
Abstract
A constant metabolism and energy supply are crucial to all organs, particularly the brain. Age-dependent neurodegenerative diseases, such as Parkinson’s disease (PD), are associated with alterations in cellular metabolism. These changes have been recognized as a novel hot topic that may provide new insights to help identify risk in the pre-symptomatic phase of the disease, understand disease pathogenesis, track disease progression, and determine critical endpoints. Nuclear receptor-related factor 1 (NURR1), an orphan member of the nuclear receptor superfamily of transcription factors, is a major risk factor in the pathogenesis of PD, and changes in NURR1 expression can have a detrimental effect on cellular metabolism. In this review, we discuss recent evidence that suggests a vital role of NURR1 in dopaminergic (DAergic) neuron development and the pathogenesis of PD. The association between NURR1 and cellular metabolic abnormalities and its implications for PD therapy have been further highlighted.
Similar content being viewed by others

Background
Parkinson’s disease (PD) is the second most common neurodegenerative disease, which has increased from 2.5 million cases in 1990 to over 6 million cases in 2016 [1, 2]. The number of people at risk for developing PD is predicted to rise to 14.2 million by 2040 [3]. Environmental factors in genetically predisposed individuals are thought to contribute to the pathogenesis of this multifaceted disease [4]. However, multiple mechanisms and pathway dysfunctions accelerate the pathogenesis of PD, including oxidative stress, mitochondrial dysfunction, cellular calcium (Ca2+) imbalance, neuroinflammation, and other neurotransmitter system deficits [5, 6]. The main neuropathological hallmarks of PD include the substantial loss of dopaminergic (DAergic) neurons within the pars compacta of the substantia nigra (SNpc), and the development of intracytoplasmic α-synuclein-containing Lewy bodies, resulting in diminished facilitation of voluntary movement [7,8,9].
Cellular metabolic status and mammalian gene expression interact under the critical regulation of various signaling pathways, transcription factors (TFs), and epigenetic remodelers that affect cell fate during development [10,11,12,13]. Each cell type in the brain has a distinct metabolic profile and sustained cellular function over time; however, only a limited degree of metabolic flexibility can react to external stimuli [14, 15]. Cellular metabolic changes that exceed an adaptability threshold will endanger brain cellular resistance and function. Metabolic alterations in protein, glucose, lipid, and dopamine (DA) have been noticed in PD [16,17,18]. Growing evidence demonstrates that PD patients have altered unsaturated fatty acids (FAs), bile acid, steroid hormones, glucose, and amino acid metabolisms [18,19,20,21]. Changes in DA-related metabolites in PD-derived midbrain DAergic neurons have recently been reported, along with a significant increase in the expression of the DA-related genes, such as phenylalanine hydroxylase, tyrosine hydroxylase (TH), catechol-O-methyltransferase, and monoamine oxidase A and B [17]. These findings suggest that exploring the metabolic abnormalities is imperative in understanding PD’s pathogenic mechanism.
TFs bind at a particular location and time to a specific gene sequence and regulate the expression of the target gene, allowing them to control the development processes of the cells. Nuclear receptor-related factor 1 (NURR1) is one of these TFs required for the differentiation, maturation, and maintenance of DAergic neurons during their development [22, 23]. In the present review, we first build a framework for NURR1 involvement in PD and then detail the participation of NURR1 in modulating metabolic states and individual metabolites to control the epigenetic landscape and cellular identity. Overall, understanding the roles of NURR1 in cellular metabolic abnormalities in PD could be crucial for developing timely and tolerable NURR1-targeting modalities for PD therapy.
Role of nuclear receptor-related factor 1 in dopaminergic neuron development and Parkinson’s disease
The nervous system is developed through the tremendous proliferation and differentiation of neural stem cells. Their fate is determined by precise signal patterning and TFs that allow for the correct regionalization, differentiation, and functional integration of new cells [24, 25]. NURR1, as a member of the “zinc finger” TF superfamily, is expressed early in embryogenesis at E10.5 and is detectable throughout the life [26, 27]. The nuclear receptor 4A (NR4A) subfamily is comprised of three nuclear receptors: NUR77 (NR4A1), NURR1 (NR4A2), and NOR1 (NR4A3). Although the deoxyribonucleic acid (DNA) binding domains in the three family members of NR4A have high similarity, their biological functions are quite different [28,29,30,31]. Many organs and tissues, including the brain, pancreas, liver, muscles, and fat, express the NR4A family members [31,32,33,34,35]. NURR1 is found in various central nervous system regions, including the cortex, hippocampus, brain stem, spinal cord, and olfactory bulb [36, 37]. The Nurr1-deficient (Nurr1−/−) mice are unable to develop midbrain DAergic neurons and die shortly after birth [38]. Those mice show impaired motor function and significant DAergic neuron loss in the SNpc and ventral tegmental area (VTA) [39]. The expression of NURR1 in the midbrain DAergic neurons decreases with age, which coincides with the increased morbidity of PD [40, 41].
NURR1 and its transcriptional targets were downregulated in DAergic neurons with a high level of the disease-causing protein α-synuclein in the midbrain [42]. In vitro, α-synuclein-induced activation of protein phosphatase 2A leads to inhibition of the phosphorylation activity of TH and aromatic amino acid decarboxylase (AADC) [43]. Furthermore, DA content is reduced in cells transfected with the A53T mutant α-synuclein [43, 44]. Previous research in the PD mouse model found that Nurr1 is co-localized with TH+ neurons in the SNpc, VTA, retrorubral field, and olfactory bulb [45]. NURR1 interacts with other factors to regulate the expression of TH, AADC, and vesicular monoamine transporter 2 (VMAT2), which are essential in the synthesis, storage, and release of DA [46,47,48,49]. There is a significant correlation between NURR1 activity and the stabilization of the NURR1 ligand-binding domain (LBD) [50]. Additionally, NURR1 is a critical factor for the specification of DA neurotransmitter identity by activating TH gene transcription [51, 52]. NURR1 mutations and polymorphisms that cause either reduced expression or dysfunction have been linked to familial and sporadic PD [53]. The determinant roles of NURR1 in the DAergic neuron genesis and PD development lead to an opportunity to develop novel therapeutics.
Cellular metabolism changes in Parkinson’s disease and potential roles of nuclear receptor-related factor 1
Neural cells are prone to cellular metabolic abnormalities due to their high specialization and reliance on metabolic balance. Here, we highlight NURR1’s role in the metabolic abnormalities associated with PD.
The impact of nuclear receptor-related factor 1 in α-synuclein-mediated dopamine cellular metabolism impairment
α-Synuclein is a 140 amino acid protein that is ubiquitously expressed in the brain, particularly throughout the neocortex, hippocampus, SNpc, thalamus, olfactory bulb, and cerebellum, where it plays a crucial role in synaptic function and plasticity [54,55,56,57]. α-Synuclein in presynaptic terminals is unfolded but misfolds under certain conditions [55, 58]. α-Synuclein is a well-established presynaptic protein [57, 59], and it is initially described as a nuclear protein [60, 61]. Various α-synuclein species have been identified in the nucleus of neuronal cells from the brains of PD patients [62,63,64] and in the animal and cellular models of PD [62, 65, 66]. Furthermore, α-synuclein significantly affects transcription and other cellular processes such as DNA integrity and the ER-Golgi system [67, 68]. In vitro, Outeiro et al. observed a reduction in toxicity of accumulated high molecular weight α-synuclein species after relocating them in the nucleus [69]. Thus, the α-synuclein nuclear localization, phosphorylation, and transcriptional regulation via DNA binding may be critical for cell homeostasis, division, and differentiation. Unni et al. also demonstrated that α-synuclein modulates DNA repair, suggesting that cytoplasmic α-synuclein aggregation may cause a loss of function, leading to increased DNA double-strand breaks and neuronal programmed cell death [70].
α-Synuclein is involved in several steps required to trigger exocytosis in the presynaptic terminal [57]. Under pathological conditions, toxic α-synuclein species trigger the dysregulation of several synaptic proteins, leading to functional impairment of the presynaptic terminal in the brains of animal models of PD and PD patients [71,72,73]. As previously stated, α-synuclein not only negatively modulates the activity of enzymes responsible for DA synthesis, but also impairs the transport and uptake of DA by altering the activity of VMAT2 and the DA transporter (DAT) [74,75,76,77,78]. The interaction of α-synuclein with surface DAT affects transporter function, which can change the synaptic availability of DA and substantially affect the membrane microenvironment near the transporter, which may impact DA neuron homeostasis [79]. These events may lead to a cycle of α-synuclein accumulation and dysregulated DA that leads to synaptopathy and neurodegeneration.
We have demonstrated that mutations in the first exon of NURR1 (−291Tdel and − 245 T → G) are linked to familial PD [53]. Furthermore, we found that NURR1 expression was significantly reduced in the PD postmortem SNpc tissues with α-synuclein inclusions compared to age-matched healthy controls (HC) [42]. NURR1 expression was normal in the SNpc neurons without inclusions and in the hippocampal neurons of PD patients, demonstrating that this change is region-specific [42]. A decrease in SNpc NURR1 expression was also observed in some progressive supranuclear palsy and Alzheimer’s disease patients [42], indicating that reducing NURR1 in DAergic neurons is linked to intracellular pathology in synucleinopathies and tauopathies. NURR1 expression in the SNpc of α-synuclein homozygous mice significantly decreased with age [80]. Similar findings were achieved when the α-synuclein preformed fibril was injected into the putamen of non-human primates [81]. Furthermore, NURR1 expression was decreased in PD patients’ peripheral blood lymphocytes (PBLs) compared to HC and neurological disease controls [82]. The reduction in NURR1 expression in PBLs suggests systemic involvement of NURR1 in PD, which might potentially be used to identify patients with PD associated with central DAergic system impairments [82].
