
Abstract
Background
Adult T-cell Lymphoma/Leukemia (ATLL) is characterized by the malignant proliferation of T-cells in Human T-Lymphotropic Virus Type 1 and a high mortality rate. Considering the emerging roles of microRNAs (miRNAs) in various malignancies, the analysis of high-throughput miRNA data employing computational algorithms helps to identify potential biomarkers.
Methods
Weighted gene co-expression network analysis was utilized to analyze miRNA microarray data from ATLL and healthy uninfected samples. To identify miRNAs involved in the progression of ATLL, module preservation analysis was used. Subsequently, based on the target genes of the identified miRNAs, the STRING database was employed to construct protein–protein interaction networks (PPIN). Real-time quantitative PCR was also performed to validate the expression of identified hub genes in the PPIN network.
Results
After constructing co-expression modules and then performing module preservation analysis, four out of 15 modules were determined as ATLL-specific modules. Next, the hub miRNA including hsa-miR-18a-3p, has-miR-187-5p, hsa-miR-196a-3p, and hsa-miR-346 were found as hub miRNAs. The protein–protein interaction networks were constructed for the target genes of each hub miRNA and hub genes were identified. Among them, UBB, RPS15A, and KMT2D were validated by Reverse-transcriptase PCR in ATLL patients.
Conclusion
The results of the network analysis of miRNAs and their target genes revealed the major players in the pathogenesis of ATLL. Further studies are required to confirm the role of these molecular factors and to discover their potential benefits as treatment targets and diagnostic biomarkers.
Similar content being viewed by others


Background
Adult T-cell lymphoma/leukemia (ATLL) affects the mature CD4 and CD25 lymphocyte lineage and has one of the poorest prognosis among hematologic malignancies [1]. The incidence of ATLL in the United States is about five individuals out of one million; however, it varies in diverse regions, with the incidence increasing up to about 27 in 100,000 in endemic zones [2]. According to the Shimoyama criteria, ATLL is divided into acute, chronic, lymphomatous, and smoldering subtypes [3]. The disease primarily occurs in the fifth and sixth decades of life [4] with poor response to conventional treatments [5]. Acute and lymphomatous types can shorten median survival to 6.2 and 10.2 months, respectively [4].
Human T-lymphotropic virus type 1 (HTLV-1) is a retrovirus and the causative agent of ATLL and a progressive chronic neurologic disorder, HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP), which results in progressive weakness of the lower extremities and significant morbidity [6,7,8]. There is a 4–7% risk of developing ATLL in HTLV-1-infected individuals [9]. Its genome contains tax and Hbz, two regulatory genes [10, 11]. Hbz is encoded from the antisense strand of the genome [12]. Tax and Hbz play key roles in the formation of persistent infection and initiation of T cell oncogenesis. Tax interacts with several genes and signaling pathways of an infected host like nuclear factor-kB (NF-κB) pathway, Rat sarcoma virus (RAS) signaling pathway, and the mammalian target of rapamycin (mTOR) pathway [11]. Despite a large number of studies and promising discoveries on the topic, the exact molecular mechanisms responsible for HTLV-1-related tumorigenesis are not well known [13]. Yet, there are several studies searching for anomalies in the area of epigenetics [14,15,16]. MicroRNAs (miRNAs) belong to a wide group of non-coding RNAs that mainly govern gene expression [17]. These small single-stranded RNAs also regulate a broad spectrum of biological processes through mRNA silencing and degradation [18]. The upregulation of miRNAs in cancer cells may lead to carcinogenesis by interdicting tumor suppressor genes. These miRNAs are known as oncogenic miRNAs (oncomiRs) [19]. On the other hand, there are miRNAs with tumor suppressor function that can result in the prevention of cancer progression by interfering with the expression of proto-oncogenes [20]. These miRNAs are identified as tumor suppressor miRNAs [21]. So far, miRNAs have been mentioned to be dysregulated in HTLV-1 infected cells, most likely influencing important signaling pathways such as pathways in cancer, WNT signaling pathway, MAPK signaling pathway, and other important pathways involved in carcinogenesis [10].
Owing to the innovations in methods of extracting genome-wide assays for biological samples in recent years, it is now possible to investigate cellular physiology and the pathophysiology of diseases in a systematic manner. One strategy in biological system analysis is to organize and study genes based on their interactions [22]. In this regard, several approaches may be utilized [23,24,25,26,27]. This study used weighted gene co-expression network analysis (WGCNA) for the purpose of classifying miRNAs into modules based on their correlations and then surveying the co-expressed miRNAs in each module. It helps to find major miRNAs with high connectivity with others that probably have critical functions in the progression of the disease.
