC) domain-containing demethylase] may be upregulated to maintain H3K9 demethylation in HF [152].
Diagnostic values
Practical application
NPs reflect cardiac stress and function, increase drastically in patients with HF, and have powerful diagnostic value for various forms of HF [153]. Plasma NP levels are also used to evaluate HF severity [154, 155]. Plasma ANP levels differ according to atrial pressure, whereas plasma BNP levels reflect ventricular overload. BNP (half-life: 22 minutes) has been shown to have greater stability than ANP (half-life: 2 minutes) [156]. Both BNP and NT-proBNP are removed by kidney, and BNP is also degraded by NEP and NPR-C. BNP has shorter half-life than NT-proBNP (half-life: 70 minutes). Thus, BNP and NT-proBNP are preferred to other NPs as gold standard for HF diagnosis, and established as rule-out tests of HF based on clinical guidelines [157, 158]. LV hypertrophy and dysfunction lead to higher levels of ANP and BNP. Thus, elevated ANP and BNP levels can be used to identify LV hypertrophy and dysfunction in general population and hospitalized patients [159]. Rapid assay for NPs can not only discriminate the origin of acute dyspnea (acute HF versus bronchial asthma), but also manage the patients with chronic HF [160,161,162]. Plasma NT-proBNP level 300 pg/ml is appropriate for ruling out acute HF. Age-dependent cutoff levels of plasma NT-proBNP are appropriate for ruling in acute HF: 450 pg/ml in patients < 50 years of age, 900 pg/ml in patients ≥ 50 years of age, and 1800 pg/ml in patients > 75 years of age [163]. Plasma BNP level 100 pg/ml and 400 pg/ml are appropriate for ruling out and ruling in acute HF, respectively. Cutoff levels of plasma NT-proBNP and BNP are 125 pg/ml and 35 pg/ml to chronic HF, respectively [164].
Confounding factors
Although applying BNP and NT-proBNP as diagnostic biomarkers of HF has brought significant improvement in treating HF, several confounding factors, such as aging, obesity, anemia, sepsis, hypertension, MI, cardiac hypertrophy, pulmonary hypertension, atrial fibrillation, diabetes mellitus, renal failure, liver cirrhosis, severe burn and cancer chemotherapy, limit their accuracy [165]. Plasma NP levels have been inversely related to body mass index in epidemiological investigations [166, 167]. Plasma NPs have lower levels in patients with diabetes mellitus, insulin resistance or metabolic syndrome, perhaps contributing to HF risk [168, 169]. Plasma BNP has higher levels in patients with hypertension or LV hypertrophy than in those without them [170, 171]. Plasma BNP has higher levels in patients with LV concentric hypertrophy than in those with LV eccentric hypertrophy or in those with normal LV structure and hypertension [172]. BP-lowering therapy reduces BNP levels and LV mass [173, 174].
Coronary artery disease
Plasma NPs have higher levels in patients with acute coronary syndrome or exercise-induced myocardial ischemia but without ventricular dilation [175]. Plasma ANP levels rise until admission and then decline in patients with acute MI. Plasma BNP levels rise until 12-24 hours after acute MI, then decline and peak once more after 5-7 days [176]. Height of the second peak is an useful indicator of LV remodeling [177]. Although there is a gradual decrease, plasma BNP levels are still increased in chronic phase, showing LV damage and remodeling [178]. However, plasma BNP has almost normal levels if early coronary reperfusion successfully prevents LV remodeling.
Chronic kidney disease
Renal function has systematical effects on NP system. In patients with chronic kidney disease (CKD), plasma NP levels are elevated as compensatory protection of renal function. CKD is always related to cardiovascular abnormalities. However, due to unclear mechanisms, there are elevated BNP levels in patients with CKD but without cardiovascular abnormalities [179]. Plasma NP levels may be regulated both by synthetic/secretory rate from heart and by extraction rate from blood. Key mechanism may not be an increase in extraction rate from kidney. Other mechanisms include the decreases in functional renal mass, second messenger synthesis and clearance receptor degradation in kidney [180, 181]. Elevated NP levels in CKD are correlated with a counter-regulatory response directed from heart to kidney, suggesting NPs as potential biomarkers of LV remodeling in patients with CKD [181]. Plasma NP levels in CKD reflect the stress on cardiac wall caused by LV hypertrophy or dysfunction [182].
End-stage renal disease
Plasma BNP and NT-proBNP have higher levels in patients with end-stage renal disease. There is a decrease of about 20–40% in plasma BNP levels after hemodialysis (HD). Peritoneal dialysis (PD) may not alter plasma BNP levels [183]. HD promotes fluid clearance and alleviates volume overload, leading to reduced wall stress and NP release [184]. Plasma BNP has lower levels in patients with PD than in those with HD, supporting that PD may lead to slower ultrafiltration rate, higher urine output, better hemodynamic condition, less cardiac load and lower BP levels than HD [185]. However, it remains inconclusive and needs more researches [186]. Meanwhile, due to reduced ultrafiltration rate, continuous volume overload and more LV hypertrophy, patients with automated PD (APD) may have higher BNP levels than those with continuous ambulatory PD (CAPD) [187]. Moreover, plasma BNP levels have a significant potential to identify LV hypertrophy or dysfunction in patients with different dialysis and renal transplant [188,189,190].
Practical application in CKD
Renal function limits current use of NPs in patients with CKD [191]. Plasma NP levels in patients with CKD are related to CKD severity, and cutoff levels are increased as CKD stages advance. Plasma BNP levels rise to almost 200 pg/ml in patients with CKD but without HF. Compared with plasma BNP levels, plasma NT-proBNP levels may be more strongly correlated with GFR and affected by age-related decrease in GFR, suggesting careful use of NT-proBNP in elderly with CKD [192]. In patients with CKD, plasma NT-proBNP levels > 1200 pg/ml suggest chronic HF in patients < 50 years of age and > 4502 pg/ml in patients between 50 and 75 years old [180]. It remains unclear if elevated NP levels in CKD effectively reflect the activation of NP system and effects on target organ. Elevated NP levels may have reduced ability to activate NP system and affect target organ in CKD. NP resistance in CKD may be caused by downregulated NPR-A expression in renal medulla and upregulated NPR-C expression in renal cortex [193]. NP resistance in CKD results in the invalidity of NP infusion in protecting renal function and treating cardiorenal syndrome in patients with HF [194].
Point-of-care systems
Previous assays for BNP are invasive and time-consuming with the discomfort caused by venipuncture. Some ideal point-of-care (POC) systems have been developed to allow rapid and repeated assays for BNP from capillary blood, like measuring blood sugar from fingertip [195]. A system at bedside would not only be useful for repeated assays at home, but also achieve routine monitoring in a remote way. Additionally, POC systems would also be used in the hospitals for BNP-guided therapy of HF and rapid triage of dyspnea. Two POC systems for BNP use either venipuncture ethylenediaminetetraacetic acid (EDTA) plasma (Alere Triage) or EDTA whole blood (Abbott i-STAT) [196,197,198,199]. In contrast, Alere Heart Check BNP Test is the first POC system easily used as a rapid assay for BNP from untreated fingertip capillary whole blood. Previous studies have demonstrated the safety and feasibility of Alere Heart Check BNP Test at home [200,201,202].
Current bioactivity assays
Current immunoassays can not reflect the bioactivity of NPs [203]. Rabbit aortic strip test (RAST) measures inhibitory activity of recombinant form of human BNP (rhBNP) on the tension of isolated aortic strip to determine its bioactivity [204]. However, it is variable, rough, laborious and time-consuming with the isolation of fresh issues from sacrificed rabbits. As the well-characterized pathways activated by rhBNP, cGMP has been quantified as alternative assays in human umbilical vein endotheliocyte (HUVEC) or rat pheochromocytoma cell-12 (PC-12) by radioimmunoassays [205]. However, HUVEC is primary cell and PC-12 tends to differentiate. Both HUVEC and PC-12 are not stable in culture. These intracellular cGMP measurements have limited accuracy, precision and reproducibility. They are tedious with the preparation of cell lysate and depend on standard curve by radioimmunoassays [206]. One stable cell-based assay is based on the NPR-A in HEK 293 cell. This single high-responsiveness clone is simplified by detecting the cGMP in culture supernatant with non-radioactive material in a high-throughput manner [207]. Although this assay has a significant potential to monitor endogenous activities of NPs, it needs more researches involving different NP forms and clinical stages [203].
Therapeutic values
Mechanisms of therapy
Mortality rate remains high in patients with HF even with the best current therapies. This mandates a continuing search for new therapies. NPs counteract RAAS by inhibiting renin secretion through second messenger cGMP, and NP augment on top of RAAS blockade may have synergistic effects on HF [208]. NPR-A suppression activates the RAAS and impairs the kidney [209]. Although HF develops with progressive activation of NPs, this response is apparently insufficient to counteract vascular constriction and sodium retention of RAAS and SNS [7]. Synthetic ANP has an attenuated renal response (natriuresis and diuresis) in patients with HF, suggesting NPR dysregulation in these patients with RAAS activation [62]. However, synthetic ANP has other significant roles, such as hemodynamic improvement and RAAS inhibition, in patients with HF [210]. Elevated NP levels maintain sodium balance in early stage of HF, and NPR-A suppression in HF causes sodium retention [211, 212]. NPs suppress angiotensin-II-induced vasoconstriction, angiotensin-II-stimulated proximal tubule sodium reabsorption, angiotensin-II-enhanced aldosterone secretion and endothelin secretion [213]. Moreover, most of plasma BNP measured with current immunoassays is less active in patients with HF. Thus, HF induces an attenuated response to elevated BNP levels, and represents a deficiency of active BNP caused by abnormal processing of NPs [214, 215].
Increasing NP bioactivity
Novel therapies are under development based on an augment in cardioprotective effects of NPs to re-balance neuroendocrine dysregulation in HF [216]. Current NP-augmenting strategies include the synthesis of NPs or agonists to increase NP bioactivity and inhibition of NEP to reduce NP breakdown [217, 218]. Nesiritide, a rhBNP, has been shown to induce hemodynamic and clinical improvements in Vasodilatation in the Management of Acute CHF (VMAC) and other trials [219]. Nesiritide is successfully approved by Food and Drug Administration (FDA) and routinely used for both acute and chronic HF. However, nesiritide has been questioned in two subsequent meta-analyses to worsen renal function and increase mortality rate [220]. Other studies, such as Acutely Decompensated Heart Failure Registry (ADHERE) trial, have not confirmed these unfavorable effects of nesiritide [221, 222]. Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND-HF) trial has reported that nesiritide has no significant relationship with mortality rate, nor is it related to worsening renal function [223]. Neutral conclusions may be correlated with nesiritide dose. Large dose of nesiritide is strongly vasodilative, causes severe hypotension and neutralizes beneficial roles. Recent studies have reported that small dose of nesiritide, particularly when administered through subcutaneous route, induces hemodynamic and clinical improvements, without increasing nephrotoxicity and mortality, thus reopening the debates about its usefulness in patients with HF [224]. Long-term antiapoptotic, antiremodeling and antihypertrophic actions of nesiritide are beneficial if NPR-A could be activated chronically [225]. Nesiritide administered twice daily for eight weeks through subcutaneous route has improved clinical symptoms and reduced LV mass in patients with HF [226]. Additionally, nesiritide has been suggested to protect LV function in patients with MI [227]. Carperitide, recombinant form of human ANP, has alleviated clinical symptoms and been recommended in Japan for acute decompensated HF [125]. But short half-life limits clinical application of carperitide. Moreover, oral forms of ANP and BNP are too unstable to be routinely used in HF. CNP is unsuitable for treating HF due to relatively short half-life and no renal-enhancing action. In Safety and Efficacy of an Intravenous Placebo-controlled Randomised Infusion of Ularitide (SIRIUS I and II), synthetic urodilatin (ularitide) has induced hemodynamic and clinical improvements, without worsening renal function and obvious BP change, in patients with acute decompensated HF [228, 229]. Another Phase III Trial of Ularitide Efficacy and Safety in Acute Heart Failure (TRUE-AHF) has shown that ularitide reduces systolic BP and cardiac stress as indicated by plasma NT-proBNP levels, but has no effect on clinical composite end point, cardiovascular mortality and myocardial injury as indicated by cardiac troponin T levels [230].
Designer NPs
Severe hypotension and short half-life make recombinant agents, including nesiritide, carperitide and ularitide, not very suitable for clinical use. Designer NPs are developed by altering genetic forms or aa structures of native NPs. These hybrid peptides have normal binding to NPR-A and increased resistance to degradation [7]. DNP is firstly discovered in snake venom, and much about DNP remains unclear in humans. Cenderitide-NP (CD-NP) is not easy to be degraded as a 37-aa hybrid NP designed by fusing native CNP-22 with 15-aa C-terminal of DNP. This first-generation designer NP retains vasodilative, antifibrotic and antihypertrophic roles of CNP, and natriuretic and diuretic roles of DNP [231]. Both NPR-A and NPR-B can be effectively activated by CD-NP to increase more GFR and cause less hypotension than nesiritide, with reduced atrial pressure and improved cardiac-unloading effect [232]. FDA has provided a fast-track designation for CD-NP in Phase II trials [233]. CU-NP has been designed by fusing 17-aa ring structure of native CNP with C- and N-terminal of urodilatin [234]. As an experimental agent in early stage, CU-NP exerts cardiac-unloading, renal-enhancing and RAAS-suppressing effects through activating cGMP. CU-NP has direct antihypertrophic effect through inhibiting sodium-hydrogen exchanger 1 (NHE-1)/calcineurin pathway [235]. Mutant ANP (M-ANP) has been designed as a 40-aa peptide by fusing native ANP with 12-aa extension to C-terminal [236]. M-ANP has exerted beneficial cardiac and renal effects, such as boosting natriuresis and diuresis, regulating BP and GFR, inhibiting RAAS and SNS, and promoting antifibrosis and antiproliferation in experiments [237]. Novel NPs are currently under clinical development programs for further trials [238]. An alternative RNA spliced transcript for BNP (AS-BNP) has a unique 34-aa C-terminal, with remaining structure of native BNP [205]. ANX-042 has been designed as a 42-aa peptide by fusing 16 aa from C-terminal of AS-BNP and 26 aa from native BNP. ANX-042 can activate cGMP to boost natriuresis and diuresis and suppress renin and angiotensin-II, but not activate cGMP to relax blood vessels. As a designer NP in a first-in-human trial, FDA has suggested ANX-042 as an investigated new drug for HF with renal protection and less hypotension [239]. CNP analog (BMN111) is one of the most promising therapy for achondroplasia, and obviously improves skeletal parameters in animal experiments [240].