In addition to the critical role in the developing and reprogramming DAergic neurons, NURR1 has been shown to preserve and protect DAergic neurons in several animal and cellular models of PD [81, 83,84,85]. Furthermore, bexarotene, a retinoid X receptor (RXR) ligand that forms heterodimers with NURR1, has been demonstrated to co-regulate NURR1 target genes, including the TH receptor component [86]. This is supported by the finding that the induction of Nurr1 expression in primary DAergic neurons expressing α-synuclein restores the dysregulated gene functions [86].
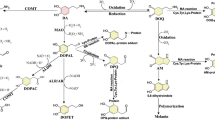
Our recent work has demonstrated that α-synuclein can modulate the transcription activity of the Nurr1 promoter region, between − 605 and − 418 bp, which contains the nuclear factor kappa B (NF-κB) binding site [87]. Furthermore, overexpression of α-synuclein (wild type or A53T mutant) reduces the NF-κB binding quantity to the Nurr1 promoter, resulting in decreased transcription of Nurr1 [87] and potentially inducing proteasome-dependent NURR1 degradation in the midbrain DAergic neurons [88]. Overexpression of α-synuclein inhibits NF-κB expression and increases glycogen synthase kinase 3β (GSK-3β) protein levels in the DAergic neurons, implying that the pathological effects may be mediated by the NF-κB signaling pathway [89]. Ji et al. found that prostaglandin E2 (PGE2) stimulation of E-type prostaglandin receptor 1 upregulates the expression of NURR1 via the activation of NF-κB signaling pathways [90], indicating that α-synuclein can suppress the expression of endogenous NURR1. Interestingly, NURR1 has a considerable inhibitory effect on α-synuclein transcription [91]. Therefore, NURR1 and α-synuclein may regulate each other (Fig. 1A).

NURR1 roles in the metabolism of α-synuclein, lipids, glucose, and mitochondria. Sharp arrows (positive regulation), rounded arrows (negative regulation) (A) α-Synuclein, and NURR1 have a detrimental impact on each other. α-Synuclein promotes inflammatory mediators and free radicals, and they, in turn, exacerbate α-synuclein accumulation, creating a vicious cycle, and NURR1 could interrupt this vicious cycle. B NURR1 activates GLUT4 transcription and induces genes involved in glucose and glycogen metabolism; Simultaneously, NURR1 expression could be inhibited by high glucose. Fasting and glucagon treatment induce Nurr1 expression (C) NURR1 in DAergic neurons positively regulates many nuclear-encoded mitochondrial genes and protects cells against the mitochondrial membrane and reactive oxygen species (D) Activating NURR1 promotes the oxidation of FAs, also up-regulates FABP5 expression. Furthermore, unsaturated FAs activate transcriptional function of NURR1. NURR1 expression and nuclear translocation are increased in response to a lipotoxic insult of palmitate. Abbreviations: NURR1: Nuclear receptor-related factor 1; FABP5: Fatty acid-binding protein 5; GLUT: Glucose transporter; Pygm: Phosphorylase glycogen muscle; Phka1: Phosphorylase kinase α 1; Pgam: Phosphoglycerate mutase 2; PGE2: Prostaglandin E2; NFR1,2: Nuclear respiratory factors 1 and 2; PGC-1α: Peroxisome proliferator-activated receptor-gamma coactivator1-alpha
The interplay of Ca2+, cytosolic DA (DAcyt), and α-synuclein contribute to the selective vulnerability of SNpc neurons in PD [92], and DAergic neurons in SNpc also depend on Ca2+ channel pacemaking [93, 94]. Therefore, various strategies for preventing neuronal death in PD can potentially be employed, including inhibiting Cav1.3 channel activity [95] and blocking the toxicity of DA-α-synuclein interactions [96]. Sulzer et al. found that levodopa (L-DOPA) increases DAcyt in the SNpc neurons 2 to 3-folds higher than VTA neurons. This response is dependent on dihydropyridine-sensitive L-type Ca2+ channels, resulting in greater susceptibility of SNpc neurons to L-DOPA-induced neurotoxicity [92]. Additionally, in hemiparkinsonian rats, Steece-Collier et al. demonstrated that genetic silencing of the striatal L-type Ca2+ channel prevented and reversed L-DOPA-induced dyskinesia (LID) [97]. Furthermore, Sellnow et al. found that ectopic induction of striatal NURR1 might induce LID behavior and associated neuropathology [98]. Therefore, NURR1 may play a crucial role in regulating the transcriptional and plasticity activities of Cav1.3 [98]. Cav1.3 activity, mediated by calcineurin, regulates NURR1 expression [99]. Moreover, NURR1 might be vital to modulate the interaction of DAcyt, Ca2+, and α-synuclein, thereby avoiding the selective death of SNpc neurons [92, 97,98,99,100]. Understanding this pathway may help identify drug targets and their future development to avoid neurotoxic and synaptic plasticity changes.
Neuroinflammation processes significantly contribute to PD pathogenesis. A meta-analysis [101] reported that a single-nucleotide variation within the human leukocyte antigen locus increases the risk of developing PD, implying an immune-related susceptibility. Furthermore, epidemiological studies [102] revealed a negative correlation between the incidence of PD and the use of anti-inflammatory medications, particularly nonsteroidal anti-inflammatory drugs, supporting the hypothesis that inflammation may promote underlying PD processes. α-Synuclein can activate microglia, produce inflammatory mediators, and trigger oxidative stress [103,104,105]. The generation of inflammatory mediators and increased levels of free radicals can exacerbate α-synuclein accumulation, producing a vicious cycle of continuous progression in PD pathogenesis (Fig. 1A) [105,106,107]. Microglia, astrocytes, and macrophages express Nurr1 mRNA and NURR1 protein under basal conditions and can elevate Nurr1 levels when activated [103, 108,109,110].
According to the report by Sajiao et al., NURR1 protects DAergic neurons from neurotoxicity and inflammation via inhibiting the expression of proinflammatory mediators in microglia and astrocytes (Fig. 1A) [111]. NURR1 is also a vital component of a negative feedback loop in microglia and astrocytes by recruiting a corepressor for element-1–silencing transcription factor to NF-κB target genes [111]. They reported that TH+ DAergic neurons’ survival rate decreased in response to inflammatory stimuli during Nurr1-deficiency [111]. Oh et al. found that NURR1 may exert its anti-inflammatory effects through modulating the expression of RAS guanyl-releasing protein 1 in lipopolysaccharide (LPS)-induced inflammatory cell model [112]. Enhancing NURR1 expression has been reported to reduce oxidative stress and protect DAergic neurons by decreasing apoptosis-related proteins and increasing antioxidant proteins [113, 114]. As a result, scavenging free radicals and regulating the generation of inflammatory mediators is one of the keys to preventing and delaying the progression of PD.
These findings imply that synuclein pathological changes may be driven by at least part of the cellular metabolic pathogenesis of PD. NURR1 is one of the targets directly or indirectly affected by the inclusion bodies’ pathological changes in PD. NURR1 may potentially protect DAergic neurons from α-synuclein in various ways, thereby delaying or blocking the progression of PD neuropathology.
Regulating effects of nuclear receptor-related factor 1 in the altered energy metabolism of Parkinson’s disease
The brain represents ~ 2% of the bodyweight of the average adult human, but it consumes 20% of the daily energy source [115, 116]. Given that one of the common features of PD pathogenesis is an energy deficit and decreased adenosine triphosphate (ATP) levels, oxidative phosphorylation and glycolysis could be critical pathological and therapeutic avenues for this neurodegenerative disease [117,118,119,120,121]. Neurons primarily rely on oxidative phosphorylation in mitochondria to meet such energy needs, with glycolysis accounting for a tiny portion of their energy supply [122]. Even though PD’s main metabolic pathological drive is from SNpc oxidative phosphorylation, there is increasing information on PD’s cortical brain metabolic pathology [123,124,125]. DA-deficiency in the SNpc causes an imbalance in both the indirect (inhibitory) and direct (activating) pathways of the cortico–basal ganglia–cortical circuit, which may lead to hypokinesia in PD [126,127,128,129]. Consequently, several imaging studies have demonstrated changes in brain metabolism in PD, and in the early stages of the disease, the cortex may exhibit a widespread low-metabolic state [130, 131]. Furthermore, lipoxidation, glycoxidation, and lipid peroxidation markers are elevated in the cerebral cortex of PD patients [132].
