
Abstract
Background
Human T-lymphotropic virus 1 (HTLV-1) infection may lead to the development of Adult T-cell leukemia/lymphoma (ATLL). To further elucidate the pathophysiology of this aggressive CD4+ T-cell malignancy, we have performed an integrated systems biology approach to analyze previous transcriptome datasets focusing on differentially expressed miRNAs (DEMs) in peripheral blood of ATLL patients.
Methods
Datasets GSE28626, GSE31629, GSE11577 were used to identify ATLL-specific DEM signatures. The target genes of each identified miRNA were obtained to construct a protein-protein interactions network using STRING database. The target gene hubs were subjected to further analysis to demonstrate significantly enriched gene ontology terms and signaling pathways. Quantitative reverse transcription Polymerase Chain Reaction (RTqPCR) was performed on major genes in certain pathways identified by network analysis to highlight gene expression alterations.
Results
High-throughput in silico analysis revealed 9 DEMs hsa-let-7a, hsa-let-7g, hsa-mir-181b, hsa-mir-26b, hsa-mir-30c, hsa-mir-186, hsa-mir-10a, hsa-mir-30b, and hsa-let-7f between ATLL patients and healthy donors. Further analysis revealed the first 5 of DEMs were directly associated with previously identified pathways in the pathogenesis of HTLV-1. Network analysis demonstrated the involvement of target gene hubs in several signaling cascades, mainly in the MAPK pathway. RT-qPCR on human ATLL samples showed significant upregulation of EVI1, MKP1, PTPRR, and JNK gene vs healthy donors in MAPK/JNK pathway.
Discussion
The results highlighted the functional impact of a subset dysregulated microRNAs in ATLL on cellular gene expression and signal transduction pathways. Further studies are needed to identify novel biomarkers to obtain a comprehensive mapping of deregulated biological pathways in ATLL.
Similar content being viewed by others

Introduction
Human T lymphotropic virus 1 (HTLV-1) is a single-stranded positive-strand RNA virus, which primarily infects CD4+ T-cells in humans [1]. At least 5-10 million individuals have been virus carriers around the globe in several endemic foci including southern Japan and the Caribbean [2]. A subpopulation of individuals infected with HTLV-1 (6% of male and of 2% female subjects) develop Adult T-cell leukemia/lymphoma (ATLL) after a long latency period of 4 to 6 decades [3, 4]. ATLL is a malignant T-cell neoplasm characterized by pleomorphic leukemic cells with hyper segmented nuclei, which are immunophenotypically comparable to regulatory T-cells [5]. This aggressive peripheral T-cell malignancy is associated with a poor prognosis and numerous clinical complications, such as hypercalcemia and immunodeficiency [4]. Since the initial description of ATLL with aggressive subtypes (acute, lymphomatous, and chronic with unfavorable prognostic factors) having a survival rate of less than one year and despite numerous modalities and therapeutic approaches, the median survival rate has not been improved significantly [4, 6]. The oncogenic properties of the viral products are substantiated through countless experiments [7, 8]. However, the low prevalence of ATLL among HTLV-1 carriers and the long latency period suggests that factors such as host genetic susceptibility and environmental factors may influence the development of the disease.
The pX region of HTLV-1 genome encodes two important regulatory proteins HBZ and Tax [9]. The substantial role of Tax and HBZ in HTLV-1 leukemogenesis is evident in the induction of T-cell lymphoma by transgenic expression of each of these transcripts in animal models [10, 11]. Interactions of HTLV-1 Tax and HBZ proteins with host machinery lead to diverse changes in cellular behavior through alterations of signal transduction pathways and gene expression marked by modulation of NF-κB, MAPK, AP-1, JAK/STAT, mTOR, IRFs, TGF-β, and p53 signaling pathways in HTLV-1 infected cells [12, 13]. The complex interactions of Tax and HBZ with host cellular pathways lead to increased proliferation and immune escape of the infected T-cells [14]. The leukemogenic effects of HTLV-1 transcripts are partially explained by their introduction of DNA instability and impairing DNA damage repair, which are signified by various mutations in immortalized ATLL cells [9, 15]. Many of these mutations also converge on pathways already dysregulated by direct interaction of Tax [16], which suggests their role in compensation of loss of Tax expression in the chronic infected cells and their capacity to progress and maintain the leukemic state during the later stages of infection [13, 17]. The higher rate of proliferation and greater survival advantage of HTLV-1 infected cells harboring Tax-mimicking mutations and negative selection of other clones in ATLL may explain the narrower and more uniform clonality of ATLL CD4+ T-cells compared to those of HTLV-1 associated myelopathy/Tropic spastic paraparesis (HAM/TSP) patients and asymptomatic carriers [18].