Reducing NP degradation
Although NP breakdown can be blocked by affecting NPR-C and inhibiting IDE, the more commonly used approach to reduce NP degradation is NEP inhibition. However, there are plentiful substrates of NEP, such as angiotensin-I, angiotensin-II, bradykinin, substance P, adrenomedullin, endothelin-1, opioid peptide, insulin β-chain, glucagon, oxytocin, chemotactic peptide, neurotensin, enkephalins, gastrin and amyloid-β peptide. NEP inhibition has potential to increase levels of these substrates, leading to conflicting effects on kidney and vessels [7]. Moreover, NEP hydrolyzes angiotensin-I to angiotensin-(1-7), which counteracts angiotensin-II. As the first pure NEP inhibitor, candoxatril is stable when administered orally. However, due to its effects on other systems, candoxatril has no benefit for patients with hypertension or HF [241]. Candoxatril is characterized by both NP augment (elevated NP levels and natriuresis) and RAAS activation (elevated angiotensin-II levels and vasoconstriction), leading to unaltered vascular resistance and unavailable antihypertensive role.
Dual ACE/NEP inhibitors
Pure NEP inhibitors have disappointing clinical effects, which may be improved by combining RAAS blockade. Due to no improvement in clinical symptoms and an increase in aplastic anemia, it is unpractical to choose an addition of NEP inhibitors (ecadotril) to standard therapies including angiotensin-converting enzyme (ACE) inhibitors [242]. Dual ACE/NEP inhibitor (vasopeptidase inhibitor, sampatrilat) has shown beneficial effects, but then been dropped due to short half-life [243]. Omapatrilat (BMS-186716) has almost affinity and inhibition for NEP and ACE. In experimental HF and hypertension models, omapatrilat has not only improved clinical symptoms and survival, but also relieved cardiac dysfunction and hypertension [244]. Moreover, omapatrilat has improved cardiac function and remodeling, and decreased cardiac hypertrophy and fibrosis in mice with MI [245]. Omapatrilat has been evaluated in patients with HF or hypertension in Omapatrilat Cardiovascular Treatment Versus Enalapril (OCTAVE), Inhibition of Metalloprotease by Omapatarilat in a Randomized Exercise and Symptoms Study of Heart Failure (IMPRESS) and Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE) trials [246,247,248]. Omapatrilat has more obviously lowered vascular resistance and BP levels than candoxatril. As bradykinin is degraded by both NEP and ACE, simultaneous inhibition of them by omapatrilat increases bradykinin levels that favor the development of angioedema. Compared with enalapril, omapatrilat has increased angioedema and hypotension, and shown no superior benefit in patients with HF or hypertension, precluding its clinical use and final approval of FDA [249].
Triple ACE/ECE/NEP inhibitors
Endothelin-1 is a multifunctional vasoconstrictor and contributes to HF progression [250]. Most endothelin-1 receptor antagonists have no prognostic improvement in patients with acute and chronic HF. In experimental HF model, endothelin-converting enzyme (ECE) inhibition has suppressed endothelin-1 synthesis, improved cardiorenal function and reduced the neurohormones, such as renin, angiotensin-II and aldosterone [251]. NPs may be degraded by ECE. Thus, ECE inhibition may simultaneously augment the NPs and suppress the endothelin [252]. ECE inhibition has induced hemodynamic improvement in patients with HF [253]. However, there is no long-term study about ECE inhibition. Dual ECE/NEP inhibitor SLV-306 (daglutril) has not only lowered LV pressure in patients with HF, but also improved cardiac function and remodeling in rats with LV hypertrophy [254]. Moreover, daglutril has inhibited BP elevation and increased NP levels in healthy humans [255]. Another dual ECE/NEP inhibitor (SLV-338) has improved cardiac fibrosis in experiment [256]. Triple ACE/ECE/NEP inhibitors may inhibit the synthesis of angiotensin-II and endothelin-1, and enhance the effects of NPs and bradykinin. In experimental HF model, triple ACE/ECE/NEP inhibition has been superior to ACE inhibition and dual ECE/NEP inhibition in improving LV structure and function [257]. However, the development of triple ACE/ECE/NEP inhibitors may be obstructed by negative conclusions about endothelin-1 receptor antagonists from large HF trials and practical concerns about the safety with ACE/NEP inhibitors. It needs to be emphasized that endothelin-1 receptor antagonism and ECE inhibition should be distinguished in further human trials.
Development of LCZ696
Angiotensin receptor blocker NEP inhibitor (ARNI, LCZ696) is a major advance in the therapies for HF in the last 15 years. Molecular moieties of NEP inhibitor prodrug sacubitril (AHU377), and valsartan, an angiotensin receptor blocker (ARB), are present in this single molecule in 1:1 molar ratio (sacubitril/valsartan). Sacubitril (AHU377) becomes active NEP inhibitor LBQ657 after cleaving ethyl ester. ARNI preserves ACE mechanism for bradykinin degradation [258]. ARNI augments beneficial effects of NPs and inhibits harmful effects of angiotensin-II. As the first-in-class ARNI, LCZ696 has improved cardiac dysfunction, fibrosis, remodeling and hypertrophy in an animal model [245]. Compared with valsartan alone, LCZ696 has more effectively lowered BP levels, without increased angioedema, in patients with hypertension [259]. In patients with HF with reduced ejection fraction (HFrEF), LCZ696 has more effectively reduced all-cause, cardiovascular and sudden death, prevented HF progression and hospitalization, and improved life quality and renal function than enalapril in Prospective comparison of ARNI with ACE inhibitor to Determine Impact on Global Mortality and morbidity in Heart Failure (PARADIGM-HF) trial [260,261,262,263]. LCZ696 has recently been approved by FDA for treating HFrEF. But translating the results of this trial into guideline recommendation has raised some concerns [264]. Firstly, this study has been discontinued ahead of schedule due to overwhelming benefit of LCZ696, and there is a doubt about efficacy and safety of LCZ696 used for longer time [265]. Secondly, LCZ696 was administered twice daily at a dose of 200 mg (160 mg of valsartan), and enalapril was administered twice daily at a dose of 10 mg. Both two doses are target doses in most HF guidelines but higher than many patients with HFrEF may tolerate. Thirdly, more symptomatic postural hypotension in LCZ-696 group limited its clinical use, particularly in patients with borderline BP before therapy. It is necessary to observe this agent according to baseline BP in further studies. Fourthly, bradykinin levels have previously been shown to increase with ARB, and there were more patients with angioedema in LCZ-696 group [266]. Fifthly, amyloid-β peptide is a key peptide in Alzheimer disease, and NEP may block its breakdown to induce Alzheimer disease [267]. However, Alzheimer disease and cancer were not increased using LCZ696, and cognitive decline related to vascular diseases may be reduced by LCZ696. Finally, drug denials have already been increased, and applying LCZ696 would be further complicated by cost. Compared with valsartan, LCZ696 has not only reduced NT-proBNP levels and caused GFR elevation, but also improved overall clinical status and decreased left atrial pressure in Prospective comparison of ARNI with ARB on Management Of heart failUre with preserved ejectioN fracTion (PARAMOUNT) trial [268]. Whether it would benefit patients with HF with preserved ejection fraction (HFpEF) is currently being tested in ongoing Efficacy and Safety of LCZ696 Compared to Valsartan on Morbidity and Mortality in Heart Failure Patients With Preserved Ejection Fraction (PARAGON-HF, NCT01920711) trial. ARNI class has sparked considerable excitement in other cardiovascular diseases including hypertension [269]. Prospective comparison of Angiotensin Receptor neprilysin inhibitor with Angiotensin receptor blocker MEasuring arterial sTiffness in the eldERly (PARAMETER) trail is also underway to compare the relationships of LCZ696 and olmesartan with central BP in patients with resistant hypertension [270].
Mechanisms of LCZ696
It remains unclear about the mechanisms responsible for the superiority of LCZ696 over ACE inhibitor. Considering elevated levels of plasma BNP and urinary cGMP, systemic vasodilation and renal natriuresis could be important mechanisms. But it remains uncertain whether its superiority is direct effect on heart or indirect effect secondary to beneficial effects of this agent on vessels (lower BP levels) and kidney (less renal injury) [271]. Lower NT-proANP and troponin levels support that LCZ696 directly reduces myocardial stretch or ischemia. LCZ696 reduces proteinuria, focal segmental glomerulosclerosis and retinopathy, and plays beneficial effects on microvascular and renal complications. Without the evidence in PARADIGM-HF, further studies are required to fully address this issue. Meanwhile, positive effects of BNP on cardiac regeneration may also play an important role, and should be fully addressed in ongoing experimental and clinical studies. Exogenous BNP or NEP inhibition may induce endogenous cardiac regeneration, and achieve the therapies for HF and MI [4]. In experimental hypertension models, either alone or combined with MI or diabetes mellitus, ARNI has improved cardiac hypertrophy and fibrosis in a BP-independent manner. Since sacubitril is largely cleared in kidney, drug accumulation may occur in patients with impaired renal function, and hypotension is a potential adverse effect in patients with CKD.
BNP-guided therapy
Serial assays for BNP over time are clinically applied to the management of HF. BNP-guided therapy can assess the effectiveness and adjust the doses of drugs for HF, and improve the survival in patients with HFrEF or HFpEF [272]. United Kingdom-based economic model of BNP-guided therapy has been developed in patient with chronic HF [273]. BNP-guided therapy is cost-effective in younger patients (< 75 years) with HFrEF. It is potentially cost-effective in younger patients (< 75 years) with HFpEF and older patients (≥ 75 years) with HFrEF, but more evidence is required, particularly with respect to the frequency, duration and target for BNP monitoring [274]. Ongoing Guiding Evidence Based Therapy Using Biomarker Intensified Treatment in Heart Failure (GUIDE-IT) trial would be very important in providing better evidence in patients with HFrEF [275]. Additionally, NEP inhibitors may increase BNP levels and lower NT-proBNP levels, and require different monitoring strategies in BNP-guided therapy [261].
Prognostic values
Plasma NP levels have prognostic values in patients with cardiovascular diseases. Previous studies on NP infusion, experimental animals and population genetics have demonstrated inverse correlations of plasma NP levels with different cardiovascular diseases. But epidemiological and clinical investigations of NPs as prognostic biomarkers have yielded positive correlations of plasma NP levels with poor prognosis [276]. As counter-regulatory hormones secreted after cardiac stretch, NPs have this paradox should be no surprise. In epidemiological investigations, elevated NP levels even in regular limit have been commonly observed in patients with subclinical cardiovascular diseases. In Framingham Offspring Study and Copenhagen, elevated NP levels have been significantly related to major adverse cardiovascular events and mortality rate in population without obvious cardiovascular diseases [277]. In patients with stable coronary artery disease, acute coronary syndrome or HF, elevated NP levels have also been significantly associated with cardiovascular events and mortality rate [154, 278, 279]. If plasma BNP or NT-proBNP levels do not fall off after the therapies for HF, patients with HF have more hospital admission and higher mortality rate. In patients with HF, NT-proBNP has higher levels, better accuracy, longer half-life and lower variation than BNP, and may be a better biomarker of HF progression and mortality rate [280]. Moreover, NPs are reliable predictors of all-cause and cardiovascular death independently of other clinical and biochemical risk factors, and have a potential to guide the therapy and predict the prognosis in patients with CKD [281,282,283].
Conclusion
NPs play central roles in the regulation of HF. Both BNP and NT-proBNP are useful biomarkers to not only make the diagnosis and assess the severity of HF, but also guide the therapy and predict the prognosis in patients with HF. Current NP-augmenting strategies include the synthesis of NPs or agonists to increase NP bioactivity and inhibition of NEP to reduce NP breakdown. Nesiritide has been established as an available therapy, and ARNI has obtained extremely encouraging results with decreased morbidity and mortality. Novel pharmacological approaches based on NPs may promote a therapeutic shift from suppressing the RAAS and SNS to re-balancing neuroendocrine dysregulation in patients with HF.
Abbreviations
- Aa:
-
Amino acid
- ANP:
-
Atrial natriuretic peptide
- APD:
-
Automated peritoneal dialysis
- ARNI:
-
Angiotensin receptor blocker NEP inhibitor
- BNP:
-
B-type natriuretic peptide
- BP:
-
Blood pressure
- CAPD:
-
Continuous ambulatory peritoneal dialysis
- CD-NP:
-
Cenderitide-NP
- cGMP:
-
Cyclic guanosine monophosphate
- CKD:
-
Chronic kidney disease
- CPCs:
-
Cardiac precursor cells
- CNP:
-
C-type natriuretic peptide
- C-terminal:
-
Carboxy-terminal
- DPP-4:
-
Dipeptidyl peptidase-4
- GC:
-
Guanylate cyclase
- GFR:
-
Glomerular filtration rate
- GTP:
-
Guanosine triphosphate
- HD:
-
Hemodialysis
- HF:
-
Heart failure
- IDE:
-
Insulin degrading enzyme
- LV:
-
Left ventricle
- miR:
-
micro-RNA
- NEP:
-
Neutral endopeptidase
- NO:
-
Nitric oxide
- NP:
-
Natriuretic peptide
- NPP:
-
Natriuretic peptide precursor
- NPR:
-
Natriuretic peptide receptor
- N-terminal:
-
Amino-terminal
- NT-proANP:
-
N-terminal proANP
- NT-proBNP:
-
N-terminal proBNP
- PD:
-
Peritoneal dialysis
- PDE:
-
Phosphodiesterase
- PKG:
-
Protein kinase
- POC:
-
Point-of-care
- RAAS:
-
Renin-angiotensin-aldosterone system
- rhBNP:
-
Recombinant form of human BNP
- SNS:
-
Sympathetic nervous system
- 5’-FR:
-
5’-flanking region
- 5’-UTR:
-
5’-untranslated region
References
Owens AT, Brozena SC, Jessup M. New Management Strategies in Heart Failure. Circ Res. 2016;118(3):480–95.