Considering ATLL’s rapid progression and the fact that it is refractory to conventional chemotherapy [28], there is a great need to have a better understanding of the pathogenesis by finding factors that have key roles in the development of the disease. The present study investigates the co-expressed dysregulated microRNAs (miRNAs) involved in the ATLL pathogenesis using weighted gene co-expression analysis. A number of target genes of the affected miRNAs are also studied to delineate and confirm the signaling pathways that may be implicated in the development and progression of ATLL.
Methods
Microarray dataset and data preprocessing
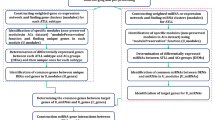
Under the accession number GSE31629 [29], microRNA expression dataset of 62 samples including 40 ATLL patients and 20 healthy individuals was retrieved from the gene expression omnibus (GEO) database [30]. The data derived from peripheral blood mononuclear cells (PBMCs) and CD4+ T cells were normalized with quantile normalization. Missing values were handled using the "goodSamplesGenes" function from the WGCNA package, version 1.71 [31, 32]. Subsequently, sample clustering was used to identify outliers. The summary of the methodology is presented in Fig. 1.

The flowchart of the methodology
Construction of co-expression network and module detection
The weighted co-expression network was constructed using the WGCNA package” version 1.71 in the R environment [33]. To do so, an adjacency matrix was built using the following formula:
where β is the exponent and Xi and Xj are the expression values of miRNAi and miRNAj, respectively. The soft-thresholding approach was applied to obtain a scale-free topology. Using the “picksoftthreshold” function of the WGCNA package, β = 4 was determined to be the optimal exponent value.
In the next step, the topological overlap measure (TOM) was determined as the measure of miRNAs connectivity in the network. Eventually, the co-expressed gene groups (modules) were detected by hierarchical clustering with parameters of minModuleSize = 15, and a threshold of 0.25 was chosen to merge the close modules.
Identification of non-preserved modules
The non-preserved modules of ATLL in the healthy samples were determined through module preservation analysis. For this purpose, the “modulePreservation” function [34] and permutation-based statistics were applied to measure medianRank and Zsummary scores [35, 36]. Zsummary is the average of Z-scores calculated for connectivity and density measures [37]. Herein, a module with Zsummary < 2 and medianRank > 12 was considered a non-preserved module.
Finding hub miRNAs and their target genes
To find the hub miRNA in each non-specific module, the function of “chooseTopHubInEachModule” in the WGCNA package was utilized. In order to find the experimentally validated genes for the mentioned hub miRNAs, the miRTarBase database was explored. miRTarBase is a collection of nearly 20 million experimentally validated miRNA–target interactions (MTIs) among 4630 miRNAs and 27,172 mRNAs [38].
Protein–protein interactions (PPIs) and enrichment analysis
The association between proteins was determined using the STRING database [39]. Due to the large size of PPINs, degree analysis was implemented to select genes with more connectivity which are also known as hub genes. The degree of a protein is its connection number to other proteins in the network. To survey the biological process of the identified target genes, the Enrichr webtool was utilized [40].
Sample preparation
This study was conducted on 10 ATLL samples from patients who had been admitted to the Shariati Hospital, Tehran, Iran, and 10 normal samples from blood donors who were referred to the Blood Transfusion Organization of Alborz. Peripheral blood mononucleolar cells (PBMCs) were isolated from EDTA-treated blood samples using a Ficoll-Paque density gradient (Cederline corporation, Canada). Total RNA was extracted from PBMCs with RNA Extraction RNJia Kit-ROJE Technology and the cDNA was synthesized using the RT-ROSET kit (ROJE, Iran). This study was approved by the Ethical Committee of Biomedical Research at Alborz University of Medical Sciences (IR.ABZUMS.REC.1399.342).
Quantitative real-time PCR
To confirm the HTLV-1 infection in the ATLL patients, the LTR and HBZ genes’ expressions were evaluated by PCR and subsequent agarose gel electrophoresis [41, 42]. Quantitative Real-Time PCR was performed on cDNAs utilizing specific primers and SYBR Green-based RT-qPCR (TaKaRa, Otsu, Japan). To measure the expression rate of UBB, RPS15A, and KMT2D genes in ATLL and normal groups, the PCR was done on a Rotor-Gene Q-6000 machine (Qiagen, Germany) following the manufacturer’s instructions. According to five 5-point standard curves, that had been already prepared for target and reference genes, the gene expression of UBB, RPS15A, KMT2D, and RPLP0 (reference gene) were analyzed. The normalized gene expression was calculated as follows: Normalized index = copy number of the gene of interest/copy number of the reference gene.
Statistical analysis
The Mann–Whitney U Test was used to compare gene expression levels between the normal and ATLL groups using GraphPad Prism software v8 (GraphPad Software, Inc., San Diego, CA, USA). The data is displayed as mean ± standard error of the mean (SEM). The results were considered statistically significant if the P-value < 0.05.