The ATLL specific genomic signature is not only reflected by the pre-transcriptional and transcriptional modification of gene expression. Infection with HTLV-1 virus and development of ATLL has been associated with significant dysregulation of microRNA (miRNAs) transcriptome in host cells, despite HTLV-1 not having any genome-encoded miRNAs [14, 19,20,21]. Indeed, the global downregulation of miRNAs in HTLV-1 infected cells by EZH2-induced trimethylated H3K27 histone (H3K27me3) has been cited as a crucial step in the development of ATLL [22]. Furthermore, the expression of DICER1 gene is also reduced in both HAM/TSP [12] and ATLL patients [23] further contributing to lower mature miRNAs in HTLV-1 infected cells. This transcriptomic profile is associated with a poor prognosis in several other malignancies such as hepatocellular carcinoma [24] and invasive breast carcinoma [25]. HTLV-1 is also associated with deregulation of numerous single miRNAs in infected cells which interfere with various biological processes, especially cell cycle regulation [26]. Analysis of these specific gene regulation signatures may reveal key molecular targets for novel treatment modalities in targeted cancer therapies for ATLL.
Here we have conducted an integrated approach to analyze gene expression profiling studies to elucidate the anomalies in miRNA gene regulation system in infected cells through high-throughput analysis of previous transcriptomic datasets in the literature. The enriched pathways and highlighted genes in this study revealed novel disruptions in cell signaling cascades which were then confirmed by real time-PCR.
Materials and Methods
Database search and inclusion of eligible datasets
We searched the public domains, Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo) and Array Express (https://www.ebi.ac.uk/arrayexpress) by the end of 2018 to find datasets relevant to the expression levels of miRNA in ATLL patients and healthy donors. Fig. 1 highlights the keywords used and the overall flowchart in data gathering. The inclusion criterion was research studies with human miRNA microarray datasets and samples derived from peripheral blood of ATLL patients and healthy donors. Duplicate results and studies in which the donors were receiving treatment for ATLL and those with healthy HTLV-1 carriers as controls were excluded.

Search strategy and exclusion criteria of the study. Out of 6764 entries in Gene Expression Omnibus (GEO) and 4070 entries in ArrayExpress, 8 datasets remained after applying the exclusion criteria. Subsequently, 3 datasets fit the inclusion criteria, which were included in this study
Pre-processing and differential expression analysis
The selected datasets were pre-processed using GEOquerry package implemented in R programming language (Version 4.0.3). The datasets were normalized using log2 transformation in Affy package and were then integrated with MetaDE package in R programming language to identify DEMs. The proportion of DEMs with a p-value of less than 0.001 were considered for further analysis. Subsequently, the gene targets for selected DEMs were determined by miRDB online database (http://mirdb.org/miRDB/) [27]. The associated genes were included with a target score of larger than 20.
Network construction and pathway enrichment analysis
The STRING database version 11.0 was employed to construct protein-protein interactions network (PPIN) for gene targets of each DEMs based on literature sources for protein-protein interactions. The interactions included for network construction in this study include physical and functional interactions, high-throughput experiments, co-expression, genomic context, databases, and text-mining. The primary PPINs analysis was conducted using NetworkAnalyzer in Cytoscape 3.5.1 [28]. Genes with higher degree and betweenness centrality measures were computed to determine hub genes. Lastly, the PPINs were reconstructed and visualized using Gephi (0.9.1) based on calculated gene hubs.
Gene enrichment analysis was carried out to enrich hub genes in each DEMs in EnrichR web tool using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways [29, 30]. A combined score of higher than 0.4 was considered as cut-off to analyze the PPINs.
Patient population and sample collection
The blood samples were collected from 8 ATLL patients and 10 healthy subjects between 2019 and 2020 from Shariati Hospital, Tehran, Iran. All samples were collected after acquiring informed consent from the patients or their next of kin when appropriate. A standardized clinical checklist, comprising demographic information and the diagnosis of ATLL was evaluated by a trained hematologist. None of the included patients were receiving chemotherapy and/or anti-cancer drugs. All methods were carried out in accordance with the relevant guidelines and regulations. The enzyme-linked immunosorbent assay (ELISA, Diapro, Italy) was used to perform the serology test for HTLV-1. PCR was then employed to confirm the serology results [31]. The inclusion criteria for healthy donors were participants with no active acute infectious disease, no concurrent drug use, and no diagnosed genetic abnormalities or defects. This investigation was approved by the Ethics Committee of Biomedical Research at Alborz University of Medical Sciences (IR.NIMAD.REC.1397.473).