Wong PC, Guo J, Zhang A. The renal and cardiovascular effects of natriuretic peptides. Adv Physiol Educ. 2017;41(2):179–85.
Gupta DK, Wang TJ. Natriuretic Peptides and Cardiometabolic Health. Circ J. 2015;79(8):1647–55.
Rosenblatt-Velin N, Badoux S, Liaudet L. Pharmacological Therapy in the Heart as an Alternative to Cellular Therapy: A Place for the Brain Natriuretic Peptide? Stem Cells Int. 2016;2016:5961342.
Zois NE, Bartels ED, Hunter I, Kousholt BS, Olsen LH, Goetze JP. Natriuretic peptides in cardiometabolic regulation and disease. Nat Rev Cardiol. 2014;11(7):403–12.
Ichiki T, Schirger JA, Huntley BK, Brozovich FV, Maleszewski JJ, Sandberg SM, Sangaralingham SJ, Park SJ, Burnett JC Jr. Cardiac fibrosis in end-stage human heart failure and the cardiac natriuretic peptide guanylyl cyclase system: regulation and therapeutic implications. J Mol Cell Cardiol. 2014;75:199–205.
Volpe M, Carnovali M, Mastromarino V. The natriuretic peptides system in the pathophysiology of heart failure: from molecular basis totreatment. Clin Sci (Lond). 2016;130(2):57–77.
de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Reprinted from Life Sci. 28:89-94, 1981. J Am Soc Nephrol. 2001;12(2):403–9.
Ogawa Y, Itoh H, Nakao K. Molecular biology and biochemistry of natriuretic peptide family. Clin Exp Pharmacol Physiol. 1995;22(1):49–53.
Knowlton KU, Rockman HA, Itani M, Vovan A, Seidman CE, Chien KR. Divergent pathways mediate the induction of ANF transgenes in neonatal and hypertrophic ventricular myocardium. J Clin Invest. 1995;96(3):1311–8.
Yan W, Wu F, Morser J, Corin WQ. a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. Proc Natl Acad Sci U S A. 2000;97(15):8525–9.
Sugawara A, Nakao K, Morii N, Yamada T, Itoh H, Shiono S, Saito Y, Mukoyama M, Arai H, Nishimura K, et al. Synthesis of atrial natriuretic polypeptide in human failing hearts. Evidence for altered processing of atrial natriuretic polypeptide precursor and augmented synthesis of beta-human ANP. J Clin Invest. 1988;81(6):1962–70.
van Kimmenade RR, Januzzi JL Jr. The evolution of the natriuretic peptides-Current applications in human and animal medicine. J Vet Cardiol. 2009;11(Suppl 1):S9–21.
Nishikimi T, Kuwahara K, Nakao K. Current biochemistry, molecular biology, and clinical relevance of natriuretic peptides. J Cardiol. 2011;57(2):131–40.
Yandle TG, Richards AM. B-type natriuretic peptide circulating forms: analytical and bioactivity issues. Clin Chim Acta. 2015;448:195–205.
Zachariah JP, Aliku T, Scheel A, Hasan BS, Lwabi P, Sable C, Beaton AZ. Amino-terminal pro-brain natriuretic peptide in children with latent rheumatic heart disease. Ann Pediatr Cardiol. 2016;9(2):120–5.
Minamino N, Horio H, Nishikimi T. Natriuretic peptides in the cardiovascular system. In: Kastin AJ, editor. The handbook of biologically active peptides. 1st ed. New York. London: Academic Press; 2006. p. 1217–25.
Abassi Z, Karram T, Ellahamc S, Winaver J, Hoffman A. Implications of the natriuretic peptide system in the pathogenesis of heart failure: diagnostic and therapeutic importance. Pharmacol Ther. 2004;102(3):223–41.
Ma KK, Ogawa T, de Bold AJ. Selective upregulation of cardiac brain natriuretic peptide at the transcriptional and translational levels by pro-inflammatory cytokines and by conditioned medium derived from mixed lymphocyte reactions via p38 MAP kinase. J Mol Cell Cardiol. 2004;36(4):505–13.
Clerico A, Vittorini S, Passino C. Circulating forms of the b-type natriuretic peptide prohormone: pathophysiologic and clinical considerations. Adv Clin Chem. 2012;58:31–44.
Ichiki T, Huntley BK, Burnett JC Jr. BNP molecular forms and processing by the cardiac serine protease corin. Adv Clin Chem. 2013;61:1–31.
Nishikimi T, Minamino N, Masashi I, Takeda Y, Tadokoro K, Shibasaki I, Fukuda H, Horiuchi Y, Oikawa S, Ieiri T, Matsubara M, Ishimitsu T. Diversity of molecular forms of plasma brain natriuretic peptide in heart failure-different proBNP-108 to BNP-32 ratios in atrial and ventricular overload. Heart. 2010;96(6):432–9.
Nishikimi T, Kuwahara K, Nakagawa Y, Kangawa K, Minamino N, Nakao K. Complexity of molecular forms of B-type natriuretic peptide in heart failure. Heart. 2013;99(10):677–9.
Liang F, O’Rear J, Schellenberger U, Tai L, Lasecki M, Schreiner GF, Apple FS, Maisel AS, Pollitt NS, Protter AA. Evidence for functional heterogeneity of circulating B-type natriuretic peptide. J Am Coll Cardiol. 2007;49(10):1071–8.
Menon SG, Mills RM, Schellenberger U, Saqhir S, Protter AA. Clinical implications of defective B-type natriuretic peptide. Clin Cardiol. 2009;32(12):E36–41.
Dickey DM, Potter LR. ProBNP(1-108) is resistant to degradation and activates guanylyl cyclase-A with reduced potency. Clin Chem. 2011;57(9):1272–8.
Semenov AG, Postnikov AB, Tamm NN, Seferian KR, Karpova NS, Bloshchitsyna MN, Koshkina EV, Krasnoselsky MI, Serebryanaya DV, Katrukha AG. Processing of pro-Brain natriuretic peptide is suppressed by O-glycosylation in the region close to the cleavage site. Clin Chem. 2009;55(3):489–98.
Semenov AG, Tamm NN, Seferian KR, Postnikov AB, Karpova NS, Serebryanaya DV, Koshkina EV, Krasnoselsky MI, Katrukha AG. Processing of pro-B-type natriuretic peptide: furin and corin as candidate convertases. Clin Chem. 2010;56(7):1166–76.
Peng J, Jiang J, Wang W, Qi X, Sun XL, Wu Q. Glycosylation and processing of pro-B-type natriuretic peptide in cardiomyocytes. Biochem Biophys Res Commun. 2011;411(3):593–8.
Huntley BK, Sandberg SM, Heublein DM, Sangaralingham SJ, Burnett JC Jr, Ichiki T. Pro-B-type natriuretic peptide-1-108 processing and degradation in human heart failure. Circ Heart Fail. 2015;8(1):89–97.
Vodovar N, Seronde MF, Laribi S, Gayat E, Lassus J, Boukef R, Nouira S, Manivet P, Samuel JL, Logeart D, Ishihara S, Cohen Solal A, Januzzi JL Jr, Richards AM, Launay JM, Mebazaa A. GREAT Network. Post-translational modifications enhance NT-proBNP and BNP production in acute decompensated heart failure. Eur Heart J. 2014;35(48):3434–41.
Nakagawa Y, Nishikimi T, Kuwahara K, Fujishima A, Oka S, Tsutamoto T, Kinoshita H, Nakao K, Cho K, Inazumi H, Okamoto H, Nishida M, Kato T, Fukushima H, Yamashita JK9, Wijnen WJ, Creemers EE, Kangawa K, Minamino N, Nakao K, Kimura T. MiR30-GALNT1/2 Axis-Mediated Glycosylation Contributes to the Increased Secretion of Inactive Human Prohormone for Brain Natriuretic Peptide (proBNP) From Failing Hearts. J Am Heart Assoc. 2017;6(2). pii: e003601.
Nishikimi T, Okamoto H, Nakamura M, Ogawa N, Horii K, Nagata K, Nakagawa Y, Kinoshita H, Yamada C, Nakao K, Minami T, Kuwabara Y, Kuwahara K, Masuda I, Kangawa K, Minamino N, Nakao K. Direct immunochemiluminescent assay for proBNP and total BNP in human plasma proBNP and total BNP levels in normal and heart failure. PLoS One. 2013;8(1):e53233.
Seferian KR, Tamm NN, Semenov AG, Mukharyamova KS, Tolstaya AA, Koshkina EV, Kara AN, Krasnoselsky MI, Apple FS, Esakova TV, Filatov VL, Katrukha AG. The brain natriuretic peptide (BNP) precursor is the major immunoreactive form of BNP in patients with heart failure. Clin Chem. 2007;53(5):866–73.
Heublein DM, Huntley BK, Boerrigter G, Cataliotti A, Sandberg SM, Redfield MM, Burnett JC Jr. Immunoreactivity and guanosine 3’, 5’-cyclic monophosphate activating actions of various molecular forms of human B-type natriuretic peptide. Hypertension. 2007;49(5):1114–9.
Scirica BM, Braunwald E, Raz I, Cavender MA, Morrow DA, Jarolim P, Udell JA, Mosenzon O, Im K, Umez-Eronini AA, Pollack PS, Hirshberg B, Frederich R, Lewis BS, McGuire DK, Davidson J, Steg PG, Bhatt DL. Heart failure, saxagliptin, and diabetes mellitus: observations from the SAVOR-TIMI 53 randomized trial. Circulation. 2015;132(15):e198.
Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369(14):1317–26.
Fadini GP, Avogaro A, Degli Esposti L, Russo P, Saragoni S, Buda S, Rosano G, Pecorelli S, Pani L. Risk of hospitalization for heart failure in patients with type 2 diabetes newly treated with DPP-4 inhibitors or other oral glucose-lowering medications: a retrospective registry study on 127,555 patients from the Nationwide OsMed Health-DB Database. Eur Heart J. 2015;36(36):2454–62.
Chang CH, Chang YC, Lin JW, Caffrey JL, LC W, Lai MS, Chuang LM. No increased risk of hospitalization for heart failure for patients treated with dipeptidyl peptidase-4 inhibitors in Taiwan. Int J Cardiol. 2016;220:14–20.
SM O, Chen HT, Kuo SC, Chen TJ, Shih CJ, Chen YT. Dipeptidyl peptidase-4 inhibitors and cardiovascular risks in patients with pre-existing heart failure. Heart. 2017;103(6):414–20.
Sato A, Yoshihisa A, Kanno Y, Takiguchi M, Miura S, Shimizu T, Nakamura Y, Yamauchi H, Owada T, Sato T, Suzuki S, Oikawa M, Yamaki T, Sugimoto K, Kunii H, Nakazato K, Suzuki H, Saitoh SI, Takeishi Y. Associations of dipeptidyl peptidase-4 inhibitors with mortality in hospitalized heart failure patients with diabetes mellitus. ESC Heart Fail. 2016;3(2):77–85.
Elgendy IY, Mahmoud AN, Barakat AF, Elgendy AY, Saad M, Abuzaid A, Wayangankar SA, Bavry AA. Cardiovascular Safety of Dipeptidyl-Peptidase IV Inhibitors: a meta-analysis of placebo-controlled randomized trials. Am J Cardiovasc Drugs. 2017;17(2):143–55.
Li L, Li S, Deng K, Liu J, Vandvik PO, Zhao P, Zhang L, Shen J, Bala MM, Sohani ZN, Wong E, Busse JW, Ebrahim S, Malaga G, Rios LP, Wang Y, Chen Q, Guyatt GH, Sun X. Dipeptidyl peptidase-4 inhibitors and risk of heart failure in type 2 diabetes: systematic review and meta-analysis of randomised and observational studies. BMJ. 2016;352:i610.
Takahashi A, Ihara M, Yamazaki S, Asanuma H, Asakura M, Kitakaze M. Impact of either GLP-1 agonists or DPP-4 inhibitors on pathophysiology of heart failure. Int Heart J. 2015;56(4):372–6.
Fadini GP, Bonora BM, Albiero M, Zaninotto M, Plebani M, Avogaro A. DPP-4 inhibition has no acute effect on BNP and its N-terminal pro-hormone measured by commercial immune-assays. A randomized cross-over trial in patients with type 2 diabetes. Cardiovasc Diabetol. 2017;16(1):22.
Wu C, Wu F, Pan J, Morser J, Wu Q. Furin-mediated processing of Pro-C-type natriuretic peptide. J Biol Chem. 2003;278(28):25847–52.
Zhang Z, Xiao Z, Diamond SL. Shear stress induction of c-type natriuretic peptide (CNP) in endothelial cells is independent of no autocrine signaling. Ann Biomed Eng. 1999;27(4):419–26.
Yasoda A, Kitamura H, Fujii T, Kondo E, Murao N, Miura M, Kanamoto N, Komatsu Y, Arai H, Nakao K. Systemic administration of c-type natriuretic peptide as a novel therapeutic strategy for skeletal dysplasias. Endocrinology. 2009;150(7):3138–44.
Mangiafico S, Costello-Boerrigter LC, Andersen IA, Cataliotti A, Burnett JC Jr. Neutral endopeptidase inhibition and the natriuretic peptide system: an evolving strategy in cardiovascular therapeutics. Eur Heart J. 2013;34(12):886–893c.
Dickey DM, Burnett JC Jr, Potter LR. Novel bifunctional natriuretic peptides as potential therapeutics. J Biol Chem. 2008;283(50):35003–9.