Results
Construction of weighted gene co-expression network
To identify the co-expressed miRNAs, the calculation of adjacency and TOM matrices, construction of hierarchical clustering, and finally merging of close clusters were performed. As a result, 15 modules were identified. Figure 2 displays the cluster dendrogram and modules before and after merging the clusters. Each unique color specifies an inimitable module. A list of co-expressed miRNAs in each module and their connectivity (degree) scores are mentioned in Additional file 1.

Dendrogram of clustered genes based on (1-TOM). The colors reveal the cluster (module) membership acquired before and after merging the modules. A total of 14 modules were identified after merging
Determination of specific modules for the ATLL samples
To detect the specific non-preserved modules of ATLL samples, the medianRank and Zsummary scores were calculated for each module. The Zsummary < 2 and medianRank > 12 were considered as criteria to find the non-preserved modules. Among 15 identified modules, four modules including green, salmon, cyan, and brown modules were identified as specific modules for ATLL (Fig. 3).

The medianRank (plot on the left) and Zsummary (plot on the right) against module size specifies the non-preserved modules (Zsummary < 2 and medianRank > 12)
Finding hub miRNA and their target genes
As the result, “chooseTopHubInEachModule” function identified hsa-miR-346, hsa-miR-187-5p, hsa-miR-196a-3p, and hsa-miR-18a-3p as hubs in the green, salmon, cyan, and brown modules, respectively. Target genes of each miRNA were obtained from miRTarBase for further analysis. The target genes are mentioned in Additional file 2.
PPINs and enrichment analysis
The PPINs between the proteins were constructed by submitting the target genes of hub miRNAs belonging to the specific modules in the STRING database (Fig. 4). Further network analysis revealed the degree of each protein. The protein with a higher degree is demonstrated as red. Based on this analysis, UBB and RPS2 (hsa-miR-18a-3p); RPL13A and RPS15A (hsa-miR-196a-3p); BCL6, TERT, KMT2D (hsa-miR-346); CDKN1B and AGO2 (hsa-miR-187-5p) were identified as hub proteins (Table 1). Subsequently, biological process enrichment was performed to find the function of each hub genes. The enrichment results are presented in Fig. 5.

The protein–protein networks among the target genes of a hsa-miR-18a-3p, b hsa-miR-196a-3p, c hsa-miR-346 and d hsa-miR-187-5p. The color and the size of nodes is related to their degree (red color and bigger size indicate higher values of degree)

Significant KEGG pathways enrichment results of A hsa-miR-18a-3p, B hsa-miR-346, C hsa-miR-187-5p and D hsa-miR-196-3p target genes. All of the pathways are statically significant (P value < 0.05) and are sorted based on the combined scores provided by Enrichr
Lower expression of UBB, RPS14A and KMT2D in ATLL patients compared to the normal control
The remarkable association between the highlighted miRNAs and pathways related to malignancies, immunity, and viral infections led us to examine the gene expression of some of the hub genes that their dysregulation in ATLL patients was never reported before. The results demonstrated significant downregulation of all three genes (P-value < 0.0001) in ATLL patients versus healthy controls (Fig. 6).

Quantitative results of RT-PCR show significant downregulation (P-value < 0.0001) of A UBB, B RPS15A and C KMT2D
UBB’s expression rate in the ATLL group (0.4484 ± 0.06539) was lower than in the normal group (1.952 ± 0.1515) and the difference was statistically significant with P-value < 0.0001. A significantly lower expression of RPS14A in the ATLL patients (0.5884 ± 0.02465) compared to the normal group (3.260 ± 0.1204) was also observed (P-value < 0.0001). The expression of KMT2D in the ATLL cases was 0.5740 ± 0.05896 and in the normal control was 4.204 ± 0.1548, which shows a statistically significant lower expression in the ATLL patients than in the normal cases (P-value < 0.0001).
Discussion
Weighted gene co-expression analysis is a robust method for identifying expression patterns of diseases in a variety of tissues across different pathological states. It is reasonable to expect that the specific co-expression patterns (or modules) for patient samples are likely associated with the underlying pathophysiological pathways and processes. As a corollary, module preservation analysis can be utilized to identify condition-specific or non-preserved modules.
In the present study, a number of hitherto unexplored miRNAs and genes were identified as possibly implicated in ATLL pathogenesis. All genes are miRNAs driver genes that have been found through exploring miRTarBase database. Module preservation analysis disclosed 4 specific modules for ATLL. The hub miRNAs in these modules have a substantial correlation with numerous cancer pathways in an enrichment assessment of their target genes. Subsequent in vitro analysis of the identified miRNAs' target genes demonstrated decreased expression of KMT2D and its potentially important role in ATLL pathogenesis.