Quantitative reverse transcriptase PCR and statistical analysis
Total RNA was extracted from fresh whole blood utilizing TriPure isolation reagent (Roche, Germany). cDNA was synthesized using RT-ROSET Kit (ROJETechnologies, Iran) and SYBR Green-based (TaKaRa, Otsu, Japan) and subsequently, RT-qPCR was performed, according to the manufacturers’ instructions. The following primers were utilized to determine the expression levels of JNK, EVI1, MKP, and PTPRR and to confirm HTLV-1 infected cells in the samples: EVI1 (forward primer (FP): 5′-TCGTCGCCTCATTCTGAACTGGAA-3′, reverse primer (RP): 5′-ACTGCCATTCATTCTCTCCTCCACA-3′) MKP (FP: 5′-AGCCACCATCTGCCTTGCT-3′ , RP: 5′-CCAGCCTCTGCCGAACAGT-3′ ) PTPPR (FP: 5′-CCAGCACTGTCCGAGGCAA-3′ , RP: 5′-GCAAACAGAGGTAGCGGTGGT-3′ ) JNK (FP: 5′-TGCTGTGTGGAATCAAGCACCT-3′ , RP: 5′-TCGGGTGCTCTGTAGTAGCGA-3′ ) HBZ (FP: 5′-ACGTCGCCCGGAGAAAACA-3′ , RP: 5′-CTCCACCTCGCCTTCCAACT-3′) 5’LTR (FP: 5′-GGCTCGCATCTCCCCTTCAC-3′ , RP: GAGCAAGCAGGGTCAGGCAA-3′). The relative two standard curves real-time PCR was performed on the cDNA samples using Q-6000 machine (Qiagen, Germany). The GAPDH gene was utilized to normalize the mRNA expression levels respectively, as well as to control the error between samples [32]. The output for each group was analyzed using Mann-Whitney U test for statistical difference between gene expression. A p-value of less than 0.05 was considered to be significant.
Results
After removal of redundancy, application of inclusion and exclusion criteria, and quality control using MetaQC package in R, 3 datasets namely, GSE28626 [33], GSE31629 [34], and GSE11577 [35] were selected for the DEM analysis of ATLL patients vs healthy individuals.
The primary analysis of microarray datasets identified hsa-let-7a, hsa-let-7g, hsa-mir-181b, hsa-mir-26b, hsa-mir-30c, hsa-mir-186, hsa-mir-10a, hsa-mir-30b, and hsa-let-7f as DEMs in ATLL patients compared to normal individuals. The target genes for each mentioned DEM were identified using miRDB. The analysis of networks using centrality parameters was utilized to select nodes with higher degree and betweenness as hub genes.
Network analysis and PPIN characteristics
The PPINs were constructed for each DEM separately using STRING to highlight the relationship between the target genes (Fig. 2). The networks were comprised of (a) 37 nodes and 176 edges for gene targets of hsa-let-7a, (b) 43 nodes and 167 edges for gene targets of hsa-let-7g, (c) 35 nodes and 125 edges for gene targets of hsa-mir-181b, (d) 30 nodes and 149 edges for gene targets of hsa-mir-26b, (e) 27 nodes and 98 edges for gene targets of hsa-mir-30c, (f) 36 nodes and 191 edges for gene targets of hsa-mir-186, (g) 44 nodes and 82 genes for gene targets of hsa-mir-10a, (h) 39 nodes and 217 edges for gene targets of hsa-mir-30b, and (i) 40 nodes and 257 edges for gene targets of hsa-let-7f.

Protein-protein interaction networks of the enriched hub genes of differentially expressed miRNAs. PPINs of the identified target genes of a hsa-let-7a, b hsa-let-7g, c hsa-mir-181b, d hsa-mir-26b, e hsa-mir-30c, f hsa-mir-186, g hsa-mir-10a, h hsa-mir-30b, and i hsa-let-7f is illustrated. Genes with higher degree and betweenness are demonstrated in the center in red
GO/Pathway enrichment analysis of identified genes
The Hub genes of all defined DEMs were enriched to reveal biological pathways associated with the PPINs. The analysis highlighted the following pathways for each DEM: hsa-let-7a: Endometrial cancer, Colorectal cancer, Renin secretion, Cocaine addiction, TNF signaling pathway, Human T-cell leukemia virus 1 infection, Thyroid hormone signaling pathway, Human cytomegalovirus infection, Renal cell carcinoma, Cortisol synthesis and secretion.
hsa-let-7g: Endometrial cancer, Colorectal cancer, Human T-cell leukemia virus 1 infection, Circadian rhythm, Thyroid hormone signaling pathway, Toxoplasmosis, TNF signaling pathway, Hedgehog signaling pathway, Human cytomegalovirus infection, Chronic myeloid leukemia;
hsa-mir-181b: Endocrine and other factor-regulated calcium reabsorption, Endometrial cancer, TNF signaling pathway, Renal cell carcinoma, Acute myeloid leukemia, Thyroid hormone signaling pathway, Human T-cell leukemia virus 1 infection, Toll-like receptor signaling pathway, Small cell lung cancer, Graft-versus-host disease.