Abbey SE, Potter R. Vasopressin-dependent inhibition of the C-type natriuretic peptide receptor, NPR-B/GC-B, requires elevated intracellular calcium concentrations. J Biol Chem. 2002;277(45):42423–30.
Misono KS. Natriuretic peptide receptor: structure and signaling. Mol Cell Biochem. 2002;230(1-2):49–60.
Theilig F, Wu Q. ANP-induced signaling cascade and its implications in renal pathophysiology. Am J Physiol Renal Physiol. 2015;308(10):F1047–55.
Duda T. Atrial natriuretic factor-receptor guanylate cyclase signal transduction mechanism. Mol Cell Biochem. 2010;334(1-2):37–51.
Potter LR. Domain analysis of human transmembrane guanylyl cyclase receptors: implications for regulation. Front Biosci. 2005;10:1205–20.
Rubattu S, Sciarretta S, Morriello A, Calvieri C, Battistoni A, Volpe M. NPR-C: a component of the natriuretic peptide family with implications in human diseases. J Mol Med (Berl). 2010;88(9):889–97.
Knecht M, Pagel I, Langenickel T, Philipp S, Scheuermann-Freestone M, Willnow T, Bruemmer D, Graf K, Dietz R, Willenbrock R. Increased expression of renal neutral endopeptidase in severe heart failure. Life Sci. 2002;71(23):2701–12.
Hawkridge AM, Heublein DM, Bergen HR III, Cataliotti A, Burnett JC Jr, Muddiman DC. Quantitative mass spectral evidence for the absence of circulating brain natriuretic peptide (BNP-32) in severe human heart failure. Proc Natl Acad Sci U S A. 2005;102(48):17442–7.
Flora DR, Potter LR. Prolonged atrial natriuretic peptide exposure stimulates guanylyl cyclase-A degradation. Endocrinology. 2010;151(6):2769–76.
Bae EH, Ma SK, Lee J, Kim SW. Altered regulation of renal nitric oxide and atrial natriuretic peptide systems in angiotensin II-induced hypertension. Regul Pept. 2011;170(1-3):31–7.
Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. 2006;27(1):47–72.
Gopi V, Parthasarathy A, Umadevi S, Vellaichamy E. Angiotensin-II down-regulates cardiac natriuretic peptide receptor-A mediated anti-hypertrophic signaling in experimental rat hearts. Indian J Exp Biol. 2013;51(1):48–55.
Goetz KL. Physiology and pathophysiology of atrial peptides. Am J Physiol. 1988;254(1 Pt 1):E1–15.
Matsukawa T, Miyamoto T. Angiotensin II-stimulated secretion of arginine vasopressin is inhibited by atrial natriuretic peptide in humans. Am J Physiol Regul Integr Comp Physiol. 2011;300(3):R624–9.
Inoue T, Nonoguchi H, Tomita K. Physiological effects of vasopressin and atrial natriuretic peptide in the collecting duct. Cardiovasc Res. 2001;51(3):470–80.
Veldkamp PJ, Carmines PK, Inscho EW, Navar LG. Direct evaluation of the microvascular actions of ANP in juxtamedullary nephrons. Am J Physiol. 1988;254(3 Pt 2):F440–4.
Brenner BM, Ballermann BJ, Gunning ME, Zeidel ML. Diverse biological actions of atrial natriuretic peptide. Physiol Rev. 1990;70(3):665–99.
Lanese DM, Yuan BH, Falk SA, Conger JD. Effects of atriopeptin III on isolated rat afferent and efferent arterioles. Am J Physiol. 1991;261(6 Pt 2):F1102–9.
Chen W, Gassner B, Börner S, Nikolaev VO, Schlegel N, Waschke J, Steinbronn N, Strasser R, Kuhn M. Atrial natriuretic peptide enhances microvascular albumin permeability by the caveolae-mediated transcellular pathway. Cardiovasc Res. 2012;93(1):141–51.
Elesgaray R, Caniffi C, Ierace DR, Jaime MF, Fellet A, Arranz C, Costa MA. Signaling cascade that mediates endothelial nitric oxide synthase activation induced by atrial natriuretic peptide. Regul Pept. 2008;151(1-3):130–4.
Costa MA, Elesgaray R, Balaszczuk AM, Arranz C. Role of NPR-C natriuretic receptor in nitric oxide system activation induced by atrial natriuretic peptide. Regul Pept. 2006;135(1-2):63–8.
Hamad AM, Clayton A, Islam B, Knox AJ. Guanylyl cyclases, nitric oxide, natriuretic peptides, and airway smooth muscle function. Am J Physiol Lung Cell Mol Physiol. 2003;285(5):L973–83.
Klinger JR, Warburton RR, Pietras LA, Smithies O, Swift R, Hill NS. Genetic disruption of atrial natriuretic peptide causes pulmonary hypertension in normoxic and hypoxic mice. Am J Physiol. 1999;276(5 Pt 1):L868–74.
Levin ER, Gardner DG, Samson WK. Natriuretic peptides. N Engl J Med. 1998;339(5):321–8.
Floras JS. Inhibitory effect of atrial natriuretic factor on sympathetic ganglionic neurotransmission in humans. Am J Physiol. 1995;269(2 Pt 2):R406–12.
Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82(3):769–824.
Knowles JW, Esposito G, Mao L, Hagaman JR, Fox JE, Smithies O, Rockman HA, Maeda N. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A-deficient mice. J Clin Invest. 2001;107(8):975–84.
Newton-Cheh C, Larson MG, Vasan RS, Levy D, Bloch KD, Surti A, Guiducci C, Kathiresan S, Benjamin EJ, Struck J, Morgenthaler NG, Bergmann A, Blankenberg S, Kee F, Nilsson P, Yin X, Peltonen L, Vartiainen E, Salomaa V, Hirschhorn JN, Melander O, Wang TJ. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat Genet. 2009;41(3):348–53.
Franco F, Dubois SK, Peshock RM, Shohet RV. Magnetic resonance imaging accurately estimates LV mass in a transgenic mouse model of cardiac hypertrophy. Am J Physiol. 1998;274(2 Pt 2):H679–83.
Somanna NK, Yariswamy M, Garagliano JM, Siebenlist U, Mummidi S, Valente AJ, Chandrasekar B. Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2. Cell Signal. 2015;27(10):1928–38.
Fujita S, Shimojo N, Terasaki F, Otsuka K, Hosotani N, Kohda Y, Tanaka T, Nishioka T, Yoshida T, Hiroe M, Kitaura Y, Ishizaka N, Imanaka-Yoshida K. Atrial natriuretic peptide exerts protective action against angiotensin II-induced cardiac remodeling by attenuating inflammation via endothelin-1/endothelin receptor A cascade. Heart Vessels. 2013;28(5):646–57.
Sharma GD, Nguyen HT, Antonov AS, Gerrity RG, von Geldern T, Pandey KN. Expression of atrial natriuretic peptide receptor-A antagonizes the mitogen-activated protein kinases (Erk2 and P38MAPK) in cultured human vascular smooth muscle cells. Mol Cell Biochem. 2002;233(1-2):165–73.
Nojiri T, Hosoda H, Tokudome T, Miura K, Ishikane S, Otani K, Kishimoto I, Shintani Y, Inoue M, Kimura T, Sawabata N, Minami M, Nakagiri T, Funaki S, Takeuchi Y, Maeda H, Kidoya H7, Kiyonari H, Shioi G, Arai Y, Hasegawa T, Takakura N, Hori M, Ohno Y, Miyazato M, Mochizuki N, Okumura M, Kangawa K. Atrial natriuretic peptide prevents cancer metastasis through vascular endothelial cells. Proc Natl Acad Sci U S A. 2015;112(13):4086–91.
Calderone A, Thaik CM, Takahashi N, Chang DL, Colucci WS. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growthpromoting effects of norepinephrine in cardiac myocytes and fibroblasts. J Clin Invest. 1998;101(4):812–8.
Rubattu S, Bigatti G, Evangelista A, Lanzani C, Stanzione R, Zagato L, Manunta P, Marchitti S, Venturelli V, Bianchi G. Association of atrial natriuretic peptide and type a natriuretic peptide receptor gene polymorphisms with left ventricular mass in human essential hypertension. J Am Coll Cardiol. 2006;48(3):499–505.
Scotland RS, Cohen M, Foster P, Lovell M, Mathur A, Ahluwalia A, Hobbs AJ. C-type natriuretic peptide inhibits leukocyte recruitment and platelet-leukocyte interactions via suppression of P-selectin expression. Proc Natl Acad Sci U S A. 2005;102(40):14452–7.
Sengenes C, Zakaroff-Girard A, Moulin A, Berlan M, Bouloumi’e A, Lafontan M, Galitzky J. Natriuretic peptide-dependent lipolysis in fat cells is a primate specificity. Am J Physiol Regul Integr Comp Physiol. 2002;283(1):R257–65.
Sengenes C, Bouloumie A, Hauner H, Berlan M, Busse R, Lafontan M, Galitzky J. Involvement of a cGMP-dependent pathway in the natriuretic peptide-mediated hormone-sensitive lipase phosphorylation in human adipocytes. J Biol Chem. 2003;278(49):48617–26.
Moro C, Pillard F, de Glisezinski I, Klimcakova E, Crampes F, Thalamas C, Harant I, Marques MA, Lafontan M, Berlan M. Exercise-induced lipid mobilization in subcutaneous adipose tissue is mainly related to natriuretic peptides in overweight men. Am J Physiol Endocrinol Metab. 2008;295(2):E505–13.
Engeli S, Birkenfeld AL, Badin PM, Bourlier V, Louche K, Viguerie N, Thalamas C, Montastier E, Larrouy D, Harant I, de Glisezinski I, Lieske S, Reinke J, Beckmann B, Langin D, Jordan J, Moro C. Natriuretic peptides enhance the oxidative capacity of human skeletal muscle. J Clin Invest. 2012;122(12):4675–9.
Bordicchia M, Liu D, Amri EZ, Ailhaud G, Dessì-Fulgheri P, Zhang C, Takahashi N, Sarzani R, Collins S. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest. 2012;122(3):1022–36.
Moilanen AM, Rysä J, Mustonen E, Serpi R, Aro J, Tokola H, Leskinen H, Manninen A, Levijoki J, Vuolteenaho O, Ruskoaho H. Intramyocardial BNP gene delivery improves cardiac function through distinct context-dependent mechanisms. Circ Heart Fail. 2011;4(4):483–95.
Cataliotti A, Tonne JM, Bellavia D, Martin FL, Oehler EA, Harders GE, Campbell JM, Peng KW, Russell SJ, Malatino LS, Burnett JC Jr, Ikeda Y. Long-term cardiac pro-B-type natriuretic peptide gene delivery prevents the development of hypertensive heart disease in spontaneously hypertensive rats. Circulation. 2011;123(12):1297–305.
Shaw SM, Fildes JE, Puchałka CM, Basith M, Yonan N, Williams SG. BNP directly immunoregulates the innate immune system of cardiac transplant recipients in vitro. Transpl Immunol. 2009;20(3):199–202.
Glezeva N, Collier P, Voon V, Ledwidge M, McDonald K, Watson C, Baugh J. Attenuation of monocyte chemotaxis-novel anti-inflammatory mechanism of action for the cardio-protective hormone B-type natriuretic peptide. J Cardiovasc Transl Res. 2013;6(4):545–57.
Chiurchiù V, Izzi V, D'Aquilio F, Carotenuto F, Di Nardo P, Baldini PM. Brain Natriuretic Peptide (BNP) regulates the production of inflammatory mediators in human THP-1 macrophages. Regul Pept. 2008;148(1-3):26–32.
Das BB, Raj S, Solinger R. Natriuretic peptides in cardiovascular diseases of fetus, infants and children. Cardiovasc Hematol Agents Med Chem. 2009;7(1):43–51.
Schwachtgen L, Herrmann M, Georg T, Schwarz P, Marx N, Lindinger A. Reference values of NT-proBNP serum concentrations in the umbilical cord blood and in healthy neonates and children. Z Kardiol. 2005;94(6):399–404.
Becker JR, Chatterjee S, Robinson TY, Bennett JS, Panáková D, Galindo CL, Zhong L, Shin JT, Coy SM, Kelly AE, Roden DM, Lim CC, MacRae CA. Differential activation of natriuretic peptide receptorsmodulates cardiomyocyte proliferation during development. Development. 2014;141(2):335–45.
Abdelalim EM, Tooyama I. BNP signaling is crucial for embryonic stem cell proliferation. PLoS One. 2009;4(4):e5341.
Kuhn M, Völker K, Schwarz K, Carbajo-Lozoya J, Flögel U, Jacoby C, Stypmann J, van Eickels M, Gambaryan S, Hartmann M, Werner M, Wieland T, Schrader J, Baba HA. The natriuretic peptide/guanylyl cyclase-system functions as a stress-responsive regulator of angiogenesis in mice. J Clin Invest. 2009;119(7):2019–30.
Bielmann C, Rignault-Clerc S, Liaudet L, Li F, Kunieda T, Sogawa C, Zehnder T, Waeber B, Feihl F, Rosenblatt-Velin N. Brain natriuretic peptide is able to stimulate cardiac progenitor cell proliferation and differentiation in murine hearts after birth. Basic Res Cardiol. 2015;110(1):455.
Bielmann C, Rignault-Clerc S, Liaudet L, Li F, Kunieda T, Sogawa C, Zehnder T, Waeber B, Feihl F, Rosenblatt-Velin N. NPR-A regulates selfrenewal and pluripotency of embryonic stem cells. Cell Mol Life Sci. 2015;72(9):1741–57.
Rignault-Clerc S, Bielmann C, Liaudet L, Waeber B, Feihl F, Rosenblatt-Velin N. Natriuretic Peptide Receptor B modulates the proliferation of the cardiac cells expressing the Stem Cell Antigen-1. Sci Rep. 2017;7:41936.