Human SNpc DAergic neurons have an exuberant and highly arborized axonal arborization, with upwards of a million neurotransmitter release sites per SNpc DAergic neuron [133, 134]. This characteristic can potentially inflict a significant bioenergetic load on these cells and subject them to a progressive elevation of oxidative stress [135, 136]. Additionally, the study by Giguère et al. found that SNpc DAergic neurons have 2-fold greater axonal arborization and are more susceptible to a 6-hydroxydopamine (6-OHDA) lesion in mice with the selective deletion of DA D2 receptor [137]. Several studies have implicated mitochondrial dysfunction, and cellular bioenergetic alterations as an underlying cause of PD [138, 139]. DAergic neurons demand more energy than other neuronal cell types [140, 141], rendering them more susceptible to mitochondrial dysfunction and, ultimately, cell death [142, 143]. Defects in mitochondrial respiration are corroborated by lower glucose consumption in PD patients [144] and lower pyruvate oxidation in PD patients’ fibroblasts [145], indicating lower acetyl-CoA entry into the tricarboxylic acid (TCA) cycle [144]. Mitochondrial respiration abnormalities may inhibit complex I nicotinamide adenine dinucleotide (NADH)-ubiquinone reductase of the electron transport chain (ETC), which might play a role in the pathogenesis of PD [146].
Role of nuclear receptor-related factor 1 in glucose metabolism of Parkinson’s disease
Glycolysis is one of the primary processes that glucose provides energy, and abnormal glucose metabolism is critical in PD’s molecular mechanism [147, 148]. Overexpression of α-synuclein along with paraquat exposure leads to increased glucose accumulation, impaired glycolysis activity, and mitochondrial respiration [149]. Glucose transporter (GLUT) inhibition prevents α-synuclein from potentiating paraquat toxicity. Furthermore, inhibition of the pentose phosphate pathway (PPP) protects against this synergistic toxicity [149]. Apart from its essential role in antioxidant defense and nucleic acid synthesis, PPP provides nicotinamide adenine dinucleotide phosphate for fatty acid and cholesterol biosynthesis [149,150,151].
Low expression of PPP enzymes and an inability to build antioxidant reserves are the early events in the development of sporadic PD, and mitochondrial damage in PD may be a direct result of PPP dysregulation [151]. α-Synuclein plays a vital part in altered glucose metabolism by PPP [151]. Glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme of PPP, is found throughout the body, and its expression and activity vary over a 10-fold range, with the highest level seen in the brain [152, 153]. The expression and activity of G6PD were increased when LPS was used in vitro and in vivo PD models, and this increase is linked to microglial activation and DAergic neurodegeneration [150]. G6PD knockdown or inhibition reduced the LPS-induced reactive oxygen species (ROS) over-production and NF-кB activation, thereby reducing microglial activation [150, 154]. These findings demonstrate that G6PD has a role in PPP dysfunction and neuroinflammation, leading to DAergic neurodegeneration.
Epidemiological evidence suggests a link between diabetes and PD, with hyperglycemia as one of the factors in neurodegeneration of the nigrostriatal pathway in PD [155, 156]. In addition to hyperglycemia, emerging evidence implies that insulin resistance in the brains of PD patients and impaired brain insulin signaling are potential contributors to PD pathogenesis [157]. In support of this, there is a downregulation of the insulin receptor in the SNpc and an increase in insulin resistance in patients with PD [158,159,160]. The activation of insulin signaling can modulate the degradation of α-synuclein and inhibit α-synuclein fibril formation by activating the insulin-degrading enzyme [161], which is supported by the fact that reversing insulin resistance can prevent the α-synuclein-induced toxicity [162]. In agreement with the effect of insulin signaling, postmortem analysis found that protein kinase B or Akt (PKB/AKT), a serine/threonine kinase, decreased in PD patients’ brains [163]. The inhibition of AKT signaling exaggerates DAergic cell death [164, 165], providing a further mechanistic link between impaired insulin signaling and PD. AKT phosphorylates NURR1 at Ser347, increasing protein stability [166]. Thus, the defective insulin signaling appears to be at the crux of insulin resistance and PD pathogenesis.
High glucose exposure in a mouse model of diabetes reduces NURR1 expression and nuclear translocation in Müller cells [167]. On the other hand, NURR1 agonists inhibit Müller cell activation and retinal ganglion cell loss [167, 168]. Furthermore, downregulation of NURR1 promotes high glucose-induced Müller cell activation by upregulating the NF-κB/Nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome axis [167, 169]. NURR1 agonists may have significant anti-inflammatory and neuroprotective effects on Müller cells in diabetic retinopathy [167]. Expression of NURR1 and GSK-3β are downregulated in the peripheral blood mononuclear cells (PBMCs) of type-2 diabetes (T2D) patients [168]. Furthermore, high levels of proinflammatory cytokines and low NR4A expression cause insulin resistance by inhibiting the expression of GLUT and the phosphorylation of insulin receptors [168]. Long-term insulin resistance contributes to hyperglycemia and hyperlipidemia, further downregulating NURR1 expression and resulting in a vicious cycle during T2D pathogenesis [168]. Furthermore, NLRP3 inflammasome activation increases in patients with T2D [170]. Therefore, the NURR1/NF-κB/NLRP3 inflammasome might be a potential pathway by which NURR1 regulates glucose metabolism, which is still an open field for research.
NURR1 activates GLUT4 transcription in skeletal muscle, and NURR1 overexpression strongly induces the expression of genes involved in glucose and glycogen metabolism, such as phosphorylase glycogen muscle, phosphorylase kinase α 1, and phosphoglycerate mutase (Fig. 1B) [171]. Nurr1 overexpression in skeletal muscle enhances glucose uptake, utilization, and storage. In contrast, fasting and glucagon treatment induces Nurr1 expression in hepatic cells [32, 172]. Treatment of the PBMCs with high glucose and palmitic acid inhibits NURR1 expression in a dose- and time-dependent manner [168]. Similarly, the NURR1 agonist, amodiaquine, enhanced glucose tolerance and restored insulin levels to normal in obese mice [32]. Although the underlying mechanisms are unknown, NURR1 plays a function in the physiological process of glucose metabolism that helps protect the DAergic neuron from the detrimental consequences of metabolic disturbances, thereby preventing cell death. In addition, it may promote searches to find novel therapeutic targets from a metabolic perspective.
Association of nuclear receptor-related factor 1 in mitochondrial dysfunction of Parkinson’s disease
It is well understood that mitochondria play a vital role in aerobic glycolysis. Mitochondria are the cells’ energy producers and are critical intercellular linkers with other organelles. Mitochondria control energy metabolism, biosynthesis, immunological response, and cell turnover by interacting with the endoplasmic reticulum, peroxisomes, and nucleus through signal transduction, vesicle transport, and membrane contact sites [173]. ETC is a critical component of mitochondrial energy production. During oxidative phosphorylation, NADH provided by the TCA cycle is oxidized and provides electrons to the ETC [174, 175]. Metabolic alterations and inactivation of the ETC complex are characteristics of PD; thus, poor energy metabolism is linked to PD [16, 149]. A large meta-analysis of genome-wide gene expression studies has reported that genes encoding oxidative phosphorylation proteins correspond to the functional category of most of the deregulated genes in the remaining DAergic neurons in PD [176]. NURR1 works with numerous genes associated with the DAergic neuron phenotype, including DA metabolism, neurotransmission, axonal development, mitochondrial function, and cell survival [177,178,179].
NURR1 regulates numerous nuclear-encoded mitochondrial genes positively, with over 90% of the genes creating down-regulated respiratory chains in Nurr1-ablated DAergic neurons [179]. It has been proposed that decreased NURR1 activity is linked to mitochondrial malfunction, which accelerates neurodegeneration in PD [180]. Furthermore, NURR1 regulates various proteins that play a role in mitochondrial functions, including pituitary homeobox 3 and Wnt/β-catenin, which regulate DAergic neurogenesis [22, 85, 181,182,183]. On the other hand, several studies have demonstrated that NURR1 protects cells by regulating mitochondrial genes such as sodium oxide dismutase 1 and mitochondrial translation elongation factor [177]. Recent research has also revealed that NURR1 could protect cells against the potential toxicity of mitochondrial membrane and intracellular ROS (Fig. 1C) [184].
Peroxisome proliferator-activated receptor-gamma coactivator1-alpha (PGC-1α) and nuclear respiratory factors 1 and 2 (NRF1 and NRF2) are fundamental transcriptional regulators of energy metabolism, acting as suppressors of ROS in neurons [185,186,187], as well as critical regulators of nuclear-encoded mitochondrial genes [187, 188]. Increased methylation of PGC-1α in the SNpc of α-synuclein mice can lead to decreased PGC-1α expression and mitochondrial content [189]. Previous research has shown that PGC-1α can be induced by parathyroid hormone, resulting in coactivation of the Nurr1 promoter activity in the osteoblast, suggesting another potential functional connection between NURR1 and PGC-1α, which may protect cells against the potential toxicity of oxidative stress derived from mitochondrial dysfunction [190].
Apart from fundamental roles in generating energy and the metabolism of lipids and amino acids, mitochondria is also a key player in maintaining Ca2+ homeostasis [191]. Identification of the molecular components of the mitochondrial Ca2+ uniport complex has provided crucial insight into the function mitochondrial Ca2+ influx plays in energy production under an increased workload and, paradoxically, in disease development, such as neurodegeneration [192, 193]. L-type Ca2+ channel activation is critical for spontaneous DAergic neuron pacemaking, which is then accompanied by sustained Ca2+ entry through L-type channels [194, 195]. Ca2+ entry via L-type channels increases mitochondrial oxidative stress, which is amplified by deglycase-1 gene deletion [196, 197]. Recent studies in PD zebrafish and drosophila models have shown that lowering high mitochondrial Ca2+ levels could improve neurodegeneration [198, 199]. To summarize, NURR1 may interact with other TFs essential for expressing nuclear respiratory genes, such as NRF1 and NRF2, or with the transcriptional coactivator PGC-1α, which serves as a master regulator of mitochondrial biogenesis and cellular respiration. Mitochondria and NURR1 metabolic involvement remain poorly understood; future studies are needed to determine the NURR1 metabolic pathways and the role of oxidative phosphorylation in NURR1-ablation and overexpression models.