hsa-mir-26b: Endometrial cancer, Small cell lung cancer, Renal cell carcinoma, Melanoma, Amyotrophic lateral sclerosis (ALS), Chronic myeloid leukemia, Human T-cell leukemia virus 1 infection, Renin secretion, Cocaine addiction, Colorectal cancer.
hsa-mir-30c: Renin secretion, Endocrine and other factor-regulated calcium reabsorption, TNF signaling pathway, Osteoclast differentiation, Endometrial cancer, Th17 cell differentiation, Human T-cell leukemia virus 1 infection, Cocaine addiction, Aldosterone-regulated sodium reabsorption, Renal cell carcinoma.
hsa-mir-186: Endocrine and other factor-regulated calcium reabsorption, Endometrial cancer, Renal cell carcinoma, Acute myeloid leukemia, Thyroid hormone signaling pathway, Salmonellosis, Chronic myeloid leukemia, Pancreatic cancer, Colorectal cancer, Th17 cell differentiation.
hsa-mir-10a: TNF signaling pathway, Cocaine addiction, Circadian rhythm, Small cell lung cancer, Endocrine and other factor-regulated calcium reabsorption, Endometrial cancer, Renal cell carcinoma, Chronic myeloid leukemia, Cortisol synthesis and secretion, Parathyroid hormone synthesis, secretion and action.
hsa-mir-30b: Endocrine and other factor-regulated calcium reabsorption, Endometrial cancer, Cocaine addiction, Thyroid hormone signaling pathway, Chronic myeloid leukemia, Human cytomegalovirus infection, Long-term depression, Salivary secretion, Renin secretion, Salmonella infection.
hsa-let-7f: Endometrial cancer, Renin secretion, Renal cell carcinoma, Colorectal cancer, Human cytomegalovirus infection, Thyroid hormone signaling pathway, Long-term depression, Cocaine addiction, Prostate cancer, Neurotrophin signaling pathway. The following five DEMs were directly associated with HTLV-1 related pathogenic pathways: hsa-let-7a, hsa-let-7g, hsa-mir-181b, hsa-mir-26b, hsa-mir-30c (Table 1).
Manual approach to enrich target genes with higher network connectivity also demonstrated the association of hub genes with several biological and signaling pathways including cell cycle regulation and DNA damage response (CDKN1A, RB1, SKP1, CDK6, SKP1, SKP2, CUL1, CDK1, ATM , SMC, XPO1, UBE2D1, RANBP2, ACTR1A, ESPL1, RANGAP1, ANAPC10, PPP1CC, PRKACA, RAD21, PAFAH1B1, ABL1, RHOA, TNF, RRM2, MTOR, APP, PAK1, CHEK1), MAP kinase (MAP 2K1, MAP3K1, MAPK8, MEF2A, MAP3K7, NRAS, MYC, YWHAZ, IL2, NCAM1, CUL3, LRRK2, PAK1) Phosphatidylinositol-3-kinase (PIK3CG, PIK3C2A, EDN1, PIK3CA, PIK3CD, IGF1R, PTEN) pathways among others.
Validation by qRT-PCR assay
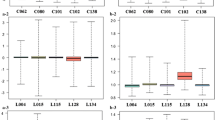
The prominent involvement of top common enriched hub genes (Table 2) in MAP kinase signaling cascade, particularly in the JNK pathway, had led us to analyze the gene expression of JNK (MAPK8) and its major regulators, namely, ecotropic viral integration site 1 (EVI1), Dual-specificity phosphatase-1 (DUSP 1/MKP), and Protein tyrosine phosphatase receptor-type R (PTPRR) in ATLL patients and healthy subjects to validate the results of the meta-analysis. The results demonstrated significant upregulation of EVI1 (p-value = 0.0062), MKP (p-value = 0.0003), PTPRR (p-value = 0.0031), and JNK (p-value < 0.0001) in ATLL patients compared to healthy controls (Fig. 3).

Gene expression of MAPK8 (JNK) and its major inhibitors in the MAPK/JNK signaling pathway. Quantitative RT-PCR revealed significant upregulation of JNK (p-value<0.0001) and its major inhibitors, EVI1 (p-value = 0.0062), PTPRR (p-value = 0.0003), and MKP1 (p-value = 0.0031). Error bars represent minimum and maximum data points in each assay. A single outlier sample with 0 value was observed in ATLL group for EVI1 PCR assay, removal of which did not affect the significance of the differential expression between the groups
Discussion
Molecular approaches have led to the identification of several DEMs and their aberrant expression in array studies. While having their own merits, the conditions of these experiments could direct to perturbations in the analysis of differential gene expression in studies restricted to limited samples, as seen in incongruity of previously reported ATLL gene expression studies [26, 36]. The high throughput analysis of previous datasets as an alternative method can solidify the results of previous studies while providing novel insights into the genetic profile of the diseases [32, 37]. Through analysis of differentially expressed miRNAs in three independent microarray datasets in this study, we highlighted 9 miRNAs associated with in peripheral blood cells of ATLL patients. Further analysis revealed more than 300 target genes for the DEMs in the literature, emphasizing the vast disruption of biological processes in ATLL.