Del Ry S, Passino C, Maltinti M, Emdin M, Giannessi D. C-type natriuretic peptide plasma levels increase in patients with chronic heart failure as a function of clinical severity. Eur J Heart Fail. 2005;7(7):1145–8.
Sangaralingham SJ, Huntley BK, Martin FL, McKie PM, Bellavia D, Ichiki T, Harders GE, Chen HH, Burnett JC Jr. The aging heart, myocardial fibrosis, and its relationship to circulating C-type natriuretic Peptide. Hypertension. 2011;57(2):201–7.
Soeki T, Kishimoto I, Okumura H, Tokudome T, Horio T, Mori K, Kangawa K. C-type natriuretic peptide, a novel antifibrotic and antihypertrophic agent, prevents cardiac remodeling after myocardial infarction. J Am Coll Cardiol. 2005;45(4):608–16.
Kohno M, Yokokawa K, Yasunari K, Kano H, Minami M, Ueda M, Yoshikawa J. Effect of natriuretic peptide family on the oxidized LDL-induced migration of human coronary artery smooth muscle cells. Circ Res. 1997;81(4):585–90.
Kairuz EM, Barber MN, Anderson CR, Kanagasundaram M, Drummond GR, Woods RL. C-type natriuretic peptide (CNP) suppresses plasminogen activator inhibitor-1 (PAI-1) in vivo. Cardiovasc Res. 2005;66:574–82.
Barton M, Bény JL, d'Uscio LV, Wyss T, Noll G, Lüscher TF. Endothelium-independent relaxation and hyperpolarization to C-type natriuretic peptide in porcine coronary arteries. J Cardiovasc Pharmacol. 1998;31(3):377–83.
Del Ry S, Cabiati M, Vozzi F, Battolla B, Caselli C, Forini F, Segnani C, Prescimone T, Giannessi D, Mattii L. Expression of C-type natriuretic peptide and its receptor NPR-B in cardiomyocytes. Peptides. 2011;32(8):1713–8.
Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, Novak L, Renfrow MB, Chen YF. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res. 2008;102(2):185–92.
Kuhn M, Voss M, Mitko D, Stypmann J, Schmid C, Kawaguchi N, Grabellus F, Baba HA. Left ventricular assist device support reverses altered cardiac expression and function of natriuretic peptides and receptors in end-stage heart failure. Cardiovasc Res. 2004;64(2):308–14.
Dickey DM, Dries DL, Margulies KB, Potter LR. Guanylyl cyclase (GC)-A and GC-B activities in ventricles and cardiomyocytes from failed and non-failed human hearts: GC-A is inactive in the failed cardiomyocyte. J Mol Cell Cardiol. 2012;52(3):727–32.
Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressureoverloaded heart. Cardiovasc Res. 2011;89(2):265–72.
Itoh T, Nagaya N, Murakami S, Fujii T, Iwase T, Ishibashi-Ueda H, Yutani C, Yamagishi M, Kimura H, Kangawa K. C-type natriuretic peptide ameliorates monocrotaline-induced pulmonary hypertension in rats. Am J Respir Crit Care Med. 2004;170(11):1204–11.
Legeai-Mallet L. C-Type Natriuretic Peptide Analog as Therapy for Achondroplasia. Endocr Dev. 2016;30:98–105.
Crozier IG, Nicholls MG, Ikram H, Espiner EA, Yandle TG, Jans S. Atrial natriuretic peptide in humans. Production and clearance by various tissues. Hypertension. 1986;8(6 Pt 2):II11–5.
Hollister AS, Rodeheffer RJ, White FJ, Potts JR, Imada T, Inagami T. Clearance of atrial natriuretic factor by lung, liver, and kidney in human subjects and the dog. J Clin Invest. 1989;83(2):623–8.
Mukoyama M, Nakao K, Hosoda K, Suga S, Saito Y, Ogawa Y, Shirakami G, Jougasaki M, Obata K, Yasue H, et al. Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J Clin Invest. 1991;87(4):1402–12.
Richards AM, Crozier IG, Yandle TG, Espiner EA, Ikram H, Nicholls MG. Brain natriuretic factor: regional plasma concentrations and correlations with haemodynamic state in cardiac disease. Br Heart J. 1993;69(5):414–7.
Kita S, Nishizawa H, Okuno Y, Tanaka M, Yasui A, Matsuda M, Yamada Y, Shimomura I. Competitive binding of musclin to natriuretic peptide receptor 3 with atrial natriuretic peptide. J Endocrinol. 2009;201(2):287–95.
Jiang W, Cai DY, Pan CS, Qi YF, Jiang HF, Geng B, Tang CS. Changes in production and metabolism of brain natriuretic peptide in rats with myocardial necrosis. Eur J Pharmacol. 2005;507(1-3):153–62.
Vanneste Y, Michel A, Dimaline R, Najdovski T, Deschodt-Lanckman M. Hydrolysis of alpha-human atrial natriuretic peptide in vitro by human kidney membranes and purified endopeptidase-24.11. Evidence for a novel cleavage site. Biochem J. 1988;254(2):531–7.
Watanabe Y, Nakajima K, Shimamori Y, Fujimoto Y. Comparison of the hydrolysis of the three types of natriuretic peptides by human kidney neutral endopeptidase 24.11. Biochem Mol Med. 1997;61(1):47–51.
Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp Pharmacol. 2009;191:341–66.
Kenny AJ, Bourne A, Ingram J. Hydrolysis of human and pig brain natriuretic peptides, urodilatin, C-type natriuretic peptide and some C-receptor ligands by endopeptidase-24.11. Biochem J. 1993;291(Pt 1):83–8.
Dickey DM, Potter LR. Human B-type natriuretic peptide is not degraded by meprin A. Biochem. Biochem Pharmacol. 2010;80(7):1007–11.
Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev. 1998;19(5):608–24.
Müller D, Schulze C, Baumeister H, Buck F, Richter D. Rat insulin-degrading enzyme: cleavage pattern of the natriuretic peptide hormones ANP, BNP, and CNP revealed by HPLC and mass spectrometry. Biochemistry. 1992;31(45):11138–43.
Smith MW, Espiner EA, Yandle TG, Charles CJ, Richards AM. Delayed metabolism of human brain natriuretic peptide reflects resistance to neutral endopeptidase. J Endocrinol. 2000;167(2):239–46.
Charles CJ, Espiner EA, Nicholls MG, Richards AM, Yandle TG, Protter A, Kosoglou T. Clearance receptors and endopeptidase 24.11: equal role in natriuretic peptide metabolism in conscious sheep. Am J Physiol. 1996;271(2 Pt 2):R373–80.
Hashimoto Y, Nakao K, Hama N, Imura H, Mori S, Yamaguchi M, Yasuhara M, Hori R. Clearance mechanisms of atrial and brain natriuretic peptides in rats. Pharm Res. 1994;11(1):60–4.
Rubattu S, Sciarretta S, Volpe M. Atrial natriuretic peptide gene variants and circulating levels: implications in cardiovascular diseases. Clin Sci (Lond). 2014;127:1–13.
Zhang S, Mao G, Zhang Y, Tang G, Wen Y, Hong X, Jiang S, Yu Y, Xu X. Association between human atrial natriuretic peptide Val7Met polymorphism and baseline blood pressure, plasma trough irbesartan concentrations, and the antihypertensive efficacy of irbesartan in rural Chinese patients with essential hypertension. Clin Ther. 2005;27(11):1774–84.
Conen D, Cheng S, Steiner LL, Buring JE, Ridker PM, Zee RY. Association of 77 polymorphisms in 52 candidate genes with blood pressure progression and incident hypertension: the Women’s Genome Health Study. J Hypertens. 2009;27(3):476–83.
Kato N, Sugiyama T, Morita H, Nabika T, Kurihara H, Yamori Y, Yazaki Y. Genetic analysis of the atrial natriuretic peptide gene in essential hypertension. Clin Sci (Lond). 2000;98(3):251–8.
Niu W. The Relationship between Natriuretic Peptide Precursor a Gene T2238C Polymorphism and Hypertension: A Meta-Analysis. Int J Hypertens. 2011;2011:653698.
Sciarretta S, Marchitti S, Bianchi F, Moyes A, Barbato E, Di Castro S, Stanzione R, Cotugno M, Castello L, Calvieri C, Eberini I, Sadoshima J, Hobbs AJ, Volpe M, Rubattu S. C2238 atrial natriuretic peptide molecular variant is associated with endothelial damage and dysfunction through natriuretic peptide receptor C signaling. Circ Res. 2013;112(10):1355–64.
Cannone V, Huntley BK, Olson TM, Heublein DM, Scott CG, Bailey KR, Redfield MM, Rodeheffer RJ, Burnett JC Jr. Atrial natriuretic peptide genetic variant rs5065 and risk for cardiovascular disease in the general community: a 9-year follow-up study. Hypertension. 2013;62(5):860–5.
Jujić A, Leosdottir M, Östling G, Gudmundsson P, Nilsson PM, Melander O, Magnusson M. A genetic variant of the atrial natriuretic peptide gene is associated with left ventricular hypertrophy in a non-diabetic population--the Malmo preventive project study. BMC Med Genet. 2013;14:64.
Cannone V, Boerrigter G, Cataliotti A, Costello-Boerrigter LC, Olson TM, McKie PM, Heublein DM, Lahr BD, Bailey KR, Averna M, Redfield MM, Rodeheffer RJ, Burnett JC Jr. A genetic variant of the atrial natriuretic peptide gene is associated with cardiometabolic protection in the general community. J Am Coll Cardiol. 2011;58(6):629–36.
Cannone V, Cefalu’ AB, Noto D, Scott CG, Bailey KR, Cavera G, Pagano M, Sapienza M, Averna MR, Burnett JC Jr. The atrial natriuretic peptide genetic variant rs5068 is associated with a favorable cardiometabolic phenotype in a Mediterranean population. Diabetes Care. 2013;36(9):2850–6.
Arora P, Wu C, Khan AM, Bloch DB, Davis-Dusenbery BN, Ghorbani A, Spagnolli E, Martinez A, Ryan A, Tainsh LT, Kim S, Rong J, Huan T, Freedman JE, Levy D, Miller KK, Hata A, Del Monte F, Vandenwijngaert S, Swinnen M, Janssens S, Holmes TM, Buys ES, Bloch KD, Newton-Cheh C, Wang TJ. Atrial natriuretic peptide is negatively regulated by microRNA-425. J Clin Invest. 2013;123(8):3378–82.
Ellis KL, Newton-Cheh C, Wang TJ, Frampton CM, Doughty RN, Whalley GA, Ellis CJ, Skelton L, Davis N, Yandle TG, Troughton RW, Richards AM, Cameron VA. Association of genetic variation in the natriuretic peptide system with cardiovascular outcomes. J Mol Cell Cardiol. 2011;50(4):695–701.
Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, Sim X, Tay WT, Chen CH, Zhang Y, Yamamoto K, Katsuya T, Yokota M, Kim YJ, Ong RT, Nabika T, Gu D, Chang LC, Kokubo Y, Huang W, Ohnaka K, Yamori Y, Nakashima E, Jaquish CE, Lee JY, Seielstad M, Isono M, Hixson JE, Chen YT, Miki T, Zhou X, Sugiyama T, Jeon JP, Liu JJ, Takayanagi R, Kim SS, Aung T, Sung YJ, Zhang X, Wong TY, Han BG, Kobayashi S, Ogihara T, Zhu D, Iwai N, JY W, Teo YY, Tai ES, Cho YS, He J. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet. 2011;43(6):531–8.