It is believed that miR-346 may be involved in both inflammatory and metabolic processes [43]; however, additional research has shown that miR-346 may also have a role in the development of malignancies of the breast, cervix, and lung, as well as follicular thyroid, cutaneous squamous cell, and nasopharyngeal carcinomas [43,44,45,46,47,48,49]. The enrichment analysis of its target genes also indicated that miR-346 is deeply associated with pathways related to viral infections and malignancies; therefore, we further studied its target hub gene, KMT2D. Lysine Methyltransferase 2D, or KMT2D, is a member of a complex of proteins associated with Set1 (COMPASS), which enables gene transcription through H3K4 methylation [50]. KMT2D plays contradictory roles in different types of malignancies, some studies stated it has pro-tumorigenic functions [51], whereas others reported tumor-suppressive characteristics [50,51,52,53]. As a tumor suppressor gene, KMT2D is known to activate Bcl6 and Sirt1 in addition to Per2, resulting in inhibitory effects on Notch and K-Ras pathways [51]. It is good to notice that Bcl6 is also a miR-346 target hub gene. Loss of KMT2D was shown to assist cell growth related to EZH1/2 in pre-tumorigenic EBV + B cell lymphoblastoid cells [54]. Our results show significant downregulation of KMT2D in ATLL patients versus healthy controls. Put it all together, these shreds of evidence are highly suggestive of its involvement in the pathogenesis of the disease, but considering its antithetical effects on cancers, more studies are needed to clarify the exact influence of KMT2D on ATLL.
In recent years miR-187 has attracted the attention of more researchers due to discoveries about its roles in various cancer types. miR-187 is likely to be involved in many cancer-related processes and characteristics such as sensitivity to drugs, proliferation, apoptosis, invasion, and migration through its regulatory effects on its targets, namely FOXA2, CRMP1, MAD2L2, STOML2, BCL6, PTRF, CYP1B1, FGF9, MAPK12, MAPK7, Bcl-2, IGF-1R [55]. Its dysregulation has been observed in many malignancies like osteosarcoma, male genitourinary tumors, prostate cancer, bladder cancer, clear cell renal cell carcinoma, colorectal cancer, hepatocellular carcinoma, oral squamous cell carcinoma, cervical cancer, ovarian cancer, breast cancer, lymphoblastic leukemia, and diffuse large b-cell lymphoma [55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73]. To our knowledge, the relationship between Adult T-Cell Leukemia/Lymphoma and miR-187 was never reported before. Our high-throughput analysis of miRNAs proposes the involvement of miR-187 in the pathogenesis of ATLL. Considering the notable effects of miR-187 on a high variety of malignancies, it seems necessary to investigate the miR-187 actions in ATLL patients.miR-18a-3p was highlighted as a tumor-suppressive agent through its inhibitory effect on K-Ras [74]. It was also reported that miR-18a-3p has a regulatory effect on glycolysis/gluconeogenesis and focal adhesion in the uterus, lung, liver, and kidney malignancies [75]. There are studies presenting pieces of evidence that show miR-18a-3p is related to nasopharyngeal and hepatocellular carcinoma, breast cancer, and glioma [76,77,78,79]. Accordingly, to do an additional survey, Ubiquitin B (UBB) was selected as it was one of the hub genes in PPIN which is also known to play key roles in basic cellular functions. UBB is one of four Ubiquitin encoding genes that is involved in the dysregulation of fundamental processes like cellular proliferation, apoptosis, and responses to DNA damage [80]. Several relationships between Ubiquitin and various types of cancers have been reported such as gynecological cancer, hepatocellular carcinoma, colorectal cancer, neuroblastoma, Esophageal Squamous Cell Carcinoma, Pediatric Medulloblastoma, seminoma, and lung cancer [80,81,82]. According to the downregulation of UBB in the RT-PCR study, the possibility of its pathogenic actions in ATLL can be ruled out.miR-196a was shown to play roles in inflammation, embryonic development, and various cancers such as pancreatic adenocarcinoma, breast cancer, leukemia, esophageal adenocarcinoma, and colorectal cancer [83]. Ribosomal protein S15A (RPS15A) has been highlighted as a candidate participating in ATLL tumorigenesis. It belongs to the 40S subunit of ribosome [84]. RPS15A was reported to be potentially involved in hepatocellular carcinoma, progression of breast cancer, lung adenocarcinoma, prostate cancer, glioblastoma, colorectal cancer, and acute myeloid leukemia as an oncogene [84,85,86,87]. RPS15A was also shown to be downregulated in ATLL patients in the PCR study, which rejects our hypothesis.
Our study have some limitations. We did not find a large miRNA dataset in databases. However, it is better that the mentioned analysis perform in a larger dataset if in it provides in future. The genes and also miRNAs should be validated in a large cohort.