The identified DEMs in this study are linked to several neoplasms and their progression and subsequent metastasis. Hsa-let-7a, has-let-7g, hsa-mir-181b, hsa-miR-26b have been observed to act as tumor suppressors in certain malignancies by reducing the levels of c-myc oncogene which is overexpressed in at least 40% of human cancers [38,39,40,41,42]. Additionally, has-let-7f also inhibits gastric cancer invasion and metastasis through interaction with MYH9 mRNA [43]. Contrarily, has-miR-30c expression increases the invasiveness of metastatic neoplastic cells and is associated with poor prognosis in breast cancer by inhibiting NOV/CCN3 regulatory proteins [44]. The dysregulation of has-miR-10a is also observed in multiple malignancies, with high expression levels in urothelial and medullary thyroid carcinoma and low expression values in chronic myeloid leukemia [45,46,47]. Moreover, miR-186-5p regulates IGF-1 expression and apoptosis in neuroblastoma cells and is also proposed as a screening biomarker in colorectal polyps and adenomas [48]. Lastly, has-let-7a-5p, hsa-miR-181b-5p, hsa-miR-26b-5p, has-miR-30c-5p were also demonstrated to be differentially expressed small RNAs in a recent study on ATLL [49].
An interesting observation in this study was the extensive upregulation of JNK apoptotic pathway in the analysis, confirmed by upregulation of MAPK8 in ATLL samples. The implication of this finding is emphasized in the context of constant activation of NF-KB pathway observed in almost all ATLL clones that normally represses the JNK pathway [8, 50]. Furthermore, TNF-induced JNK activation, which is among upregulated DEGs in this study, normally results in apoptosis and cell death in target cells [51]. Therefore, to be able to explain this inconsistency, we examined the expression levels of the major inhibitors of JNK, namely, EVI1, PTPRR, and MKP. To our knowledge, this is the first study to demonstrate such upregulation of JNK repressors in ATLL. Dysregulation of PTPRR and MKP are implicated in the development and progression of various cancers owing to their ability to regulate both p38 and JNK MAPK pathways [52,53,54,55]. EVI1 has also been recognized as one of the most aggressive oncogenes associated with human leukemias such as acute myeloid leukemia. Aberrant expression of EVI1 leads to repression of TGF-β signaling, upregulated cell proliferation, and impaired cellular differentiation [56]. The levels of EVI1 transcripts are also associated with a poor prognosis in serous epithelial ovarian cancer [57].
Counterintuitively, quantitative PCR assay revealed considerable upregulation of the analyzed JNK inhibitors. Previous studies have described upregulation of JNK pathway via Tax-mediated activation of TAK1 and MEKK1, constitutive activation of this pathway in HTLV-1 transformed cells, and their role in the virus-induced tumorigenesis [58,59,60]. Therefore, the high levels of expression of the JNK inhibitors coupled with general activation of the pathway demonstrates a complex disruption of JNK signaling cascade. Seemingly, the extensive stimulation of NF-KB pathway in ATLL interferes with the function of JNK through interaction with the various activators of JNK pathway including TNF signaling and downstream pathways leading to apoptosis [51, 61, 62]. This subverted JNK pathway is deprived of its pro-apoptotic activities and contributes to leukemogenesis by retaining its stimulation of AP1 and repression of p53 pathways which subsequently govern cell cycle and survival. Furthermore, this dysregulated pathway also promotes visceral invasion of ATLL leukemic cells by virtue of MMP-7 upregulation [63, 64]. Therapeutic targeting of JNK pathway thus proves to be an interesting topic for future studies.
Conclusion
In this high throughput meta-analysis, we identified significant disruption of genes related to cell cycle, proliferation, and signal transduction in ATLL. Subsequent in vitro assay demonstrated higher gene expression of JNK (MAPK8) and the major regulators of MAPK/JNK in ATLL vs healthy controls. The results of this study provide further insight into the dysregulated biological processes in ATLL. Further studies are needed to identify novel and reliable biomarkers, and prognostic factors and to obtain a comprehensive mapping of deregulated biological pathways in ATLL.
Availability of data and materials
The datasets analyzed during this study are available in the gene expression omnibus public repository (www.ncbi.nlm.nih.gov/geo) [33,34,–35].
Abbreviations
- HTLV-1:
-
Human T lymphotropic virus 1
- ATLL::
-
Adult T-cell Leukemia/Lymphoma
- miRNA:
-
MicroRNA
- HAM/TSP:
-
HTLV-1-associated myelopathy/Tropical spastic paraparesis
- PPIN:
-
protein-protein interactions network
References
Bangham CRM, Human T. Cell leukemia virus type 1: persistence and pathogenesis. Ann Rev Immunol. 2018;36(1):43–71.