International Consortium for Blood Pressure Genome-Wide Association Studies, Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, Smith AV, Tobin MD, Verwoert GC, Hwang SJ, Pihur V, Vollenweider P, O'Reilly PF, Amin N, Bragg-Gresham JL, Teumer A, Glazer NL, Launer L, Zhao JH, Aulchenko Y, Heath S, Sõber S, Parsa A, Luan J, Arora P, Dehghan A, Zhang F, Lucas G, Hicks AA, Jackson AU, Peden JF, Tanaka T, Wild SH, Rudan I, Igl W, Milaneschi Y, Parker AN, Fava C, Chambers JC, Fox ER, Kumari M, Go MJ, van der Harst P, Kao WH, Sjögren M, Vinay DG, Alexander M, Tabara Y, Shaw-Hawkins S, Whincup PH, Liu Y, Shi G, Kuusisto J, Tayo B, Seielstad M, Sim X, Nguyen KD, Lehtimäki T, Matullo G, Wu Y, Gaunt TR, Onland-Moret NC, Cooper MN, Platou CG, Org E, Hardy R, Dahgam S, Palmen J, Vitart V, Braund PS, Kuznetsova T, Uiterwaal CS, Adeyemo A, Palmas W, Campbell H, Ludwig B, Tomaszewski M, Tzoulaki I, Palmer ND, CARDIoGRAM consortium, CKDGen Consortium; KidneyGen Consortium, EchoGen consortium; CHARGE-HF consortium, Aspelund T, Garcia M, Chang YP, O'Connell JR, Steinle NI, Grobbee DE, Arking DE, Kardia SL, Morrison AC, Hernandez D, Najjar S, WL MA, Hadley D, Brown MJ, Connell JM, Hingorani AD, Day IN, Lawlor DA, Beilby JP, Lawrence RW, Clarke R, Hopewell JC, Ongen H, Dreisbach AW, Li Y, Young JH, Bis JC, Kähönen M, Viikari J, Adair LS, Lee NR, Chen MH, Olden M, Pattaro C, Bolton JA, Köttgen A, Bergmann S, Mooser V, Chaturvedi N, Frayling TM, Islam M, Jafar TH, Erdmann J, Kulkarni SR, Bornstein SR, Grässler J, Groop L, Voight BF, Kettunen J, Howard P, Taylor A, Guarrera S, Ricceri F, Emilsson V, Plump A, Barroso I, Khaw KT, Weder AB, Hunt SC, Sun YV, Bergman RN, Collins FS, Bonnycastle LL, Scott LJ, Stringham HM, Peltonen L, Perola M, Vartiainen E, Brand SM, Staessen JA, Wang TJ, Burton PR, Soler Artigas M, Dong Y, Snieder H, Wang X, Zhu H, Lohman KK, Rudock ME, Heckbert SR, Smith NL, Wiggins KL, Doumatey A, Shriner D, Veldre G, Viigimaa M, Kinra S, Prabhakaran D, Tripathy V, Langefeld CD, Rosengren A, Thelle DS, Corsi AM, Singleton A, Forrester T, Hilton G, CA MK, Salako T, Iwai N, Kita Y, Ogihara T, Ohkubo T, Okamura T, Ueshima H, Umemura S, Eyheramendy S, Meitinger T, Wichmann HE, Cho YS, Kim HL, Lee JY, Scott J, Sehmi JS, Zhang W, Hedblad B, Nilsson P, Smith GD, Wong A, Narisu N, Stančáková A, Raffel LJ, Yao J, Kathiresan S, O'Donnell CJ, Schwartz SM, Ikram MA, Longstreth WT Jr, Mosley TH, Seshadri S, Shrine NR, Wain LV, Morken MA, Swift AJ, Laitinen J, Prokopenko I, Zitting P, Cooper JA, Humphries SE, Danesh J, Rasheed A, Goel A, Hamsten A, Watkins H, Bakker SJ, van Gilst WH, Janipalli CS, Mani KR, Yajnik CS, Hofman A, Mattace-Raso FU, Oostra BA, Demirkan A, Isaacs A, Rivadeneira F, Lakatta EG, Orru M, Scuteri A, Ala-Korpela M, Kangas AJ, Lyytikäinen LP, Soininen P, Tukiainen T, Würtz P, Ong RT, Dörr M, Kroemer HK, Völker U, Völzke H, Galan P, Hercberg S, Lathrop M, Zelenika D, Deloukas P, Mangino M, Spector TD, Zhai G, Meschia JF, Nalls MA, Sharma P, Terzic J, Kumar MV, Denniff M, Zukowska-Szczechowska E, Wagenknecht LE, Fowkes FG, Charchar FJ, Schwarz PE, Hayward C, Guo X, Rotimi C, Bots ML, Brand E, Samani NJ, Polasek O, Talmud PJ, Nyberg F, Kuh D, Laan M, Hveem K, Palmer LJ, van der Schouw YT, Casas JP, Mohlke KL, Vineis P, Raitakari O, Ganesh SK, Wong TY, Tai ES, Cooper RS, Laakso M, Rao DC, Harris TB, Morris RW, Dominiczak AF, Kivimaki M, Marmot MG, Miki T, Saleheen D, Chandak GR, Coresh J, Navis G, Salomaa V, Han BG, Zhu X, Kooner JS, Melander O, Ridker PM, Bandinelli S, Gyllensten UB, Wright AF, Wilson JF, Ferrucci L, Farrall M, Tuomilehto J, Pramstaller PP, Elosua R, Soranzo N, Sijbrands EJ, Altshuler D, Loos RJ, Shuldiner AR, Gieger C, Meneton P, Uitterlinden AG, Wareham NJ, Gudnason V, Rotter JI, Rettig R, Uda M, Strachan DP, Witteman JC, Hartikainen AL, Beckmann JS, Boerwinkle E, Vasan RS, Boehnke M, Larson MG, Järvelin MR, Psaty BM, Abecasis GR, Chakravarti A, Elliott P, van Duijn CM, Newton-Cheh C, Levy D, Caulfield MJ, Johnson T, International Consortium for Blood Pressure Genome-Wide Association S. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478(7367):103–9.
Rame JE, Drazner MH, Post W, Peshock R, Lima J, Cooper RS, Dries DL. Corin I555(P568) allele is associated with enhanced cardiac hypertrophic response to increased systemic afterload. Hypertension. 2007;49(4):857–64.
Wong LL, Wee AS, Lim JY, Ng JY, Chong JP, Liew OW, Lilyanna S, Martinez EC, Ackers-Johnson MA, Vardy LA, Armugam A, Jeyaseelan K, Ng TP, Lam CS, Foo RS, Richards AM, Chen YT. Natriuretic peptide receptor 3 (NPR3) is regulated by microRNA-100. J Mol Cell Cardiol. 2015;82:13–21.
Kotlo KU, Hesabi B, Danziger RS. Implication of microRNAs in atrial natriuretic peptide and nitric oxide signaling in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2011;301(4):C929–37.
Wong LL, Wang J, Liew OW, Richards AM, Chen YT. MicroRNA and Heart Failure. Int J Mol Sci. 2016;17(4):502.
Hohl M, Wagner M, Reil JC, Müller SA, Tauchnitz M, Zimmer AM, Lehmann LH, Thiel G, Böhm M, Backs J, Maack C. HDAC 4 controls histone methylation in response to elevated cardiac load. J Clin Invest. 2013;123(3):1359–70.
van Veldhuisen DJ, Linssen GC, Jaarsma T, van Gilst WH, Hoes AW, Tijssen JG, Paulus WJ, Voors AA, Hillege HL. B-type natriuretic peptide and prognosis in heart failure patients with preserved and reduced ejection fraction. J Am Coll Cardiol. 2013;61(14):1498–506.
Yasue H, Yoshimura M, Sumida H, Kikuta K, Kugiyama K, Jougasaki M, Ogawa H, Okumura K, Mukoyama M, Nakao K. Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation. 1994;90(1):195–203.
Yoshimura M, Yasue H, Okumura K, Ogawa H, Jougasaki M, Mukoyama M, Nakao K, Imura H. Different secretion patterns of atrial natriuretic peptide and brain natriuretic peptide in patients with congestive heart failure. Circulation. 1993;87(2):464–9.
Semenov AG, Katrukha AG. Analytical issues with natriuretic peptides-has this been overly simplified? EJIFCC. 2016;27(3):189–207.
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):1810–52.
Troughton RW, Frampton CM, Brunner-La Rocca HP, Pfisterer M, Eurlings LW, Erntell H, Persson H, O'Connor CM, Moertl D, Karlström P, Dahlström U, Gaggin HK, Januzzi JL, Berger R, Richards AM, Pinto YM, Nicholls MG. Effect of B-type natriuretic peptide-guided treatment of chronic heart failure on total mortality and hospitalization: An individual patient meta-analysis. Eur Heart J. 2014;35(23):1559–67.
Wang TJ, Larson MG, Levy D, Benjamin EJ, Leip EP, Omland T, Wolf PA, Vasan RS. Plasma natriuretic peptide levels and the risk of cardiovascular events and death. N Engl J Med. 2004;350(7):655–63.
Jessup M, Abraham WT, Casey DE, Feldman AM, Francis GS, Ganiats TG, Konstam MA, Mancini DM, Rahko PS, Silver MA, Stevenson LW, Yancy CW. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119(14):1977–2016.
Dickstein K, Cohen-Solal A, Filippatos G, JJ MM, Ponikowski P, Poole-Wilson PA, Strömberg A, van Veldhuisen DJ, Atar D, Hoes AW, Keren A, Mebazaa A, Nieminen M, Priori SG, Swedberg K, ESC Committee for Practice Guidelines (CPG). ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur Heart J. 2008;29(19):2388–442.
Heart Failure Society of America, Lindenfeld J, Albert NM, Boehmer JP, Collins SP, Ezekowitz JA, Givertz MM, Katz SD, Klapholz M, Moser DK, Rogers JG, Starling RC, Stevenson WG, Tang WH, Teerlink JR, Walsh MN. HFSA 2010 comprehensive heart failure practice guideline. J Card Fail. 2010;16(6):e1–194.
Januzzi JL Jr, Camargo CA, Anwaruddin S, Baggish AL, Chen AA, Krauser DG, Tung R, Cameron R, Nagurney JT, Chae CU, Lloyd-Jones DM, Brown DF, Foran-Melanson S, Sluss PM, Lee-Lewandrowski E, Lewandrowski KB. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol. 2005;95(8):948–54.
JJ MM, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez-Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A, ESC Committee for Practice Guidelines. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33(14):1787–847.
Stokes NR, Dietz BW, Liang JJ. Cardiopulmonary laboratory biomarkers in the evaluation of acute dyspnea. Open Access Emerg Med. 2016;8:35–45.
Fox ER, Musani SK, Bidulescu A, Nagarajarao HS, Samdarshi TE, Gebreab SY, Sung JH, Steffes MW, Wang TJ, Taylor HA, Vasan RS. Relation of obesity to circulating B-type natriuretic peptide concentrations in blacks: the Jackson Heart Study. Circulation. 2011;124(9):1021–7.
Richards M, Di Somma S, Mueller C, Nowak R, Peacock WF, Ponikowski P, Mockel M, Hogan C, AH W, Clopton P, Filippatos GS, Anand I, Ng L, Daniels LB, Neath SX, Shah K, Christenson R, Hartmann O, Anker SD, Maisel A. Atrial fibrillation impairs the diagnostic performance of cardiac natriuretic peptides in dyspneic patients: Results from the BACH Study (Biomarkers in ACute Heart Failure). JACC Heart Fail. 2013;1(3):192–9.
Gruden G, Landi A, Bruno G. Natriuretic peptides, heart, and adipose tissue: new findings and future developments for diabetes research. Diabetes Care. 2014;37(11):2899–908.
Lazo M, Young JH, Brancati FL, Coresh J, Whelton S, Ndumele CE, Hoogeveen R, Ballantyne CM, Selvin E. NH2-terminal pro-brain natriuretic peptide and risk of diabetes. Diabetes. 2013;62(9):3189–93.
Buckley MG, Markandu ND, Miller MA, Sagnella GA, MacGregor GA. Plasma concentrations and comparisons of brain and atrial natriuretic peptide in normal subjects and in patients with essential hypertension. J Hum Hypertens. 1993;7(3):245–50.
Yamamoto K, Burnett JC Jr, Jougasaki M, Nishimura RA, Bailey KR, Saito Y, Nakao K, Redfield MM. Superiority of brain natriuretic peptide as a hormonal marker of ventricular systolic and diastolic dysfunction and ventricular hypertrophy. Hypertension. 1996;28(6):988–94.
Nishikimi T, Yoshihara F, Morimoto A, Ishikawa K, Ishimitsu T, Saito Y, Kangawa K, Matsuo H, Omae T, Matsuoka H. Relationship between left ventricular geometry and natriuretic peptide levels in essential hypertension. Hypertension. 1996;28(1):22–30.
Kohno M, Horio T, Yokokawa K, Yasunari K, Ikeda M, Minami M, Kurihara N, Takeda T. Brain natriuretic peptide as a marker for hypertensive left ventricular hypertrophy: changes during 1-year antihypertensive therapy with angiotensin-converting enzyme inhibitor. Am J Med. 1995;98(3):257–65.
Vasan RS, Benjamin EJ, Larson MG, Leip EP, Wang TJ, Wilson PW, Levy D. Plasma natriuretic peptides for community screening for left ventricular hypertrophy and systolic dysfunction: the Framingham heart study. JAMA. 2002;288(10):1252–9.
Clerico A, Recchia FA, Passino C, Emdin M. Cardiac endocrine function is an essential component of the homeostatic regulation network: physiological and clinical implications. Am J Physiol Heart Circ Physiol. 2006;290(1):H17–29.
Morita E, Yasue H, Yoshimura M, Ogawa H, Jougasaki M, Matsumura T, Mukoyama M, Nakao K. Increased plasma levels of brain natriuretic peptide in patients with acute myocardial infarction. Circulation. 1993;88(1):82–91.
Nishikimi T, Maeda N, Matsuoka H. The role of natriuretic peptides in cardioprotection. Cardiovasc Res. 2006;69(2):318–28.
Nagaya N, Goto Y, Nishikimi T, Uematsu M, Miyao Y, Kobayashi Y, Miyazaki S, Hamada S, Kuribayashi S, Takamiya M, Matsuo H, Kangawa K, Nonogi H. Sustained elevation of plasma brain natriuretic peptide levels associated with progressive ventricular remodelling after acute myocardial infarction. Clin Sci (Lond). 1999;96(2):129–36.
Tagore R, Ling LH, Yang H, Daw HY, Chan YH, Sethi SK. Natriuretic peptides in chronic kidney disease. Clin J Am Soc Nephrol. 2008;3(6):1644–51.
Anwaruddin S, Lloyd-Jones DM, Baggish A, Chen A, Krauser D, Tung R, Chae C, Januzzi JL Jr. Renal function, congestive heart failure, andamino-terminal pro-brain natriuretic peptide measurement:results from the pro-BNP Investigation of Dyspnea in the Emergency Department (PRIDE) study. J Am Coll Cardiol. 2006;47(1):91–7.
Santos-Araújo C, Leite-Moreira A, Pestana M. Clinical value of natriuretic peptides in chronic kidney disease. Nefrologia. 2015;35(3):227–33.
Satyan S, Light RP, Agarwal R. Relationships of N-terminalpro-B-natriuretic peptides and cardiac troponin T to left ventricular mass and function and mortality in asymptomatic hemodialysis patients. Am J Kidney Dis. 2007;50(6):1009–19.
Obineche EN, Pathan JY, Fisher S, Prickett TC, Yandle TG, Frampton CM, Cameron VA, Nicholls MG. Natriuretic peptide and adrenomedullin levels in chronic renal failure and effects of peritoneal dialysis. Kidney Int. 2006;69(1):152–6.
Madsen LH, Ladefoged S, Corell P, Schou M, Hildebrandt PR, Atar D. N-terminal pro brain natriuretic peptide predicts mortality in patients with end-stage renal disease in hemodialysis. Kidney Int. 2007;71(6):548–54.
Sanjuan R, Oliva SM, Blasco ML, Puchades M, Torregrosa I, Ramón RG, Carrasco AM. Plasma brain natriuretic peptide levels in cardiac function assessment in chronic dialysis patients. Cardiorenal Med. 2011;1(3):147–55.
Taskapan MC, Ulutas O, Aksoy Y, Senel S, Sahin I, Kosar F, Taskapan H. Brain natriuretic peptide and its relationship to left ventricular hypertrophy in patients on peritoneal dialysis or hemodialysis less than 3 years. Ren Fail. 2006;28(2):133–9.