Involvement of nuclear receptor-related factor 1 in the altered lipid metabolism of Parkinson’s disease
The human brain has the second-largest lipid content after adipose tissue. Lipids help maintain brain activities, such as synaptic function, making it highly vulnerable to lipid metabolic disorders [200, 201]. Lipids are involved in a multitude of aspects of PD pathology, including unique cytotoxic interactions with α-synuclein [202, 203], mutations in enzymes involved in lipid metabolism genes that increase the risk of PD development [204, 205], lipid pathway alterations [206, 207], and lipid involvement in oxidative stress and inflammation [208]. Disruption of the lipid membrane is one potential mechanism of cytotoxicity. Studies have shown that the toxicity of α-synuclein and docosahexaenoic acid (DHA) oligomers to cells is partially due to the disruption of the integrity of the lipid membrane [20, 202]. Mutations in glucocerebrosidase and sphingomyelinase 1 lead to a loss of glucocerebroside function and an increase in α-synuclein aggregation, subsequently augmenting the development of PD [209, 210]. Changes in membrane lipids have been observed in both affected and unaffected regions of PD patients’ brains and various experimental models of PD [211, 212], implying that changes in lipid metabolism or metabolic pathways may precede the development of PD. αSynuclein has some structural similarities with the class A2 lipoproteins and fatty acid-binding protein (FABP), which may play an important role in lipid metabolism [213,214,215].
Fatty tissue highly expresses NR4A members during the early stages of adipocyte differentiation [216]. Nurr1 is upregulated during extreme obesity and normalized after fat loss [217]. Furthermore, activated NURR1 can promote the oxidation of FAs to supply ATP, which could be regulated by PGE2, a critical transcriptional integrator that allows crosstalk between the PGE2 and FAs oxidation pathways [218]. Interestingly, Briand et al. found that palmitate lipotoxic insult increased NURR1 expression and nuclear translocation in the insulinoma cell line, Min6 [219], implying the involvement of NURR1 in at least some FAs induced transcriptional responses. These results contradict the previously mentioned study in which palmitic acid could reduce the expression of NURR1 in PBMCs from patients with T2D [168]. It is worth noting that Nurr1 overexpression in purified human islets decreased the expression of a cluster of genes that are involved in inflammation control [219]. Decreased NURR1 and GSK-3β phosphorylation expression levels in PBMCs were negatively correlated with interleukin-6 and tumor necrosis factor-α levels [168]; whether this downregulation results from a long-term adaptive and protective response to glucose homeostasis remains to be determined by studying tissue-specific gene knockouts. Furthermore, the unsaturated FAs can directly bind to NURR1 and activate its transcriptional function (Fig. 1D) [220, 221].
FABP, also known as intracellular lipid chaperon, may dictate the destiny of lipids that coordinate lipid trafficking and signaling and are intimately linked to metabolic and inflammatory pathways [222]. FABP5 and NURR1 are expressed in the mouse brain; however, they are not co-localized in basal conditions and are both induced in response to stress stimuli such as brain injury, seizure, or inflammation [110, 223,224,225]. In HEK293 cells, NURR1 increases retinoic acid levels by upregulating FABP5-induced signaling of peroxisome proliferator-activated receptors and activating DHA-induced RXR [223]. All these findings suggest that NURR1 can influence the signaling of other nuclear receptors by regulating the expression levels of FABP5. Moreover, in vivo and in vitro, HX600, a synthetic agonist of NURR1/RXR, can reduce microglia-expressed proinflammatory mediators and prevent inflammation-induced cell death [226]. As a result, NURR1/RXR may play a dual role in PD, providing both neuroprotection from inflammation and symptomatic relief through upregulation of TH, AADC, and guanosine-5′-triphosphate cyclohydrolase I transcription and an increase in striatal DA level [227].
Clinical and experimental evidence suggests that steroid hormones, such as estrogen [228], progesterone [229], and thyroid pituitary axis hormones [230, 231], have a role in the pathogenesis of PD. Additionally, NURR1 regulates the synthesis of hormones, including aldosterone in the adrenal cortex [232, 233], osteocalcin in osteoblasts [234], and lactotropes in the female pituitary [235]. Although previous research has suggested that NURR1 may regulate some hormone metabolisms, it is unclear if NURR1’s role in hormone metabolism impacts PD. Understanding the molecular mechanisms of their involvements in PD would enable researchers to better explore the pathometabolic processes and signalings, which may elucidate tailored therapeutic targets for this devastating disease.
Nuclear receptor-related factor 1-targeting therapy for Parkinson’s disease
Despite the extensive research breakthroughs, the current treatments for PD are primarily symptomatic relief, and there are no therapies available that can prevent or delay disease progression. It will be a big challenge to develop disease-modifying and mechanism-based approaches, although several preclinical investigations of targeted molecular therapeutics, for example, have been conducted with encouraging findings [236, 237].
A growing body of evidence from in vitro and in vivo studies has demonstrated that NURR1-activating compounds, NURR1 agonists, and Nurr1 gene therapy can enhance DA neurotransmission and inhibit the microglial and astrocytic production of neurotoxic mediators [111, 184, 238], thereby protecting DAergic neurons from cell injury [238,239,240]. Using cell-based assays, Kim et al. found that three NURR1 agonist compounds among food and drug administration-approved drugs sharing an identical chemical scaffold targeting the NURR1 LBD can be exploited as a potential mechanism-based neuroprotective therapy for PD [84]. Importantly, these compounds significantly alleviate behavioral abnormalities in the lesioned 6-OHDA rat model of PD without any inducing symptoms of dyskinesia-like behavior [84].
Moreover, NURR1 modulators targeting RXR and the Wnt/β-catenin pathway may enhance the effects of NURR1-based therapies in PD [86, 241,242,243,244]. In a subacute mouse model of 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine hydrochloride-induced PD, the herbal extract consisting of Bupleuri Radix, Moutan Cortex Radicis, and Angelica Dahuricae resulted in recovery from movement impairment [245]. This herbal extract is shown to upregulate NURR1 expression and consequently increase DA level, DAergic neurons, and fibers in the nigrostriatal projection [245].
Although the implications of NURR1 in PD treatment have not yet been thoroughly evaluated, identifying its molecular mechanisms in DAergic neuron development and cellular metabolic function may eventually help to develop individualized treatments aiming at the restoration of functional integrity of disease-specific brain pathology and reverse the decline of DAergic function in PD. Another promising strategy is identifying selective and safe NURR1 agonists to support DAergic neuron functions and reduce neuroinflammatory activity.
Conclusion and perspective
Even though research on the involvement of NURR1 in DAergic neurons began more than 20 years ago, a recent surge of evidence indicates that it plays a critical role in embryonic development and cellular metabolism. The altered metabolic state of PD patients may result in the downregulation of NURR1 expression, which increases the deposition of α-synuclein, and promotes the formation of abnormal cellular metabolism, thereby culminating in a vicious circle. The restoration or enhancement of NURR1 expression and function may disrupt this cycle, prevent cellular metabolic disorders, and delay the progression of PD. NURR1-related developmental and cellular metabolism modulation may provide crucial new therapeutic insight for PD. At the current stage of PD research, the exact mechanism of NURR1 in neuronal development, cellular metabolic disorders, and PD pathogenesis is still not fully understood, and future studies to clarify this are required.
Availability of data and materials
Not applicable.
Abbreviations
- PD:
-
Parkinson’s disease
- DA:
-
Dopamine
- DAergic:
-
Dopaminergic
- SNpc:
-
Compacta of the substantia nigra
- Ca2+ :
-
Calcium
- TFs:
-
Transcription factors
- NURR1:
-
Nuclear receptor-related factor 1
- TH:
-
Tyrosine hydroxylase
- AADC:
-
Aromatic amino acid decarboxylase
- 6-OHDA:
-
6-hydroxydopamine
- VMAT2:
-
Vesicular monoamine transporter 2
- DAT:
-
DA transporter
- NR4A:
-
Nuclear Receptor 4A
- DNA:
-
Deoxyribonucleic acid
- LBD:
-
Ligand-binding domain
- VTA:
-
Ventral tegmental area
- NF-κB:
-
Nuclear factor-kappa B
- GSK-3β:
-
Glycogen synthase kinase 3β
- DAcyt:
-
Cytosolic DA
- L-DOPA:
-
Levodopa
- LID:
-
L-DOPA-induced dyskinesia
- LPS:
-
Lipopolysaccharide
- ATP:
-
Adenosine triphosphate
- PBMCs:
-
Peripheral blood mononuclear cells
- HC:
-
Healthy control
- RXR:
-
Retinoid X receptor
- PGE2 :
-
Prostaglandin E2
- GLUT:
-
Glucose transporter
- PPP:
-
Pentose phosphate pathway
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- ROS:
-
Reactive oxygen species
- T2D:
-
Type-2 diabetes
- NLRP3:
-
Nucleotide-binding oligomerization domain-like receptor protein 3
- ATP:
-
Adenosine-5′-triphosphate
- ETC:
-
Electron transport chain
- NADH:
-
Nicotinamide adenine dinucleotide
- PGC-1α:
-
Peroxisome proliferator-activated receptor-gamma coactivator1-alpha
- NRF1 and NRF2:
-
Nuclear respiratory factors 1 and 2
- DHA:
-
Docosahexaenoic acid
- FAs:
-
Fatty acids
- TCA:
-
Tricarboxylic acid
- FABP5:
-
Fatty acid binding protein 5
References
GBD 2016 Parkinson's Disease Collaborators. Global, regional, and national burden of Parkinson's disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17:939–53.