Gessain A, Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol. 2012;3(388). https://doi.org/10.3389/fmicb.2012.00388.
Kataoka K, Koya J. Clinical application of genomic aberrations in adult T-cell leukemia/lymphoma. J Clin Exp Hematop. 2020;60(3):66–72.
Taylor GP, Matsuoka M. Natural history of adult T-cell leukemia/lymphoma and approaches to therapy. Oncogene. 2005;24(39):6047–57.
Yasunaga JI. Strategies of human T-cell leukemia virus type 1 for persistent infection: implications for leukemogenesis of adult T-cell leukemia-lymphoma. Front Microbiol. 2020;11:979.
Durer C, Babiker HM. Adult T cell leukemia. Treasure Island, FL: StatPearls Publishing Copyright © 2020. StatPearls Publishing LLC.; 2020.
Chan CP, Kok KH, Jin DY. Human T-cell leukemia virus type 1 infection and adult T-cell leukemia. Adv Exp Med Biol. 2017;1018:147–66.
Giam CZ. HTLV-1 replication and adult T cell leukemia development. Recent Results Cancer Res. 2021;217:209–43.
Matsuoka M, Yasunaga J-I. Human T-cell leukemia virus type 1: replication, proliferation and propagation by Tax and HTLV-1 bZIP factor. Curr Opin Virol. 2013;3(6):684–91.
Satou Y, Yasunaga J-I, Zhao T, Yoshida M, Miyazato P, Takai K, et al. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLOS Pathog. 2011;7(2):e1001274.
Ohsugi T, Kumasaka T, Okada S, Urano T. The tax protein of HTLV-1 promotes oncogenesis in not only immature T cells but also mature T cells. Nat Med. 2007;13(5):527–8.
Aghajanian S, Teymoori-Rad M, Molaverdi G, Mozhgani S-H. Immunopathogenesis and cellular interactions in human T-cell leukemia virus type 1 associated myelopathy/tropical spastic paraparesis. Front Microbiol. 2020;11:614940.
Grassmann R, Aboud M, Jeang K-T. Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene. 2005;24(39):5976–85.
Fochi S, Ciminale V, Trabetti E, Bertazzoni U, D’Agostino DM, Zipeto D, et al. NF-κB and microRNA deregulation mediated by HTLV-1 tax and HBZ. Pathogens. 2019;8(4):290.
Giam C-Z, Semmes OJ. HTLV-1 infection and adult T-cell leukemia/lymphoma—A tale of two proteins: tax and HBZ. Viruses. 2016;8(6):161.
Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J-I, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genetics. 2015;47(11):1304–15.
Takeda S, Maeda M, Morikawa S, Taniguchi Y, Yasunaga J-I, Nosaka K, et al. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int J Cancer. 2004;109(4):559–67.
Gillet NA, Malani N, Melamed A, Gormley N, Carter R, Bentley D, et al. The host genomic environment of the provirus determines the abundance of HTLV-1–infected T-cell clones. Blood. 2011;117(11):3113–22.
Moles R, Nicot C. The emerging role of miRNAs in HTLV-1 infection and ATLL pathogenesis. Viruses. 2015;7(7):4047–74.
Sampey G, Van Duyne R, Currer R, Das R, Narayanan A, Kashanchi F. Complex role of microRNAs in HTLV-1 infections. Front Genetics. 2012;3:295.
Ruggero K, Corradin A, Zanovello P, Amadori A, Bronte V, Ciminale V, et al. Role of microRNAs in HTLV-1 infection and transformation. Mol Aspects Med. 2010;31(5):367–82.
Yamagishi M, Fujikawa D, Watanabe T, Uchimaru K. HTLV-1-mediated epigenetic pathway to adult T-cell leukemia-lymphoma. Front Microbiol. 2018;9:1686.
Gazon H, Belrose G, Terol M, Meniane J-C, Mesnard J-M, Césaire R, et al. Impaired expression of DICER and some microRNAs in HBZ expressing cells from acute adult T-cell leukemia patients. Oncotarget. 2016;7(21):30258–75.
Kitagawa N, Ojima H, Shirakihara T, Shimizu H, Kokubu A, Urushidate T, et al. Downregulation of the microRNA biogenesis components and its association with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2013;104(5):543–51.
Kwon SY, Lee JH, Kim B, Park JW, Kwon TK, Kang SH, et al. Complexity in regulation of microRNA machinery components in invasive breast carcinoma. Pathol Oncol Res. 2014;20(3):697–705.
Pichler K, Schneider G, Grassmann R. MicroRNA miR-146a and further oncogenesis-related cellular microRNAs are dysregulated in HTLV-1-transformed T lymphocytes. Retrovirology. 2008;5(1):100.
Chen Y. Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucl Acids Res. 2020;48(D1):D127–D31.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14(1):128.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucl Acids Res. 2000;28(1):27–30.