Bavbek N, Akay H, Altay M, Uz E, Turgut F, Uyar ME, Karanfil A, Selcoki Y, Akcay A, Duranay M, Serum BNP. concentration and left ventricular mass in CAPD and automated peritoneal dialysis patients. Perit Dial Int. 2007;27(6):663–8.
Chan W, Bosch JA, Jones D, McTernan PG, Inston N, Moore S, Kaur O, Phillips AC, Borrows R. Hypervolemia and blood pressure in prevalent kidney transplant recipients. Transplantation. 2014;98(3):320–7.
Memon L, Spasojevic-Kalimanovska V, Stanojevic NB, Kotur-Stevuljevic J, Simic-Ogrizovic S, Giga V, Dopsaj V, Jelic-Ivanovic Z, Spasic S. Are levels of NT-proBNP and SDMA useful to determine diastolic dysfunction in chronic kidney disease and renal transplant patients? J Clin Lab Anal. 2013;27(6):461–70.
Gheissari A, Sabri M, Pirpiran M, Merrikhi A. Possible correlation among echocardiographic measures, serum brain natriuretic peptide, and angiotensin II levels in hypertensive kidney transplanted children. Exp Clin Transplant. 2013;11(2):128–33.
Codognotto M, Piccoli A, Zaninotto M, Mion M, Plebani M, Vertolli U, Tona F, Ruzza L, Barchita A, Boffa GM. Renal dysfunction is a confounder for plasma natriuretic peptides in detecting heart dysfunction in uremic and idiopathic dilated cardiomyopathies. Clin Chem. 2007;53(12):2097–104.
McCullough PA, Sandberg KR. B-type natriuretic peptide and renal disease. Heart Fail Rev. 2003;8(4):355–8.
Santos-Araújo C, Roncon-Albuquerque R Jr, Moreira-Rodrigues M, Henriques-Coelho T, Quelhas-Santos J, Faria B, Sampaio-Maia B, Leite-Moreira AF, Pestana M. Local modulation of the natriuretic peptide system in the rat remnant kidney. Nephrol Dial Transplant. 2009;24(6):1774–82.
Sackner-Bernstein JD, Skopicki HA, Aaronson KD. Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation. 2005;111(12):1487–91.
Cheng V, Kazanagra R, Garcia A, Lenert L, Krishnaswamy P, Gardetto N, Clopton P, Maisel AA. Rapid bedside test for B-type peptide predicts treatment outcomes in patients admitted for decompensated heart failure: a pilot study. J Am Coll Cardiol. 2001;37(2):386–91.
Vogeser M, Jacob K. B-type natriuretic peptide (BNP)-validation of an immediate response assay. Clin Lab. 2001;47(1-2):29–33.
Ro R, Thode HC Jr, Taylor M, Gulla J, Tetrault E, Singer AJ. Comparison of the diagnostic characteristics of two B-type natriuretic peptide point-of-care devices. J Emerg Med. 2011;41(6):661–7.
Shah K, Terracciano GJ, Jiang K, Maisel AS, Fitzgerald RL. Comparability of Results between Point-of-Care and Automated Instruments to Measure B-type Natriuretic Peptide. West J Emerg Med. 2010;11(1):44–8.
Heart check BNP test strip package insert 0017 spec-0363 rev. 1, 2010/09, Alere Technologies Ltd., Stirling, Scotland.
Triage® BNP test product insert, 2011, Alere Technologies Ltd., Stirling, Scotland.
Lang NN, Wong CM, Dalzell JR, Jansz S, Leslie SJ, Gardner RS. The ease of use and reproducibility of the Alere Heart Check System: a comparison of patient and healthcare professional measurement of BNP. Biomark Med. 2014;8(6):791–6.
Maisel A, Barnard D, Jaski B, Frivold G, Marais J, Azer M, Miyamoto MI, Lombardo D, Kelsay D, Borden K, Iqbal N, Taub PR, Kupfer K, Clopton P, Greenberg B. Primary results of the HABIT Trial (heart failure assessment with BNP in the home). J Am Coll Cardiol. 2013;61(16):1726–35.
Hunter I, Goetze JP. Next generation natriuretic peptide measurement. Adv Clin Chem. 2012;58:45–8.
Sun Z, Chen J, Yao H, Liu L, Wang J, Zhang J, Liu JN. Use of Ssp dnaB derived mini-intein as a fusion partner for production of recombinant human brain natriuretic peptide in Escherichia coli. Protein Expr Purif. 2005;43(1):26–32.
Pan S, Chen HH, Dickey DM, Boerrigter G, Lee C, Kleppe LS, Hall JL, Lerman A, Redfield MM, Potter LR, Burnett JC Jr, Simari RD. Biodesign of a renal-protective peptide based on alternative splicing of B-type natriuretic peptide. Proc Natl Acad Sci USA. 2009;106(27):11282–7.
Dickey DM, Barbieri KA, McGuirk CM, Potter LR. Arg13 of B-type natriuretic Peptide reciprocally modulates binding to guanylyl cyclase but not clearance receptors. Mol Pharmacol. 2010;78(3):431–5.
Yu L, Rao C, Shi X, Li Y, Gao K, Li X, Wang JA. novel bioassay for the activity determination of therapeutic human brain natriuretic peptide (BNP). PLoS One. 2012;7(11):e49934.
Han B, Hasin Y. Cardiovascular effects of natriuretic peptides and their interrelation with endothelin-1. Cardiovasc Drugs Ther. 2003;17(1):41–52.
Charloux A, Piquard F, Doutreleau S, Brandenberger G, Geny B. Mechanisms of renal hyporesponsiveness to ANP in heart failure. Eur J Clin Invest. 2003;33(9):769–78.
Lohmeier TE, Mizelle HL, Reinhart GA, Montani JP, Hord CE Jr, Didlake RH. Atrial natriuretic peptide and sodium homeostasis in compensated heart failure. Am J Physiol. 1996;271(5 Pt 2):R1353–63.
Costello-Boerrigter LC, Boerrigter G, Burnett JC Jr. Revisiting salt and water retention: new diuretics, aquaretics, and natriuretics. Med Clin North Am. 2003;87(2):475–91.
Stevens TL, Burnett JC Jr, Kinoshita M, Matsuda Y, Redfield MMA. functional role for endogenous atrial natriuretic peptide in a canine model of early left ventricular dysfunction. J Clin Invest. 1995;95(3):1101–8.
Wada A, Tsutamato T, Maeda Y, Kanamori T, Matsuda Y, Kinoshita M. Endogenous atrial natriuretic peptide inhibits endothelin-1 secretion in dogs with severe congestive heart failure. Am J Physiol. 1996;270(5 Pt 2):H1819–24.
Chen HH. Heart failure: a state of brain natriuretic peptide deficiency or resistance or both. J Am Coll Cardiol. 2007;49(10):1089–91.
Fielitz J, Dendorfer A, Pregla R, Ehler E, Zurbrügg HR, Bartunek J, Hetzer R, Regitz-Zagrosek V. Neutral endopeptidase is activated in cardiomyocytes in human aortic valve stenosis and heart failure. Circulation. 2002;105(3):286–9.
Song W, Wang H, Atrial WQ. natriuretic peptide in cardiovascular biology and disease (NPPA). Gene. 2015;569(1):1–6.
Filippatos G, Farmakis D, Parissis J, Lekakis J. Drug therapy for patients with systolic heart failure after the PARADIGM-HF trial: In need of a new paradigm of LCZ696 implementation in clinical practice. BMC Med. 2015;13:35.
Bayés-Genís A, Barallat J, Galán A, de Antonio M, Domingo M, Zamora E, Urrutia A, Lupón J. Soluble neprilysin is predictive of cardiovascular death and heart failure hospitalization in heart failure patients. J Am Coll Cardiol. 2015;65(7):657–65.
Colucci WS, Elkayam U, Horton DP, Abraham WT, Bourge RC, Johnson AD, Wagoner LE, Givertz MM, Liang CS, Neibaur M, Haught WH, LeJemtel TH. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N Engl J Med. 2000;343(4):246–53.
Partovian C, Li SX, Xu X, Lin H, Strait KM, Hwa J, Krumholz HM. Patterns of change in nesiritide use in patients with heart failure: how hospitals react to new information. JACC Heart Fail. 2013;1(4):318–24.
Abraham WT, Adams KF, Fonarow GC, Costanzo MR, Berkowitz RL, LeJemtel TH, Cheng ML, Wynne J, Scientific Advisory ADHERE. Committee and Investigators; ADHERE Study Group. In-hospital mortality in patients with acute decompensated heart failure requiring intravenous vasoactive medications: an analysis from the Acute Decompensated Heart Failure National Registry (ADHERE). J Am Coll Cardiol. 2005;46(1):57–64.
Witteles RM, Kao D, Christopherson D, Matsuda K, Vagelos RH, Schreiber D, Fowler MB. Impact of nesiritide on renal function in patients with acute decompensated heart failure and pre-existing renal dysfunction: a randomized, doubleblind, placebo-controlled clinical trial. J Am Coll Cardiol. 2007;50(19):1835–40.
O'Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, Komajda M, Massie BM, McMurray JJ, Nieminen MS, Reist CJ, Rouleau JL, Swedberg K, Adams KF Jr, Anker SD, Atar D, Battler A, Botero R, Bohidar NR, Butler J, Clausell N, Corbalán R, Costanzo MR, Dahlstrom U, Deckelbaum LI, Diaz R, Dunlap ME, Ezekowitz JA, Feldman D, Felker GM, Fonarow GC, Gennevois D, Gottlieb SS, Hill JA, Hollander JE, Howlett JG, Hudson MP, Kociol RD, Krum H, Laucevicius A, Levy WC, Méndez GF, Metra M, Mittal S, BH O, Pereira NL, Ponikowski P, Tang WH, Tanomsup S, Teerlink JR, Triposkiadis F, Troughton RW, Voors AA, Whellan DJ, Zannad F, Califf RM. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365(1):32–43.
Gottlieb SS, Stebbins A, Voors AA, Hasselblad V, Ezekowitz JA, Califf RM, O'Connor CM, Starling RC, Hernandez AF. Effects of nesiritide and predictors of urine output in acute decompensated heart failure: results from ASCEND-HF (acute study of clinical effectiveness of nesiritide and decompensated heart failure). J Am Coll Cardiol. 2013;62(13):1177–83.
Kato T, Muraski J, Chen Y, Tsujita Y, Wall J, Glembotski CC, Schaefer E, Beckerle M, Sussman MA. Atrial natriuretic peptide promotes cardiomyocyte survival by cGMP-dependent nuclear accumulation of zyxin and Akt. J Clin Invest. 2005;115(10):2716–30.
Chen HH, Glockner JF, Schirger JA, Cataliotti A, Redfield MM, Burnett JC Jr. Novel protein therapeutics for systolic heart failure: chronic subcutaneous B-type natriuretic peptide. J Am Coll Cardiol. 2012;60(22):2305–12.
Lyu T, Zhao Y, Zhang T, Zhou W, Yang F, Ge H, Ding S, Pu J, He B. Natriuretic peptides as an adjunctive treatment for acute myocardial infarction. Int Heart J. 2014;55(1):8–16.
Mitrovic V, Lüss H, Nitsche K, Forssmann K, Maronde E, Fricke K, Forssmann WG, Meyer M. Effects of the renal natriuretic peptide urodilatin (ularitide) in patients with decompensated chronic heart failure: a double-blind, placebo-controlled, ascending-dose trial. Am Heart J. 2005;150(6):1239.
Mitrovic V, Seferovic PM, Simeunovic D, Ristic AD, Miric M, Moiseyev VS, Kobalava Z, Nitsche K, Forssmann WG, Lüss H, Meyer M. Haemodynamic and clinical effects of ularitide in decompensated heart failure. Eur Heart J. 2006;27(23):2823–32.
Packer M, Holcomb R, Abraham WT, Anker S, Dickstein K, Filippatos G, Krum H, Maggioni AP, McMurray JJ, Mebazaa A, O'Connor C, Peacock F, Ponikowski P, Ruschitzka F, van Veldhuisen DJ, Holzmeister J. TRUE-AHF Investigators and Committees. Rationale for and design of the TRUE-AHF trial: the effects of ularitide on the short-term clinical course and long-term mortality of patients with acute heart failure. Eur J Heart Fail. 2017;19(5):673–81.
Lisy O, Huntley BK, McCormick DJ, Kurlansky PA, Burnett JC Jr. Design, synthesis, and actions of a novel chimeric natriuretic peptide: CDNP. J Am Coll Cardiol. 2008;52(1):60–8.
Dickey DM, Potter LR. Dendroaspis natriuretic peptide and the designer natriuretic peptide, CD-NP, are resistant to proteolytic inactivation. J Mol Cell Cardiol. 2011;51(1):67–71.
Neutel J, Rolston W, Maddock S, Goldsmith S, Koren M, Antwerp BV, Burnett J, Lieu HD. Initial experience with subcutaneous infusion of cenderitide in patients with chronic heart failure. J Am Coll Cardiol. 2012;59:E1037.
Martin FL, Sangaralingham SJ, Huntley BK, McKie PM, Ichiki T, Chen HH, Korinek J, Harders GE, Burnett JC Jr. CD-NP: a novel engineered dual guanylylcyclase activator with anti-fibrotic actions in the heart. PLoS One. 2012;7(12):e52422.
Lee CY, Boerrigter G, Chen HH, Sandberg S, Heublein D, Harty G, Burnett JC Jr. Cardiorenal and Neurohumoral Actions of a Novel Designer Natriuretic Peptide, CU-NP. In Canine Experimental Heart Failure. Circulation. 2008;118:S293.
McKie PM, Ichiki T, Burnett JC Jr. M-atrial natriuretic peptide: a novel antihypertensive protein therapy. Curr Hypertens Rep. 2012;14(1):62–9.