Conclusion
The weighted miRNA-co-expression analysis, network analysis of the target genes, enrichment analysis, and comprehensive literature review highlighted hsa-miR-18a, has-miR-187, hsa-miR-196a-3p, and hsa-miR-346 as four new miRNAs that are strongly suspected to be involved in the pathogenesis of ATLL. Further investigation is needed to assess the utility of these findings and their values as reliable biomarkers and/or therapeutic targets. The expression levels of the three hub genes of the identified miRNAs target genes, namely, UBB, RPS15A, and KMT2D also imply the significant role of the miRNAs in maintaining the dysregulated pattern of gene expression in ATLL. Likely, the downregulation of KMT2D plays an important role in ATLL carcinogenesis, but its controversial effects on different cancer types preclude a definite conclusion regarding its role.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Rodríguez-Zúñiga MJM, Cortez-Franco F, Qujiano-Gomero E. Adult T-cell leukemia/lymphoma. Rev Lit Actas dermosifiliogr. 2018;109(5):399–407.
Mehta-Shah N, Ratner L, Horwitz SM. Adult T-cell leukemia/lymphoma. J Oncol Pract. 2017;13(8):487–92.
Ghobadi MZ, Emamzadeh R, Afsaneh E. Exploration of mRNAs and miRNA classifiers for various ATLL cancer subtypes using machine learning. BMC Cancer. 2022;22(1):433.
Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the lymphoma study group (1984–87). Br J Haematol. 1991;79(3):428–37.
Malpica L, Pimentel A, Reis IM, Gotuzzo E, Lekakis L, Komanduri K, et al. Epidemiology, clinical features, and outcome of HTLV-1-related ATLL in an area of prevalence in the United States. Blood Adv. 2018;2(6):607–20.
Giam CZ, Semmes OJ. HTLV-1 infection and adult T-cell leukemia/lymphoma-A tale of two proteins: tax and HBZ. Viruses. 2016;8(6):161.
Furukawa Y, Yamashita M, Usuku K, Izumo S, Nakagawa M, Osame M. Phylogenetic subgroups of human T cell lymphotropic virus (HTLV) type I in the tax gene and their association with different risks for HTLV-I-associated myelopathy/tropical spastic paraparesis. J Infect Dis. 2000;182(5):1343–9.
Zarei Ghobadi M, Emamzadeh R. Integration of gene co-expression analysis and multi-class SVM specifies the functional players involved in determining the fate of HTLV-1 infection toward the development of cancer (ATLL) or neurological disorder (HAM/TSP). PLoS ONE. 2022;17(1): e0262739.
Satake M, Yamada Y, Atogami S, Yamaguchi K. The incidence of adult T-cell leukemia/lymphoma among human T-lymphotropic virus type 1 carriers in Japan. Leuk Lymphoma. 2015;56(6):1806–12.
Machado CB, da Cunha LS, Maués J, Pessoa F, de Oliveira MB, Ribeiro RM, et al. Role of miRNAs in human T cell leukemia virus type 1 induced T cell leukemia: a literature review and bioinformatics approach. Int J Mol Sci. 2022;23(10):5486.
Ernzen KJ, Panfil AR. Regulation of HTLV-1 transformation. Biosci Rep. 2022;42(3):BSR20211921.
Toyoda K, Matsuoka M. Functional and pathogenic roles of retroviral antisense transcripts. Front Immunol. 2022;13: 875211.
Kogure Y, Kataoka K. Genetic alterations in adult T-cell leukemia/lymphoma. Cancer Sci. 2017;108(9):1719–25.
Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: moving forward. PLoS Genet. 2018;14(6): e1007362.
Recillas-Targa F. Cancer epigenetics: an overview. Arch Med Res. 2022;53(8):732–40.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27.
Chen L, Heikkinen L, Wang C, Yang Y, Sun H, Wong G. Trends in the development of miRNA bioinformatics tools. Brief Bioinform. 2019;20(5):1836–52.
Bavelloni A, Ramazzotti G, Poli A, Piazzi M, Focaccia E, Blalock W, et al. MiRNA-210: a current overview. Anticancer Res. 2017;37(12):6511–21.
Segal M, Slack FJ. Challenges identifying efficacious miRNA therapeutics for cancer. Expert Opin Drug Discov. 2020;15(9):987–92.
Ghobadi MZ, Emamzadeh R, Mozhgani S-H. Deciphering microRNA-mRNA regulatory network in adult T-cell leukemia/lymphoma; the battle between oncogenes and anti-oncogenes. PLoS ONE. 2021;16(2): e0247713.
Otmani K, Lewalle P. Tumor suppressor miRNA in cancer cells and the tumor microenvironment: mechanism of deregulation and clinical implications. Front Oncol. 2021;11: 708765.
Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol. 2012;8:565.
Ha J, Park S. NCMD: Node2vec-based neural collaborative filtering for predicting miRNA-disease association. IEEE/ACM Trans Comput Biol Bioinform. 2022.
Ha J. MDMF: predicting miRNA-disease association based on matrix factorization with disease similarity constraint. J Pers Med. 2022;12(6):885.
Ha J, Park C, Park C, Park S. IMIPMF: Inferring miRNA-disease interactions using probabilistic matrix factorization. J Biomed Inform. 2020;102: 103358.