Mozhgani S-H, Jahantigh HR, Rafatpanah H, Valizadeh N, Mohammadi A, Basharkhah S, et al. Interferon lambda family along with HTLV-1 proviral load, tax, and HBZ implicated in the pathogenesis of myelopathy/tropical spastic paraparesis. Neurodegenerative Diseases. 2018;18:150–5.
Mozhgani S-H, Piran M, Zarei-Ghobadi M, Jafari M, Jazayeri S-M, Mokhtari-Azad T, et al. An insight to HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) pathogenesis; evidence from high-throughput data integration and meta-analysis. Retrovirology. 2019;16(1):46.
Ruggero K, Guffanti A, Corradin A, Sharma VK, De Bellis G, Corti G, et al. Small noncoding RNAs in cells transformed by human T-cell leukemia virus type 1: a role for a tRNA fragment as a primer for reverse transcriptase. J Virol. 2014;88(7):3612–22.
Yamagishi M, Nakano K, Miyake A, Yamochi T, Kagami Y, Tsutsumi A, et al. Polycomb-mediated loss of miR-31 activates NIK-dependent NF-κB pathway in adult T cell leukemia and other cancers. Cancer Cell. 2012;21(1):121–35.
Yeung ML, Yasunaga J, Bennasser Y, Dusetti N, Harris D, Ahmad N, et al. Roles for microRNAs, miR-93 and miR-130b, and tumor protein 53-induced nuclear protein 1 tumor suppressor in cell growth dysregulation by human T-cell lymphotrophic virus 1. Cancer Res. 2008;68(21):8976–85.
Bellon M, Lepelletier Y, Hermine O, Nicot C. Deregulation of microRNA involved in hematopoiesis and the immune response in HTLV-I adult T-cell leukemia. Blood. 2009;113(20):4914–7.
Ramasamy A, Mondry A, Holmes CC, Altman DG. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLOS Medicine. 2008;5(9):e184.
Liu Y, Yin B, Zhang C, Zhou L, Fan J. Hsa-let-7a functions as a tumor suppressor in renal cell carcinoma cell lines by targeting c-myc. Biochem Biophys Res Commun. 2012;417(1):371–5.
Nakajima GO, Hayashi K, Xi Y, Kudo K, Uchida K, Takasaki KEN, et al. Non-coding MicroRNAs hsa-let-7g and hsa-miR-181b are Associated with Chemoresponse to S-1 in Colon Cancer. Cancer Genomics—Proteomics. 2006;3(5):317.
Lan F-F, Wang H, Chen Y-C, Chan C-Y, Ng SS, Li K, et al. Hsa-let-7g inhibits proliferation of hepatocellular carcinoma cells by downregulation of c-Myc and upregulation of p16INK4A. Int J Cancer. 2011;128(2):319–31.
Miller DM, Thomas SD, Islam A, Muench D. Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012;18(20):5546–53.
Li J, Liang Y, Lv H, Meng H, Xiong G, Guan X, et al. miR-26a and miR-26b inhibit esophageal squamous cancer cell proliferation through suppression of c-MYC pathway. Gene. 2017;625:1–9.
Liang S, He L, Zhao X, Miao Y, Gu Y, Guo C, et al. MicroRNA let-7f inhibits tumor invasion and metastasis by targeting MYH9 in human gastric cancer. PLOS ONE. 2011;6(4):e18409.
Dobson JR, Taipaleenmäki H, Hu Y-J, Hong D, van Wijnen AJ, Stein JL, et al. hsa-mir-30c promotes the invasive phenotype of metastatic breast cancer cells by targeting NOV/CCN3. Cancer Cell Int. 2014;14(1):73.
Agirre X, Jiménez-Velasco A, San José-Enériz E, Garate L, Bandrés E, Cordeu L, et al. Down-regulation of <em>hsa-miR-10a</em> in chronic myeloid leukemia CD34<sup>+</sup> Cells increases USF2-mediated cell growth. Mol Cancer Res. 2008;6(12):1830.
Veerla S, Lindgren D, Kvist A, Frigyesi A, Staaf J, Persson H, et al. MiRNA expression in urothelial carcinomas: Important roles of miR-10a, miR-222, miR-125b, miR-7 and miR-452 for tumor stage and metastasis, and frequent homozygous losses of miR-31. Int J Cancer. 2009;124(9):2236–42.
Hudson J, Duncavage E, Tamburrino A, Salerno P, Xi L, Raffeld M, et al. Overexpression of miR-10a and miR-375 and downregulation of YAP1 in medullary thyroid carcinoma. Exp Mol Pathol. 2013;95(1):62–7.
Wang R, Bao H, Zhang S, Li R, Chen L, Zhu Y. miR-186-5p promotes apoptosis by targeting IGF-1 in SH-SY5Y OGD/R model. Int J Biol Sci. 2018;14(13):1791–9.