McKie PM, Cataliotti A, Ichiki T, Sangaralingham SJ, Chen HH, Burnett JC Jr. M-atrial natriuretic peptide and nytroglycerin in a canine model of experimental acute hypertensive heart failure: a differential actions of 2 cGMP activating therapeutics. J Am Heart Assoc. 2014;3(1):e000206.
Rolston W, Maddok S, Goldsmith S, Koren M, Bill VA, Burnett J, Lieu HD. Initial experience with subcutaneous infusion of cenderitide in chronic heart failure patients. J Am Coll Cardiol. 2012;59:E1037.
von Lueder TG, Sangaralingham SJ, Wang BH, Kompa AR, Atar D, Burnett JC Jr, Krum H. Renin-angiotensin blockade combined with natriuretic peptide system augmentation: noveltherapeutic concepts to combat heart failure. Circ Heart Fail. 2013;6(3):594–605.
Wendt DJ, Dvorak-Ewell M, Bullens S, Lorget F, Bell SM, Peng J, Castillo S, Aoyagi-Scharber M, O'Neill CA, Krejci P, Wilcox WR, Rimoin DL, Bunting S. Neutral endopeptidase-resistant C-type natriuretic peptide variant represents a new therapeutic approach for treatment of fibroblast growth factor receptor 3-related dwarfism. J Pharmacol Exp Ther. 2015;353:132–49.
Galli A, Lombardi F. Neprilysin inhibition for heart failure. N Engl J Med. 2014;371(24):2335.
Cleland JG, Swedberg K. Lack of efficacy of neutral endopeptidase inhibitor ecadotril in heart failure. The International Ecadotril Multicentre Dose-Ranging Study Investigators. Lancet. 1998;351(9116):1657–8.
Maki T, Nasa Y, Tanonaka K, Takahashi M, Takeo S. Beneficial effects of sampatrilat, a novel vasopeptidase inhibitor, on cardiac remodeling and function of rats with chronic heart failure following left coronary artery ligation. J Pharmacol Exp Ther. 2003;305(1):97–105.
Weber MA. Vasopeptidase inhibitors. Lancet. 2001;358(9292):1525–32.
von Lueder TG, Wang BH, Kompa AR, Huang L, Webb R, Jordaan P, Atar D, Krum H. Angiotensin receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail. 2015;8(1):71–8.
Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E. Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs Enalapril (OCTAVE) trial. Am J Hypertens. 2004;17(2):103–11.
Packer M, Califf RM, Konstam MA, Krum H, McMurray JJ, Rouleau JL, Swedberg K. Comparison of omapatrilat and enalapril in patients with chronic heart failure: the Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE). Circulation. 2002;106(8):920–6.
Rouleau JL, Pfeffer MA, Stewart DJ, Isaac D, Sestier F, Kerut EK, Porter CB, Proulx G, Qian C, Block AJ. Comparison of vasopeptidase inhibitor, omapatrilat, and lisinopril on exercise tolerance and morbidity in patients with heart failure: IMPRESS randomised trial. Lancet. 2000;356(9230):615–20.
Armstrong PW, Lorell BH, Nissen S, Borer J. Omapatrilat. Circulation. 2002;106(6):e9011–2.
von Lueder TG, Kjekshus H, Edvardsen T, ØIe E, Urheim S, Vinge LE, Ahmed MS, Smiseth OA, Attramadal H. Mechanisms of elevated plasma endothelin-1 in CHF: congestion increases pulmonary synthesis and secretion of endothelin-1. Cardiovasc Res. 2004;63(1):41–50.
Wada A, Ohnishi M, Tsutamoto T, Fujii M, Matsumoto T, Yamamoto T, Wang X, Kinoshita M. Chronic effects of an endothelin-converting enzyme inhibitor on cardiorenal and hormonal function in heart failure. Clin Sci (Lond). 2002;103(Suppl 48):254S–7S.
Nakayama K, Emoto N, Suzuki Y, Vignon-Zellweger N, Yagi K, Hirata K. Physiological relevance of hydrolysis of atrial natriuretic peptide by endothelin-converting enzyme-1. Kobe J Med Sci. 2012;58(1):E12–8.
Mulder P, Barbier S, Monteil C, Jeng AY, Henry JP, Renet S, Thuillez C. Sustained improvement of cardiac function and prevention of cardiac remodeling after long-term dual ECE-NEP inhibition in rats with congestive heart failure. J Cardiovasc Pharmacol. 2004;43(4):489–94.
Emoto N, Raharjo SB, Isaka D, Masuda S, Adiarto S, Jeng AY, Yokoyama M, Dual ECE. NEP inhibition on cardiac and neurohumoral function during the transition from hypertrophy to heart failure in rats. Hypertension. 2005;45(6):1145–52.
Seed A, Kuc RE, Maguire JJ, Hillier C, Johnston F, Essers H, de Voogd HJ, McMurray J, Davenport AP. The dual endothelin converting enzyme/neutral endopeptidase inhibitor SLV-306 (daglutril), inhibits systemic conversion of big endothelin-1 in humans. Life Sci. 2012;91(13-14):743–8.
Kalk P, Sharkovska Y, Kashina E, von Websky K, Relle K, Pfab T, Alter M, Guillaume P, Provost D, Hoffmann K, Fischer Y, Hocher B. Endothelinconverting enzyme/neutral endopeptidase inhibitor SLV338 prevents hypertensive cardiac remodeling in a blood pressure-independent manner. Hypertension. 2011;57(4):755–63.
Mellin V, Jeng AY, Monteil C, Renet S, Henry JP, Thuillez C, Mulder P. Triple ACE-ECE-NEP inhibition in heart failure: a comparison with ACE and dual ECE-NEP inhibition. J Cardiovasc Pharmacol. 2005;46(3):390–7.
Braunwald E. The path to an angiotensin receptor antagonist-neprilysin inhibitor in the treatment of heart failure. J Am Coll Cardiol. 2015;65(10):1029–41.
Ruilope LM, Dukat A, Böhm M, Lacourcière Y, Gong J, Lefkowitz MP. Blood-pressure reduction with LCZ696, a novel dual-acting inhibitor of the angiotensin II receptor and neprilysin: a randomised, double-blind, placebo-controlled, active comparator study. Lancet. 2010;375(9722):1255–66.
McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR. PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371(11):993–1004.
Vardeny O, Miller R, Solomon SD. Combined neprilysin and renin-angiotensin system inhibition for the treatment of heart failure. JACC Heart Fail. 2014;2(6):663–70.
Desai AS, McMurray JJ, Packer M, Swedberg K, Rouleau JL, Chen F, Gong J, Rizkala AR, Brahimi A, Claggett B, Finn PV, Hartley LH, Liu J, Lefkowitz M, Shi V, Zile MR, Solomon SD. Effect of the angiotensin-receptor-neprilysin inhibitor LCZ696 compared with enalapril on mode of death in heart failure patients. Eur Heart J. 2015;36(30):1990–7.
Packer M, McMurray JJ, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile M, Andersen K, Arango JL, Arnold JM, Bělohlávek J, Böhm M, Boytsov S, Burgess LJ, Cabrera W, Calvo C, Chen CH, Dukat A, Duarte YC, Erglis A, Fu M, Gomez E, Gonzàlez-Medina A, Hagège AA, Huang J, Katova T, Kiatchoosakun S, Kim KS, Kozan Ö, Llamas EB, Martinez F, Merkely B, Mendoza I, Mosterd A, Negrusz-Kawecka M, Peuhkurinen K, Ramires FJ, Refsgaard J, Rosenthal A, Senni M, Sibulo AS Jr, Silva-Cardoso J, Squire IB, Starling RC, Teerlink JR, Vanhaecke J, Vinereanu D, Wong RC. PARADIGM-HF Investigators and Coordinators. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation. 2015;131(1):54–61.
Minguet J, Sutton G, Ferrero C, Gomez T, Bramlage P. LCZ696: A new paradigm for the treatment of heart failure? Expert Opin Pharmacother. 2015;16(3):435–46.
Henderson SJ, Andersson C, Narwal R, Janson J, Goldschmidt TJ, Appelkvist P, Bogstedt A, Steffen AC, Haupts U, Tebbe J, Freskgård PO, Jermutus L, Burrell M, Fowler SB, Webster CI. Sustained peripheral depletion of amyloid-β with a novel form of neprilysin does not affect central levels of amyloid-β. Brain. 2014;137(Pt 2):553–64.
Campbell DJ, Krum H, Esler MD. Losartan increases bradykinin levels in hypertensive humans. Circulation. 2005;111(3):315–20.
Grimm MO, Mett J, Stahlmann CP, Haupenthal VJ, Zimmer VC, Hartmann T. Neprilysin and Aβ Clearance: Impact of the APP Intracellular Domain in NEP Regulation and Implications in Alzheimer’s Disease. Front Aging Neurosci. 2013;5:98.
Solomon SD, Zile M, Pieske B, Voors A, Shah A, Kraigher-Krainer E, Shi V, Bransford T, Takeuchi M, Gong J, Lefkowitz M, Packer M, McMurray JJ. Prospective comparison of ARNI with ARB on Management Of heart failUre with preserved ejectioN fracTion (PARAMOUNT) Investigators. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet. 2012;380(9851):1387–95.
Bavishi C, Messerli FH, Kadosh B, Ruilope LM, Kario K. Role of neprilysin inhibitor combinations in hypertension: insights from hypertension and heart failure trials. Eur Heart J. 2015;36(30):1967–73.
Williams B, Cockcroft JR, Kario K, Zappe DH, Cardenas P, Hester A, Brunel P, Zhang J. Rationale and study design of the Prospective comparison of Angiotensin Receptor neprilysin inhibitor with Angiotensin receptor blocker MEasuring arterial sTiffness in the eldERly (PARAMETER) study. BMJ Open. 2014;4(2):e004254.
Bodey F, Hopper I, Krum H. Neprilysin inhibitors preserve renal function in heart failure: A meta-analysis of randomised controlled trials. Int J Cardiol. 2015;179:329–30.
Guéant Rodriguez RM, Spada R, Pooya S, Jeannesson E, Moreno Garcia MA, Anello G, Bosco P, Elia M, Romano A, Alberto JM, Juillière Y, Guéant JL. Homocysteine predicts increased NT-pro-BNP through impaired fatty acid oxidation. Int J Cardiol. 2013;167(3):768–75.
Laramée P, Wonderling D, Swain S, Al-Mohammad A, Mant J. Cost-effectiveness analysis of serial measurement of circulating natriuretic peptide concentration in chronic heart failure. Heart. 2013;99(4):267–71.
Mohiuddin S, Reeves B, Pufulete M, Maishman R, Dayer M, Macleod J, McDonagh T, Purdy S, Rogers C, Hollingworth W. Model-based cost-effectiveness analysis of B-type natriuretic peptide-guided care in patients with heart failure. BMJ Open. 2016;6(12):e014010.
Felker GM, Ahmad T, Anstrom KJ, Adams KF, Cooper LS, Ezekowitz JA, Fiuzat M, Houston-Miller N, Januzzi JL, Leifer ES, Mark DB, Desvigne-Nickens P, Paynter G, Piña IL, Whellan DJ, O'Connor CM. Rationale and design of the GUIDE-IT study: guiding evidence based therapy using biomarker intensified treatment in heart failure. JACC Heart Fail. 2014;2(5):457–65.
Daniels LB, Maisel AS. Natriuretic peptides. J Am Coll Cardiol. 2007;50(25):2357–68.
Kistorp C, Raymond I, Pedersen F, Gustafsson F, Faber J, Hildebrandt P. N-terminal pro-brain natriuretic peptide, C-reactive protein, and urinary albumin levels as predictors of mortality and cardiovascular events in older adults. JAMA. 2005;293(13):1609–16.
de Lemos JA, Morrow DA, Bentley JH, Omland T, Sabatine MS, McCabe CH, Hall C, Cannon CP, Braunwald E. The prognostic value of B-type natriuretic peptide in patients with acute coronary syndromes. N Engl J Med. 2001;345(14):1014–21.
Sabatine MS, Morrow DA, de Lemos JA, Omland T, Sloan S, Jarolim P, Solomon SD, Pfeffer MA, Braunwald E. Evaluation of multiple biomarkers of cardiovascular stress for risk prediction and guiding medical therapy in patients with stable coronary disease. Circulation. 2012;125(2):233–40.
Hunt PJ, Richards AM, Nicholls MG, Yandle TG, Doughty RN, Espiner EA. Immunoreactive amino-terminal pro-brain natriureticpeptide (NT-PROBNP): a new marker of cardiac impairment. Clin Endocrinol (Oxf). 1997;47(3):287–96.
Wang AY, Lam CW, CM Y, Wang M, Chan IH, Zhang Y, Lui SF, Sanderson JE. N-terminal pro-brain natriuretic peptide: an independent risk predictor of cardiovascular congestion, mortality, and adverse cardiovascular outcomes in chronic peritoneal dialysis patients. J Am Soc Nephrol. 2007;18(1):321–30.
Khalifeh N, Haider D, Hörl WH. Natriuretic peptides in chronic kidney disease and during renal replacement therapy: an update. J Investig Med. 2009;57(1):33–9.
Paniagua R, Amato D, Mujais S, Vonesh E, Ramos A, Correa-Rotter R, Horl WH. Predictive value of brain natriuretic peptides in patients on peritoneal dialysis: results from the ADEMEX trial. Clin J Am Soc Nephrol. 2008;3(2):407–15.
Funding
This work is supported by the grants from Health Special Scientific Research Project of Chinese People’s Liberation Army (12BJZ34 and 14BJZ12), and Sanya Medical and Health Science and Technology Innovation Project (2016YW21). The funding bodies play no role in manuscript preparation and submission.
Availability of data and materials
Available.
Author information
Authors and Affiliations
Contributions
SF, PP, FW, LL: reviewed the literature and prepared the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fu, S., Ping, P., Wang, F. et al. Synthesis, secretion, function, metabolism and application of natriuretic peptides in heart failure. J Biol Eng 12, 2 (2018). https://doi.org/10.1186/s13036-017-0093-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13036-017-0093-0