Ha J, Park C, Park S. PMAMCA: prediction of microRNA-disease association utilizing a matrix completion approach. BMC Syst Biol. 2019;13(1):33.
Ha J, Park C. MLMD: metric learning for predicting miRNA-disease associations. IEEE Access. 2021;9:78847–58.
Takeuchi M, Miyoshi H, Ohshima K. Tumor microenvironment of adult T-cell leukemia/lymphoma. J Clin Exp Hematopathol JCEH. 2021;61(4):202–9.
Yamagishi M, Nakano K, Miyake A, Yamochi T, Kagami Y, Tsutsumi A, et al. Polycomb-mediated loss of miR-31 activates NIK-dependent NF-κB pathway in adult T cell leukemia and other cancers. Cancer Cell. 2012;21(1):121–35.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41(Database issue):D991–5.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559.
Langfelder P, Horvath S. Fast R functions for robust correlations and hierarchical clustering. J Stat Softw. 2012;46(11):i11.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9(1):1–13.
Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7(1): e1001057.
Zarei Ghobadi M, Emamzadeh R, Teymoori-Rad M, Mozhgani SH. Decoding pathogenesis factors involved in the progression of ATLL or HAM/TSP after infection by HTLV-1 through a systems virology study. Virol J. 2021;18(1):175.
Ghobadi MZ, Emamzadeh R, Teymoori-Rad M, Afsaneh E. Exploration of blood− derived coding and non-coding RNA diagnostic immunological panels for COVID-19 through a co-expressed-based machine learning procedure. Front Immunol. 2022;13:1001070.
Zarei Ghobadi M, Mozhgani SH, Erfani Y. Identification of dysregulated pathways underlying HTLV-1-associated myelopathy/tropical spastic paraparesis through co-expression network analysis. J Neurovirol. 2021;27(6):820–30.
Huang HY, Lin YC, Cui S, Huang Y, Tang Y, Xu J, et al. miRTarBase update 2022: an informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022;50(D1):D222–30.
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–12.
Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, et al. Gene set knowledge discovery with enrichr. Curr Protoc. 2021;1(3): e90.
Rafatpanah H, Hedayati-Moghaddam MR, Fathimoghadam F, Bidkhori HR, Shamsian SK, Ahmadi S, et al. High prevalence of HTLV-I infection in Mashhad, Northeast Iran: a population-based seroepidemiology survey. J Clin Virol Off Publ Pan Am Soc Clin Virol. 2011;52(3):172–6.
Mozhgani SH, Zarei Ghobadi M, Norouzi M, Rahimi H, Valizadeh N, Teymoori-Rad M, et al. Signaling factors potentially associated to the pathogenesis of adult T-cell leukemia/lymphoma: a network-analysis and novel findings assessment. Virus Res. 2022;319: 198875.
Du L, Borkowski R, Zhao Z, Ma X, Yu X, Xie XJ, et al. A high-throughput screen identifies miRNA inhibitors regulating lung cancer cell survival and response to paclitaxel. RNA Biol. 2013;10(11):1700–13.
Weber F, Teresi RE, Broelsch CE, Frilling A, Eng C. A limited set of human MicroRNA is deregulated in follicular thyroid carcinoma. J Clin Endocrinol Metab. 2006;91(9):3584–91.
Sun CC, Li SJ, Yuan ZP, Li DJ. MicroRNA-346 facilitates cell growth and metastasis, and suppresses cell apoptosis in human non-small cell lung cancer by regulation of XPC/ERK/Snail/E-cadherin pathway. Aging. 2016;8(10):2509–24.
Chen B, Pan W, Lin X, Hu Z, Jin Y, Chen H, et al. MicroRNA-346 functions as an oncogene in cutaneous squamous cell carcinoma. Tumour Biol J Int Soc Oncodev Biol Med. 2016;37(2):2765–71.
Yan HL, Li L, Li SJ, Zhang HS, Xu W. miR-346 promotes migration and invasion of nasopharyngeal carcinoma cells via targeting BRMS1. J Biochem Mol Toxicol. 2016;30(12):602–7.
Yang F, Luo LJ, Zhang L, Wang DD, Yang SJ, Ding L, et al. MiR-346 promotes the biological function of breast cancer cells by targeting SRCIN1 and reduces chemosensitivity to docetaxel. Gene. 2017;600:21–8.
Guo J, Lv J, Liu M, Tang H. miR-346 up-regulates argonaute 2 (AGO2) protein expression to augment the activity of other MicroRNAs (miRNAs) and contributes to cervical cancer cell malignancy. J Biol Chem. 2015;290(51):30342–50.
Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199–208.
Dhar SS, Lee MG. Cancer-epigenetic function of the histone methyltransferase KMT2D and therapeutic opportunities for the treatment of KMT2D-deficient tumors. Oncotarget. 2021;12(13):1296–308.
Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, et al. KMT2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell. 2020;37(4):599-617.e7.
Rabello DDA, Ferreira V, Berzoti-Coelho MG, Burin SM, Magro CL, Cacemiro MDC, et al. MLL2/KMT2D and MLL3/KMT2C expression correlates with disease progression and response to imatinib mesylate in chronic myeloid leukemia. Cancer Cell Int. 2018;18:26.
Yamagishi M, Hori M, Fujikawa D, Ohsugi T, Honma D, Adachi N, et al. Targeting excessive EZH1 and EZH2 activities for abnormal histone methylation and transcription network in malignant lymphomas. Cell Rep. 2019;29(8):2321-37.e7.
Peng W, Sha H, Sun X, Zou R, Zhu Y, Zhou G, et al. Role and mechanism of miR-187 in human cancer. Am J Transl Res. 2020;12(9):4873–84.
Fei D, Zhao K, Yuan H, Xing J, Zhao D. MicroRNA-187 exerts tumor-suppressing functions in osteosarcoma by targeting ZEB2. Am J Cancer Res. 2016;6(12):2859–68.
Xiao Y, Zhao Q, Du B, Chen HY, Zhou DZ. MicroRNA-187 inhibits growth and metastasis of osteosarcoma by downregulating S100A4. Cancer Invest. 2018;36(1):1–9.
Cui C, Shi X. miR-187 inhibits tumor growth and invasion by directly targeting MAPK12 in osteosarcoma. Exp Ther Med. 2017;14(2):1045–50.
Casanova-Salas I, Masiá E, Armiñán A, Calatrava A, Mancarella C, Rubio-Briones J, et al. MiR-187 targets the androgen-regulated gene ALDH1A3 in prostate cancer. PLoS ONE. 2015;10(5): e0125576.
Li Z, Lin C, Zhao L, Zhou L, Pan X, Quan J, et al. Oncogene miR-187–5p is associated with cellular proliferation, migration, invasion, apoptosis and an increased risk of recurrence in bladder cancer. Biomed Pharmacother Biomed Pharmacother. 2018;105:461–9.
Zhao J, Lei T, Xu C, Li H, Ma W, Yang Y, et al. MicroRNA-187, down-regulated in clear cell renal cell carcinoma and associated with lower survival, inhibits cell growth and migration though targeting B7–H3. Biochem Biophys Res Commun. 2013;438(2):439–44.
Wang ZS, Zhong M, Bian YH, Mu YF, Qin SL, Yu MH, et al. MicroRNA-187 inhibits tumor growth and invasion by directly targeting CD276 in colorectal cancer. Oncotarget. 2016;7(28):44266–76.
Han X, Wang X, Zhao B, Chen G, Sheng Y, Wang W, et al. MicroRNA-187 inhibits tumor growth and metastasis via targeting of IGF-1R in hepatocellular carcinoma. Mol Med Rep. 2017;16(2):2241–6.
Dou C, Liu Z, Xu M, Jia Y, Wang Y, Li Q, et al. miR-187-3p inhibits the metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma by targeting S100A4. Cancer Lett. 2016;381(2):380–90.
Liu CJ, Lin JS, Cheng HW, Hsu YH, Cheng CY, Lin SC. Plasma miR-187* is a potential biomarker for oral carcinoma. Clin Oral Invest. 2017;21(4):1131–8.
Lin SC, Kao SY, Chang JC, Liu YC, Yu EH, Tseng SH, et al. Up-regulation of miR-187 modulates the advances of oral carcinoma by targeting BARX2 tumor suppressor. Oncotarget. 2016;7(38):61355–65.
Lin M, Xue XY, Liang SZ, Li YX, Lv YY, He LH, et al. MiR-187 overexpression inhibits cervical cancer progression by targeting HPV16 E6. Oncotarget. 2017;8(38):62914–26.
Liang H, Luo R, Chen X, Zhao Y, Tan A. miR-187 inhibits the growth of cervical cancer cells by targeting FGF9. Oncol Rep. 2017;38(4):1977–84.
He CY, Yang J. miR-187 induces apoptosis of SiHa cervical carcinoma cells by downregulating Bcl-2. Genet Mol Res GMR. 2017;16(1).
Chao A, Lin CY, Lee YS, Tsai CL, Wei PC, Hsueh S, et al. Regulation of ovarian cancer progression by microRNA-187 through targeting disabled homolog-2. Oncogene. 2012;31(6):764–75.
Matamala N, Vargas MT, González-Cámpora R, Arias JI, Menéndez P, Andrés-León E, et al. MicroRNA deregulation in triple negative breast cancer reveals a role of miR-498 in regulating BRCA1 expression. Oncotarget. 2016;7(15):20068–79.
Lou Y, Liu L, Zhan L, Wang X, Fan H. miR-187-5p regulates cell growth and apoptosis in acute lymphoblastic leukemia via DKK2. Oncol Res. 2016;24(2):89–97.