Nussbaum RL, Ellis CE. Alzheimer's disease and Parkinson's disease. New Engl J Med. 2003;348:1356–64.
Dorsey ER, Bloem BR. The Parkinson Pandemic-A Call to Action. JAMA Neurol. 2018;75:9–10.
Warner TT, Schapira AH. Genetic and environmental factors in the cause of Parkinson's disease. Ann Neurol. 2003;53(Suppl 3):S16–23 discussion S23–15.
Cannon JR, Greenamyre JT. Gene-environment interactions in Parkinson's disease: specific evidence in humans and mammalian models. Neurobiol Dis. 2013;57:38–46.
Kalia LV, Lang AE. Parkinson's disease. Lancet (London, England). 2015;386:896–912.
Alexander GE. Biology of Parkinson's disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dial Clin Neurosci. 2004;6:259–80.
Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909.
Braak H, Del Tredici K. Invited Article: Nervous system pathology in sporadic Parkinson disease. Neurology. 2008;70:1916–25.
Enzo E, Santinon G, Pocaterra A, Aragona M, Bresolin S, Forcato M, et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. Embo J. 2015;34:1349–70.
Sindhu C, Samavarchi-Tehrani P, Meissner A. Transcription factor-mediated epigenetic reprogramming. J Biol Chem. 2012;287:30922–31.
Gurdon JB. Cell fate determination by transcription factors. Curr Top Dev Biol. 2016;116:445–54.
Tian L, Al-Nusaif M, Chen X, Li S, Le W. Roles of transcription factors in the development and reprogramming of the dopaminergic neurons. Int J Mole Sci. 2022;23(2):845.
Neves A, Costalat R, Pellerin L. Determinants of brain cell metabolic phenotypes and energy substrate utilization unraveled with a modeling approach. PLoS Comput Biol. 2012;8:e1002686.
Smith RL, Soeters MR, Wüst RCI, Houtkooper RH. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr Rev. 2018;39:489–517.
Anandhan A, Jacome M, Lei S, Hernandez-Franco P, Pappa A, Panayiotidis M, et al. Metabolic Dysfunction in Parkinson's Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. Brain Res Bull. 2017;133:12–30.
Ren Y, Jiang H, Pu J, Li L, Wu J, Yan Y, et al. Molecular features of parkinson's disease in patient-derived midbrain dopaminergic neurons. Movement Disord. 2022;37:70–9.
Xicoy H, Wieringa B, GJM M. The role of Lipids in Parkinson's disease. Cells. 2019:8.
Shao Y, Li T, Liu Z, Wang X, Xu X, Li S, et al. Comprehensive metabolic profiling of Parkinson's disease by liquid chromatography-mass spectrometry. Mole Neurodegeneration. 2021;16:4.
Alecu I, Bennett SAL. Dysregulated Lipid metabolism and its role in α-synucleinopathy in Parkinson's Disease. Front Neurosci. 2019;13:328.
Marques A, Dutheil F, Durand E, Rieu I, Mulliez A, Fantini ML, et al. Glucose dysregulation in Parkinson's disease: Too much glucose or not enough insulin? Parkinsonism Related Disord. 2018;55:122–7.
Saucedo-Cardenas O, Quintana-Hau JD, Le WD, Smidt MP, Cox JJ, De Mayo F, et al. Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proc National Acad Sci U.S.A. 1998;95:4013–8.
Sacchetti P, Carpentier R, Ségard P, Olivé-Cren C, Lefebvre P. Multiple signaling pathways regulate the transcriptional activity of the orphan nuclear receptor NURR1. Nucleic Acids Res. 2006;34:5515–27.
Rifes P, Isaksson M, Rathore GS, Aldrin-Kirk P, Møller OK, Barzaghi G, et al. Modeling neural tube development by differentiation of human embryonic stem cells in a microfluidic WNT gradient. Nat Biotechnol. 2020;38:1265–73.
Wen S, Li H, Liu J. Dynamic signaling for neural stem cell fate determination. Cell Adh Migr. 2009;3:107–17.
Hegarty SV, Sullivan AM, O'Keeffe GW. Midbrain dopaminergic neurons: a review of the molecular circuitry that regulates their development. Dev Biol. 2013;379:123–38.
Alavian KN, Jeddi S, Naghipour SI, Nabili P, Licznerski P, Tierney TS. The lifelong maintenance of mesencephalic dopaminergic neurons by Nurr1 and engrailed. J Biomed Sci. 2014;21:27.
Zárraga-Granados G, Muciño-Hernández G, Sánchez-Carbente M, Villamizar-Gálvez W, Peñas-Rincón A, Arredondo C, et al. The nuclear receptor NR4A1 is regulated by SUMO modification to induce autophagic cell death. PloS one. 2020;15:e0222072.
Torii T, Kawarai T, Nakamura S, Kawakami H. Organization of the human orphan nuclear receptor Nurr1 gene. Gene. 1999;230:225–32.
Hisaoka M, Ishida T, Imamura T, Hashimoto H. TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2004;40:325–8.
Kanzleiter T, Schneider T, Walter I, Bolze F, Eickhorst C, Heldmaier G, et al. Evidence for Nr4a1 as a cold-induced effector of brown fat thermogenesis. Physiol Genom. 2005;24:37–44.
Amoasii L, Sanchez-Ortiz E, Fujikawa T, Elmquist J, Bassel-Duby R, Olson E. NURR1 activation in skeletal muscle controls systemic energy homeostasis. Proc Natl Acad Sci U S A. 2019;116:11299–308.
Zhao Y, Bruemmer D. NR4A orphan nuclear receptors: transcriptional regulators of gene expression in metabolism and vascular biology. Arterioscler Thromb Vasc Biol. 2010;30:1535–41.
Martínez-González J, Cañes L, Alonso J, Ballester-Servera C, Rodríguez-Sinovas A, Corrales I, et al. NR4A3. 2021;22.
Safe S, Jin UH, Morpurgo B, Abudayyeh A, Singh M, Tjalkens RB. Nuclear receptor 4A (NR4A) family - orphans no more. J Steroid Biochem Mole Biol. 2016;157:48–60.
Zetterström RH, Williams R, Perlmann T, Olson L. Cellular expression of the immediate early transcription factors Nurr1 and NGFI-B suggests a gene regulatory role in several brain regions including the nigrostriatal dopamine system. Brain Res Mole Brain Res. 1996;41:111–20.
Li Y, Cong B, Ma C, Qi Q, Fu L, Zhang G, et al. Expression of Nurr1 during rat brain and spinal cord development. Neurosc Letters. 2011;488:49–54.
Zetterström RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science (New York, NY). 1997;276:248–50.
Le W, Conneely OM, Zou L, He Y, Saucedo-Cardenas O, Jankovic J, et al. Selective agenesis of mesencephalic dopaminergic neurons in Nurr1-deficient mice. Exp Neurol. 1999;159:451–8.
Chu Y, Kompoliti K, Cochran EJ, Mufson EJ, Kordower JH. Age-related decreases in Nurr1 immunoreactivity in the human substantia nigra. J Comparative Neurol. 2002;450:203–14.
Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, et al. Incidence of Parkinson's disease: variation by age, gender, and race/ethnicity. Am J Epidemiol. 2003;157:1015–22.
Chu Y, Le W, Kompoliti K, Jankovic J, Mufson EJ, Kordower JH. Nurr1 in Parkinson's disease and related disorders. J Comparative Neurol. 2006;494:495–514.
Tehranian R, Montoya SE, Van Laar AD, Hastings TG, Perez RG. Alpha-synuclein inhibits aromatic amino acid decarboxylase activity in dopaminergic cells. J Neurochem. 2006;99:1188–96.
Baptista MJ, O'Farrell C, Daya S, Ahmad R, Miller DW, Hardy J, et al. Co-ordinate transcriptional regulation of dopamine synthesis genes by alpha-synuclein in human neuroblastoma cell lines. J Neurochem. 2003;85:957–68.
Bäckman C, Perlmann T, Wallén A, Hoffer BJ, Morales M. A selective group of dopaminergic neurons express Nurr1 in the adult mouse brain. Brain Res. 1999;851:125–32.
Eells JB, Misler JA, Nikodem VM. Reduced tyrosine hydroxylase and GTP cyclohydrolase mRNA expression, tyrosine hydroxylase activity, and associated neurochemical alterations in Nurr1-null heterozygous mice. Brain Res Bull. 2006;70:186–95.