Nascimento A, Valadão de Souza DR, Pessôa R, Pietrobon AJ, Nukui Y, Pereira J, et al. Global expression of noncoding RNome reveals dysregulation of small RNAs in patients with HTLV-1–associated adult T-cell leukemia: a pilot study. Infectious Agents Cancer. 2021;16(1):4.
Verzella D, Pescatore A, Capece D, Vecchiotti D, Ursini MV, Franzoso G, et al. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Disease. 2020;11(3):210.
Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, et al. Inhibition of JNK activation through NF-κB target genes. Nature. 2001;414(6861):313–7.
Bang Y-J, Kwon JH, Kang SH, Kim JW, Yang YC. Increased MAPK activity and MKP-1 overexpression in human gastric adenocarcinoma. Biochem Biophys Res Commun. 1998;250(1):43–7.
Wang J, Zhou J-Y, Wu GS. ERK-dependent MKP-1–mediated cisplatin resistance in human ovarian cancer cells. Cancer Res. 2007;67(24):11933–41.
Munkley J, Lafferty NP, Kalna G, Robson CN, Leung HY, Rajan P, et al. Androgen-regulation of the protein tyrosine phosphatase PTPRR activates ERK1/2 signalling in prostate cancer cells. BMC Cancer. 2015;15(1):1–11.
Su P, Lin Y, Huang R, Liao Y, Lee H, Wang H, et al. Epigenetic silencing of PTPRR activates MAPK signaling, promotes metastasis and serves as a biomarker of invasive cervical cancer. Oncogene. 2013;32(1):15–26.
Nucifora G, Laricchia-Robbio L, Senyuk V. EVI1 and hematopoietic disorders: history and perspectives. Gene. 2006;368:1–11.
Nanjundan M, Nakayama Y, Cheng KW, Lahad J, Liu J, Lu K, et al. Amplification of MDS1/EVI1 and EVI1, located in the 3q26.2 amplicon, is associated with favorable patient prognosis in ovarian cancer. Cancer Res. 2007;67(7):3074.
Boxus M, Twizere J-C, Legros S, Dewulf J-F, Kettmann R, Willems L. The HTLV-1 tax interactome. Retrovirology. 2008;5(1):76.
Xu X, Heidenreich O, Kitajima I, McGuire K, Li Q, Su B, et al. Constitutively activated JNK is associated with HTLV-1 mediated tumorigenesis. Oncogene. 1996;13(1):135–42.
Yin MJ, Christerson LB, Yamamoto Y, Kwak YT, Xu S, Mercurio F, et al. HTLV-I Tax protein binds to MEKK1 to stimulate IkappaB kinase activity and NF-kappaB activation. Cell. 1998;93(5):875–84.
De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, et al. Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature. 2001;414(6861):308–13.
Papa S, Bubici C, Zazzeroni F, Pham CG, Kuntzen C, Knabb JR, et al. The NF-κB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease. Cell Death Differen. 2006;13(5):712–29.
Nakachi S, Nakazato T, Ishikawa C, Kimura R, Mann DA, Senba M, et al. Human T-cell leukemia virus type 1 Tax transactivates the matrix metalloproteinase 7 gene via JunD/AP-1 signaling. Biochim Biophys Acta Mol Cell Res. 2011;1813(5):731–41.
Wagner EF, Nebreda ÁR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–49.
Acknowledgments
Research reported in this publication was supported by Elite Researcher Grant Committee under award number [977505] from the National Institutes for Medical Research Development (NIMAD), Tehran, Iran.
Funding
This work was funded and made possible by the grant provided by the Elite Researcher Grant Committee under award number [977505] from the National Institutes for Medical Research Development (NIMAD), Tehran, Iran
Author information
Authors and Affiliations
Contributions
SS, ND, MN, CRP, S-HM, and S-MJ conceptualized the study. SS, ND, S-HM, and MM performed the bioinformatics analysis. S-HM and SA conducted the statistical analysis. S-HM, S-MJ, SA, and AS wrote and edited the manuscript. ME, FH-S, CRP, and SP did the systematic search and literature review. MO and FA contributed with the blood samples. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interest
The authors declare that they have no competing interests.
Ethics approval and consent to participate
This work was approved by the Ethics Committee of Biomedical Research at Alborz University of Medical Sciences (IR.NIMAD.REC.1397.473)
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shahrzad Shadabi and Nargess Delrish are co-first authors of the paper.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shadabi, S., Delrish, N., Norouzi, M. et al. Comprehensive high-throughput meta-analysis of differentially expressed microRNAs in transcriptomic datasets reveals significant disruption of MAPK/JNK signal transduction pathway in Adult T-cell leukemia/lymphoma. Infect Agents Cancer 16, 49 (2021). https://doi.org/10.1186/s13027-021-00390-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13027-021-00390-3