Huang F, Jin Y, Wei Y. MicroRNA-187 induces diffuse large B-cell lymphoma cell apoptosis via targeting BCL6. Oncol Lett. 2016;11(4):2845–50.
Rodriguez-Aguayo C, Monroig PDC, Redis RS, Bayraktar E, Almeida MI, Ivan C, et al. Regulation of hnRNPA1 by microRNAs controls the miR-18a-K-RAS axis in chemotherapy-resistant ovarian cancer. Cell Discov. 2017;3:17029.
Shu J, Silva B, Gao T, Xu Z, Cui J. Dynamic and modularized MicroRNA regulation and its implication in human cancers. Sci Rep. 2017;7(1):13356.
Li C, Feng SY, Chen L. SET7/9 promotes H3K4me3 at lncRNA DRAIC promoter to modulate growth and metastasis of glioma. Eur Rev Med Pharmacol Sci. 2020;24(23):12241–50.
Chen W, Cao R, Su W, Zhang X, Xu Y, Wang P, et al. Simple and fast isolation of circulating exosomes with a chitosan modified shuttle flow microchip for breast cancer diagnosis. Lab Chip. 2021;21(9):1759–70.
Wang L, Zhou L, Hou J, Meng J, Lin K, Wu X, et al. Three novel circRNAs upregulated in tissue and plasma from hepatocellular carcinoma patients and their regulatory network. Cancer Cell Int. 2021;21(1):72.
Sun K, Wang H, Xu X, Wei X, Su J, Zhu K, et al. Tumor-educated platelet miR-18a-3p as a novel liquid-biopsy biomarker for early diagnosis and chemotherapy efficacy monitoring in nasopharyngeal carcinoma. Front Oncol. 2021;11: 736412.
Oh C, Park S, Lee EK, Yoo YJ. Downregulation of ubiquitin level via knockdown of polyubiquitin gene Ubb as potential cancer therapeutic intervention. Sci Rep. 2013;3:2623.
Huang P, Guo YD, Zhang HW. Identification of Hub genes in pediatric medulloblastoma by multiple-microarray analysis. J Mol Neurosci MN. 2020;70(4):522–31.
Chen Y, Qi C, Xia L, Li G. Identification of novel genetic etiology and key molecular pathways for seminoma via network-based studies. Int J Oncol. 2017;51(4):1280–90.
Chen C, Zhang Y, Zhang L, Weakley SM, Yao Q. MicroRNA-196: critical roles and clinical applications in development and cancer. J Cell Mol Med. 2011;15(1):14–23.
Zhao X, Shen L, Feng Y, Yu H, Wu X, Chang J, et al. Decreased expression of RPS15A suppresses proliferation of lung cancer cells. Tumour Biol J Int Soc Oncodev Biol Med. 2015;36(9):6733–40.
Yao Y, Liu Y, Lv X, Dong B, Wang F, Li J, et al. Down-regulation of ribosomal protein S15A inhibits proliferation of human glioblastoma cells in vivo and in vitro via AKT pathway. Tumour Biol J Int Soc Oncodev Biol Med. 2016;37(4):4979–90.
Xu M, Wang Y, Chen L, Pan B, Chen F, Fang Y, et al. Down-regulation of ribosomal protein S15A mRNA with a short hairpin RNA inhibits human hepatic cancer cell growth in vitro. Gene. 2014;536(1):84–9.
Wan BS, Wang XY, Tiang J, Zhou C, Lin J, Wang Z. Ribosomal protein RPS15A augments proliferation of colorectal cancer RKO cells via regulation of BIRC3, p38 MAPK and Chk1. Eur Rev Med Pharmacol Sci. 2021;25(11):3967–80.
Acknowledgements
Not applicable.
Funding
None.
Author information
Authors and Affiliations
Contributions
MZG, AS, MF, SS and ZS conceptualized the work and contributed to drafting the manuscript. AH, PH, SK, MH and SA contributed to drafting and revising the manuscript. MZG performed the bioinformatic analysis. MZG and S-HM supervised the project and performed the analyses. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All methods were performed in accordance with the Declaration of Helsinki. Informed consent for study participation was obtained from all subjects. This study was approved by the ethics committee of Alborz University of medical sciences, Alborz, Karaj, Iran. (IR.ABZUMS.REC.1399.342).
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
List of co-expressed miRNAs and their connectivity scores in each module.
Additional file 2.
The target genes of non-preserved modules.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shayeghpour, A., Forghani-Ramandi, MM., Solouki, S. et al. Identification of novel miRNAs potentially involved in the pathogenesis of adult T-cell leukemia/lymphoma using WGCNA followed by RT-qPCR test of hub genes. Infect Agents Cancer 18, 12 (2023). https://doi.org/10.1186/s13027-023-00492-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13027-023-00492-0