Chen XX, Qian Y, Wang XP, Tang ZW, Xu JT, Lin H, et al. Nurr1 promotes neurogenesis of dopaminergic neuron and represses inflammatory factors in the transwell coculture system of neural stem cells and microglia. CNS Neurosci Therapeut. 2018;24:790–800.
Kadkhodaei B, Ito T, Joodmardi E, Mattsson B, Rouillard C, Carta M, et al. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J Neurosci. 2009;29:15923–32.
Jankovic J, Chen S, Le WD. The role of Nurr1 in the development of dopaminergic neurons and Parkinson's disease. Progress Neurobiol. 2005;77:128–38.
Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, Liu X, et al. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature. 2003;423:555–60.
Kim KS, Kim CH, Hwang DY, Seo H, Chung S, Hong SJ, et al. Orphan nuclear receptor Nurr1 directly transactivates the promoter activity of the tyrosine hydroxylase gene in a cell-specific manner. J Neurochem. 2003;85:622–34.
Kim TE, Seo JS, Yang JW, Kim MW, Kausar R, Joe E, et al. Nurr1 represses tyrosine hydroxylase expression via SIRT1 in human neural stem cells. PloS one. 2013;8:e71469.
Le WD, Xu P, Jankovic J, Jiang H, Appel SH, Smith RG, et al. Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet. 2003;33:85–9.
Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci U S A. 2008;105:19666–71.
Wang C, Zhao C, Li D, Tian Z, Lai Y, Diao J, et al. Versatile Structures of α-Synuclein. Front Mole Neurosci. 2016;9:48.
Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48.
Burré J. The Synaptic Function of α-Synuclein. J Parkinson's Dis. 2015;5:699–713.
Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D. In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J Biol Chem. 2013;288:6371–85.
Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron. 2013;79:1044–66.
Mori F, Tanji K, Yoshimoto M, Takahashi H, Wakabayashi K. Immunohistochemical comparison of alpha- and beta-synuclein in adult rat central nervous system. Brain Res. 2002;941:118–26.
Yu S, Li X, Liu G, Han J, Zhang C, Li Y, et al. Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience. 2007;145:539–55.
Siddiqui A, Chinta SJ, Mallajosyula JK, Rajagopolan S, Hanson I, Rane A, et al. Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: implications for Parkinson's disease. Free Radical Biol Med. 2012;53:993–1003.
Garcia-Esparcia P, Hernández-Ortega K, Koneti A, Gil L, Delgado-Morales R, Castaño E, et al. Altered machinery of protein synthesis is region- and stage-dependent and is associated with α-synuclein oligomers in Parkinson's disease. Acta Neuropathol Commu. 2015;3:76.
Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–15.
Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, et al. Formation of toxic oligomeric alpha-synuclein species in living cells. PloS one. 2008;3:e1867.
Zhong SC, Luo X, Chen XS, Cai QY, Liu J, Chen XH, et al. Expression and subcellular location of alpha-synuclein during mouse-embryonic development. Cell Mole Neurobiol. 2010;30:469–82.
Paiva I, Pinho R, Pavlou MA, Hennion M, Wales P, Schütz AL, et al. Sodium butyrate rescues dopaminergic cells from alpha-synuclein-induced transcriptional deregulation and DNA damage. Human Mole Genet. 2017;26:2231–46.
Paiva I, Jain G, Lázaro DF, Jerčić KG, Hentrich T, Kerimoglu C, et al. Alpha-synuclein deregulates the expression of COL4A2 and impairs ER-Golgi function. Neurobiol Dis. 2018;119:121–35.
Pinho R, Paiva I, Jercic KG, Fonseca-Ornelas L, Gerhardt E, Fahlbusch C, et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Human Mole Genet. 2019;28:31–50.
Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham CM, Boutros SW, et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep. 2019;9:10919.
Yavich L, Tanila H, Vepsäläinen S, Jäkälä P. Role of alpha-synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24:11165–70.
Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–95.
Gaugler MN, Genc O, Bobela W, Mohanna S, Ardah MT, El-Agnaf OM, et al. Nigrostriatal overabundance of α-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012;123:653–69.
Lotharius J, Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–42.
Wersinger C, Sidhu A. Attenuation of dopamine transporter activity by alpha-synuclein. Neuroscience Letters. 2003;340:189–92.
Guo JT, Chen AQ, Kong Q, Zhu H, Ma CM, Qin C. Inhibition of vesicular monoamine transporter-2 activity in alpha-synuclein stably transfected SH-SY5Y cells. Cell Mole Neurobiol. 2008;28:35–47.
Bridi JC, Hirth F. Mechanisms of α-Synuclein Induced Synaptopathy in Parkinson's Disease. Front Neurosci. 2018;12:80.
Bellucci A, Navarria L, Falarti E, Zaltieri M, Bono F, Collo G, et al. Redistribution of DAT/α-synuclein complexes visualized by "in situ" proximity ligation assay in transgenic mice modelling early Parkinson's disease. PloS One. 2011;6:e27959.
Swant J, Goodwin JS, North A, Ali AA, Gamble-George J, Chirwa S, et al. α-Synuclein stimulates a dopamine transporter-dependent chloride current and modulates the activity of the transporter. J Biol Chem. 2011;286:43933–43.
Argyrofthalmidou M, Spathis AD, Maniati M, Poula A, Katsianou MA, Sotiriou E, et al. Nurr1 repression mediates cardinal features of Parkinson's disease in α-synuclein transgenic mice. Human Mole Genet. 2021;30:1469–83.
Chu Y, Muller S, Tavares A, Barret O, Alagille D, Seibyl J, et al. Intrastriatal alpha-synuclein fibrils in monkeys: spreading, imaging and neuropathological changes. Brain. 2019;142:3565–79.
Le W, Pan T, Huang M, Xu P, Xie W, Zhu W, et al. Decreased NURR1 gene expression in patients with Parkinson's disease. J Neurol Sci. 2008;273:29–33.
Hammond SL, Safe S, Tjalkens RB. A novel synthetic activator of Nurr1 induces dopaminergic gene expression and protects against 6-hydroxydopamine neurotoxicity in vitro. Neuroscience Letters. 2015;607:83–9.
Kim CH, Han BS, Moon J, Kim DJ, Shin J, Rajan S, et al. Nuclear receptor Nurr1 agonists enhance its dual functions and improve behavioral deficits in an animal model of Parkinson's disease. Proc Natl Acad Sci U S A. 2015;112:8756–61.
Paliga D, Raudzus F, Leppla S, Heumann R, Neumann S. Lethal Factor Domain-Mediated Delivery of Nurr1 Transcription Factor Enhances Tyrosine Hydroxylase Activity and Protects from Neurotoxin-Induced Degeneration of Dopaminergic Cells. Mole Neurobiol. 2019;56:3393–403.
Volakakis N, Tiklova K, Decressac M, Papathanou M, Mattsson B, Gillberg L, et al. Nurr1 and Retinoid X Receptor Ligands Stimulate Ret Signaling in Dopamine Neurons and Can Alleviate α-Synuclein Disrupted Gene Expression. J Neurosci. 2015;35:14370–85.
Jia C, Qi H, Cheng C, Wu X, Yang Z, Cai H, et al. α-Synuclein Negatively Regulates Nurr1 Expression Through NF-κB-Related Mechanism. Front Mole Neurosci. 2020;13:64.
Lin X, Parisiadou L, Sgobio C, Liu G, Yu J, Sun L, et al. Conditional expression of Parkinson's disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci. 2012;32:9248–64.
Yuan Y, Jin J, Yang B, Zhang W, Hu J, Zhang Y, et al. Overexpressed alpha-synuclein regulated the nuclear factor-kappaB signal pathway. Cell Mole Neurobiol. 2008;28:21–33.
Ji R, Sanchez CM, Chou CL, Chen XB, Woodward DF, Regan JW. Prostanoid EP1 receptors mediate up-regulation of the orphan nuclear receptor Nurr1 by cAMP-independent activation of protein kinase A, CREB and NF-κB. Br J Pharmacol. 2012;166:1033–46.
Yang Y, Latchman D. Nurr1 transcriptionally regulates the expression of alpha-synuclein. Neuroreport. 2008;19:867–71.
Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–29.
Ortner NJ. Voltage-Gated Ca (2+) Channels in Dopaminergic Substantia Nigra Neurons: Therapeutic Targets for Neuroprotection in Parkinson's Disease? Front Synaptic Neurosci. 2021;13:636103.
Putzier I, Kullmann PH, Horn JP, Levitan ES. Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J Neurosci. 2009;29:15414–9.
Surmeier DJ. Calcium, ageing, and neuronal vulnerability in Parkinson's disease. Lancet Neurol. 2007;6:933–8.
Sulzer D. alpha-synuclein and cytosolic dopamine: stabilizing a bad situation. Nat med. 2001;7:1280–2.
Steece-Collier K, Stancati JA, Collier NJ, Sandoval IM, Mercado NM, Sortwell CE, et al. Genetic silencing of striatal CaV1.3 prevents and ameliorates levodopa dyskinesia. Move Disord. 2019;34:697–707.
Sellnow RC, Steece-Collier K, Altwal F, Sandoval IM, Kordower JH, Collier TJ, et al. Striatal Nurr1 Facilitates the Dyskinetic State and Exacerbates Levodopa-Induced Dyskinesia in a Rat Model of Parkinson's Disease. J Neurosci. 2020;40:3675–91.
Tokuoka H, Hatanaka T, Metzger D, Ichinose H. Nurr1 expression is regulated by voltage-dependent calcium channels and calcineurin in cultured hippocampal neurons. Neurosci Letters. 2014;559:50–5.
Steece-Collier K, Collier TJ, Lipton JW, Stancati JA, Winn ME, Cole-Strauss A, et al. Striatal Nurr1, but not FosB expression links a levodopa-induced dyskinesia phenotype to genotype in Fisher 344 vs. Lewis hemiparkinsonian rats. Exp Neurol. 2020;330:113327.
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. 2014;46:989–93.
Noyce AJ, Bestwick JP, Silveira-Moriyama L, Hawkes CH, Giovannoni G, Lees AJ, et al. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol. 2012;72:893–901.
Shao QH, Yan WF, Zhang Z, Ma KL, Peng SY, Cao YL, et al. Nurr1: A vital participant in the TLR4-NF-κB signal pathway stimulated by α-synuclein in BV-2 cells. Neuropharmacology. 2019;144:388–99.
Chatterjee K, Roy A, Banerjee R, Choudhury S, Mondal B, Halder S, et al. Inflammasome and α-synuclein in Parkinson's disease: a cross-sectional study. J Neuroimmunol. 2020;338:577089.
Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, et al. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–10.
Gruden M, Yanamandra K, Kucheryanu V, Bocharova O, Sherstnev V, Morozova-Roche L, et al. Correlation between protective immunity to α-synuclein aggregates, oxidative stress and inflammation. Neuroimmunomodulation. 2012;19:334–42.
La Vitola P, Balducci C, Baroni M, Artioli L, Santamaria G, Castiglioni M, et al. Peripheral inflammation exacerbates α-synuclein toxicity and neuropathology in Parkinson's models. Neuropathol Appl Neurobiol. 2021;47:43–60.
Fan X, Luo G, Ming M, Pu P, Li L, Yang D, et al. Nurr1 expression and its modulation in microglia. Neuroimmunomodulation. 2009;16:162–70.
Barish GD, Downes M, Alaynick WA, Yu RT, Ocampo CB, Bookout AL, et al. A Nuclear Receptor Atlas: macrophage activation. Mole Endocrinol (Baltimore, Md). 2005;19:2466–77.
Pei L, Castrillo A, Chen M, Hoffmann A, Tontonoz P. Induction of NR4A orphan nuclear receptor expression in macrophages in response to inflammatory stimuli. J Biol Chem. 2005;280:29256–62.
Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59.
Oh M, Kim SY, Gil JE, Byun JS, Cha DW, Ku B, et al. Nurr1 performs its anti-inflammatory function by regulating RasGRP1 expression in neuro-inflammation. Sci Rep. 2020;10:10755.
Sun C, Wang Y, Mo M, Song C, Wang X, Chen S, et al. Minocycline protects against rotenone-induced neurotoxicity correlating with upregulation of Nurr1 in a Parkinson's disease rat model. Biomed Res Int. 2019;2019:6843265.
Sousa KM, Mira H, Hall AC, Jansson-Sjöstrand L, Kusakabe M, Arenas E. Microarray analyses support a role for Nurr1 in resistance to oxidative stress and neuronal differentiation in neural stem cells. Stem cells (Dayton, Ohio). 2007;25:511–9.
Yellen G. Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J Cell Biol. 2018;217:2235–46.
Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–58.
Morais VA, Haddad D, Craessaerts K, De Bock PJ, Swerts J, Vilain S, et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science (New York, NY). 2014;344:203–7.
Muddapu VR, Chakravarthy VS. Influence of energy deficiency on the subcellular processes of Substantia Nigra Pars Compacta cell for understanding Parkinsonian neurodegeneration. Sci Rep. 2021;11:1754.
Uittenbogaard M, Chiaramello A. Mitochondrial biogenesis: a therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr Pharm Des. 2014;20:5574–93.
Bell SM, Burgess T, Lee J, Blackburn DJ, Allen SP, Mortiboys H. Peripheral Glycolysis in Neurodegenerative Diseases. Int J Mole Sci. 2020;21(23):8924.
Cai R, Zhang Y, Simmering JE, Schultz JL, Li Y, Fernandez-Carasa I, et al. Enhancing glycolysis attenuates Parkinson's disease progression in models and clinical databases. J Clin Invest. 2019;129:4539–49.
Bolaños JP, Almeida A, Moncada S. Glycolysis: a bioenergetic or a survival pathway? Trends Biochem Sci. 2010;35:145–9.
Eberling JL, Richardson BC, Reed BR, Wolfe N, Jagust WJ. Cortical glucose metabolism in Parkinson's disease without dementia. Neurobiol Aging. 1994;15:329–35.
Ferrer I. Early involvement of the cerebral cortex in Parkinson's disease: convergence of multiple metabolic defects. Progress Neurobiol. 2009;88:89–103.
Han R, Liang J, Zhou B. Glucose Metabolic Dysfunction in Neurodegenerative Diseases-New Mechanistic Insights and the Potential of Hypoxia as a Prospective Therapy Targeting Metabolic Reprogramming. Int J Mole Sci. 2021;22(11):5887.
Braak H, Del Tredici K. Cortico-basal ganglia-cortical circuitry in Parkinson's disease reconsidered. Exp Neurol. 2008;212:226–9.
Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–75.
Alexander GE, Crutcher MD, DeLong MR. Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, "prefrontal" and "limbic" functions. Prog Brain Res. 1990;85:119–46.
Wichmann T, DeLong MR. Models of basal ganglia function and pathophysiology of movement disorders. Neurosurg Clin N Am. 1998;9:223–36.
Borghammer P, Chakravarty M, Jonsdottir KY, Sato N, Matsuda H, Ito K, et al. Cortical hypometabolism and hypoperfusion in Parkinson's disease is extensive: probably even at early disease stages. Brain Struct Funct. 2010;214:303–17.
Jokinen P, Scheinin N, Aalto S, Någren K, Savisto N, Parkkola R, et al. [(11) C]PIB-, [(18) F]FDG-PET and MRI imaging in patients with Parkinson's disease with and without dementia. Parkinsonism Relat Disord. 2010;16:666–70.
Dalfó E, Portero-Otín M, Ayala V, Martínez A, Pamplona R, Ferrer I. Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J Neuropathol Exp Neurol. 2005;64:816–30.
Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Move Disord. 2012;27:1478–83.
Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29:444–53.
Ren Y, Liu W, Jiang H, Jiang Q, Feng J. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J Biol Chem. 2005;280:34105–12.
Giguère N, Burke Nanni S, Trudeau LE. On cell loss and selective vulnerability of neuronal populations in Parkinson's disease. Front Neurol. 2018;9:455.
Giguère N, Delignat-Lavaud B, Herborg F, Voisin A, Li Y, Jacquemet V, et al. Increased vulnerability of nigral dopamine neurons after expansion of their axonal arborization size through D2 dopamine receptor conditional knockout. PLoS Genet. 2019;15:e1008352.
Jansen IE, Ye H, Heetveld S, Lechler MC, Michels H, Seinstra RI, et al. Discovery and functional prioritization of Parkinson's disease candidate genes from large-scale whole exome sequencing. Genome Biol. 2017;18:22.
Milanese C, Payán-Gómez C, Galvani M, Molano González N, Tresini M, Nait Abdellah S, et al. Peripheral mitochondrial function correlates with clinical severity in idiopathic Parkinson's disease. Move Disord. 2019;34:1192–202.
Mamelak M. Parkinson's Disease, the Dopaminergic Neuron and Gammahydroxybutyrate. Neurol Ther. 2018;7:5–11.
Bossers K, Meerhoff G, Balesar R, van Dongen JW, Kruse CG, Swaab DF, et al. Analysis of gene expression in Parkinson's disease: possible involvement of neurotrophic support and axon guidance in dopaminergic cell death. Brain pathology (Zurich, Switzerland). 2009;19:91–107.
Juárez Olguín H, Calderón Guzmán D, Hernández García E, Barragán Mejía G. The role of dopamine and its dysfunction as a consequence of oxidative stress. Oxid Med Cell Longev. 2016;2016:9730467.
Kim TY, Leem E, Lee JM, Kim SR. Control of reactive oxygen species for the prevention of Parkinson's disease: the possible application of flavonoids. Antioxidants (Basel). 2020;9(7):583.
Jadiya P, Garbincius JF, Elrod JW. Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol Commu. 2021;9:124.
Mytilineou C, Werner P, Molinari S, Di Rocco A, Cohen G, Yahr MD. Impaired oxidative decarboxylation of pyruvate in fibroblasts from patients with Parkinson's disease. J Neural Transm Park Dis Dement Sect. 1994;8:223–8.
Parker WD Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol. 1989;26:719–23.
Tang BL. Glucose, glycolysis, and neurodegenerative diseases. J Cell Physiol. 2020;235:7653–62.
Błaszczyk JW. Energy metabolism decline in the aging brain-pathogenesis of neurodegenerative disorders. Metabolites. 2020;10(11):450.
Powers R, Lei S, Anandhan A, Marshall DD, Worley B, Cerny RL, et al. Metabolic investigations of the molecular mechanisms associated with parkinson's disease. Metabolites. 2017;7(2):22.
Tu D, Gao Y, Yang R, Guan T, Hong JS, Gao HM. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J Neuroinflammation. 2019;16:255.
Dunn L, Allen G, Mamais A, Ling H, Li A, Duberley K, et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson's disease. Neurobiol Aging. 2014;35:1111–5.
D'Urso M, Mareni C, Toniolo D, Piscopo M, Schlessinger D, Luzzatto L. Regulation of glucose 6-phosphate dehydrogenase expression in CHO-human fibroblast somatic cell hybrids. Somatic Cell Genet. 1983;9:429–43.
Battistuzzi G, D'Urso M, Toniolo D, Persico GM, Luzzatto L. Tissue-specific levels of human glucose-6-phosphate dehydrogenase correlate with methylation of specific sites at the 3′ end of the gene. Proc Natl Acad Sci U S A. 1985;82:1465–9.
Fang J, She J, Lin F, Wu JC, Han R, Sheng R, et al. RRx-001 Exerts Neuroprotection Against LPS-Induced Microglia Activation and Neuroinflammation Through Disturbing the TLR4 Pathway. Front Pharmacol. 2022;13:889383.
Sergi D, Renaud J, Simola N, Martinoli MG. Diabetes, a Contemporary Risk for Parkinson's Disease: Epidemiological and Cellular Evidences. Front Aging Neurosci. 2019;11:302.
Santiago JA, Potashkin JA. Shared dysregulated pathways lead to Parkinson's disease and diabetes. Trends Mole Med. 2013;19:176–86.
Athauda D, Foltynie T. Insulin resistance and Parkinson's disease: A new target for disease modification? Prog Neurobiol. 2016;145–146:98–120.
Moroo I, Yamada T, Makino H, Tooyama I, McGeer PL, McGeer EG, et al. Loss of insulin receptor immunoreactivity from the substantia nigra pars compacta neurons in Parkinson's disease. Acta Neuropathol. 1994;87:343–8.
Takahashi M, Yamada T, Tooyama I, Moroo I, Kimura H, Yamamoto T, et al. Insulin receptor mRNA in the substantia nigra in Parkinson's disease. Neurosci Letters. 1996;204:201–4.
Duarte AI, Moreira PI, Oliveira CR. Insulin in central nervous system: more than just a peripheral hormone. J Aging Res. 2012;2012:384017.
Sharma SK, Chorell E, Steneberg P, Vernersson-Lindahl E, Edlund H, Wittung-Stafshede P. Insulin-degrading enzyme prevents α-synuclein fibril formation in a nonproteolytical manner. Sci Rep. 2015;5:12531.
Kao SY. Rescue of alpha-synuclein cytotoxicity by insulin-like growth factors. Biochem Biophys Res Commu. 2009;385:434–8.
Malagelada C, Jin ZH, Greene LA. RTP801 is induced in Parkinson's disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci. 2008;28:14363–71.
Xu Y, Liu C, Chen S, Ye Y, Guo M, Ren Q, et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson's disease. Cell Sign. 2014;26:1680–9.
Kim SR, Ries V, Cheng HC, Kareva T, Oo TF, Yu WH, et al. Age and α-synuclein expression interact to reveal a dependence of dopaminergic axons on endogenous Akt/PKB signaling. Neurobiol Dis. 2011;44:215–22.
Jo AY, Kim MY, Lee HS, Rhee YH, Lee JE, Baek KH, et al. Generation of dopamine neurons with improved cell survival and phenotype maintenance using a degradation-resistant nurr1 mutant. Stem Cells (Dayton, Ohio). 2009;27:2238–46.
Li W, Liu X, Tu Y, Ding D, Yi Q, Sun X, et al. Dysfunctional Nurr1 promotes high glucose-induced Müller cell activation by up-regulating the NF-κB/NLRP3 inflammasome axis. Neuropeptides. 2020;82:102057.
Xu Y, Huang Q, Zhang W, Wang Y, Zeng Q, He C, et al. Decreased expression levels of Nurr1 are associated with chronic inflammation in patients with type 2 diabetes. Mole Med Rep. 2015;12:5487–93.
Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mole Immunol. 2021;18:1141–60.
Lee H-M, Kim J-J, Kim HJ, Shong M, Ku BJ, Jo E-K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204.
Amoasii L, Holland W, Sanchez-Ortiz E, Baskin K, Pearson M, Burgess S, et al. A MED13-dependent skeletal muscle gene program controls systemic glucose homeostasis and hepatic metabolism. Genes Dev. 2016;30:434–46.
Pei L, Waki H, Vaitheesvaran B, Wilpitz D, Kurland I, Tontonoz P. NR4A orphan nuclear receptors are transcriptional regulators of hepatic glucose metabolism. Nat Med. 2006;12:1048–55.
Xia M, Zhang Y, Jin K, Lu Z, Zeng Z, Xiong W. Communication between mitochondria and other organelles: a brand-new perspective on mitochondria in cancer. Cell Biosci. 2019;9:27.
van Horssen J, van Schaik P, Witte M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci Lett. 2019;710:132931.
Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102.
Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med. 2010;2:52ra73.
Decressac M, Volakakis N, Björklund A, Perlmann T. NURR1 in Parkinson disease--from pathogenesis to therapeutic potential. Nat Rev Neurol. 2013;9:629–36.
Heng X, Jin G, Zhang X, Yang D, Zhu M, Fu S, et al. Nurr1 regulates Top IIβ and functions in axon genesis of mesencephalic dopaminergic neurons. Mole Neurodegener. 2012;7:4.
Kadkhodaei B, Alvarsson A, Schintu N, Ramsköld D, Volakakis N, Joodmardi E, et al. Transcription factor Nurr1 maintains fiber integrity and nuclear-encoded mitochondrial gene expression in dopamine neurons. Proc Natl Acad Sci U S A. 2013;110:2360–5.
Bantle CM, Hirst WD, Weihofen A, Shlevkov E. Mitochondrial dysfunction in astrocytes: a role in parkinson's disease? Front Cell Dev Biol. 2020;8:608026.
Castelo-Branco G, Wagner J, Rodriguez FJ, Kele J, Sousa K, Rawal N, et al. Differential regulation of midbrain dopaminergic neuron development by Wnt-1, Wnt-3a, and Wnt-5a. Proc Natl Acad Sci U S A. 2003;100:12747–52.
Singh S, Mishra A, Mohanbhai SJ, Tiwari V, Chaturvedi RK, Khurana S, et al. Axin-2 knockdown promote mitochondrial biogenesis and dopaminergic neurogenesis by regulating Wnt/β-catenin signaling in rat model of Parkinson's disease. Free Radic Biol Med. 2018;129:73–87.
L'Honoré A, Commère PH, Ouimette JF, Montarras D, Drouin J, Buckingham M. Redox regulation by Pitx2 and Pitx3 is critical for fetal myogenesis. Dev Cell. 2014;29:392–405.
Jodeiri Farshbaf M, Forouzanfar M, Ghaedi K, Kiani-Esfahani A, Peymani M, Shoaraye Nejati A, et al. Nurr1 and PPARγ protect PC12 cells against MPP(+) toxicity: involvement of selective genes, anti-inflammatory, ROS generation, and antimitochondrial impairment. Mol Cell Biochem. 2016;420:29–42.
Liang H, Ward WF. PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ. 2006;30:145–51.
Han B, Jiang W, Liu H, Wang J, Zheng K, Cui P, et al. Upregulation of neuronal PGC-1α ameliorates cognitive impairment induced by chronic cerebral hypoperfusion. Theranostics. 2020;10:2832–48.
Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–24.
Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006;3:333–41.
Su X, Chu Y, Kordower JH, Li B, Cao H, Huang L, et al. PGC-1α Promoter Methylation in Parkinson's Disease. PloS One. 2015;10:e0134087.
Nervina JM, Magyar CE, Pirih FQ, Tetradis S. PGC-1alpha is induced by parathyroid hormone and coactivates Nurr1-mediated promoter activity in osteoblasts. Bone. 2006;39:1018–25.
Giorgi C, Agnoletto C, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion. 2012;12:77–85.
Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–72.
Liao Y, Dong Y, Cheng J. The function of the mitochondrial calcium uniporter in neurodegenerative disorders. Int J Mole Sci. 2017;18(2):248.
Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci. 2007;27:645–56.
Goldberg JA, Guzman JN, Estep CM, Ilijic E, Kondapalli J, Sanchez-Padilla J, et al. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson's disease. Nat Neurosci. 2012;15:1414–21.
Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700.
Foehring RC, Zhang XF, Lee JC, Callaway JC. Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J Neurophysiol. 2009;102:2326–33.
Lee KS, Huh S, Lee S, Wu Z, Kim AK, Kang HY, et al. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc Natl Acad Sci U S A. 2018;115:E8844–e8853.
Soman SK, Bazała M, Keatinge M, Bandmann O, Kuznicki J. Restriction of mitochondrial calcium overload by mcu inactivation renders a neuroprotective effect in zebrafish models of Parkinson's disease. Biol Open. 2019;8(10):bio044347.
Zhang X, Alshakhshir N, Zhao L. Glycolytic Metabolism, Brain Resilience, and Alzheimer's Disease. Front Neurosci. 2021;15:662242.
Poitelon Y, Kopec AM, Belin S. Myelin fat facts: an overview of lipids and fatty acid metabolism. Cells. 2020;9(4):812.
Fecchio C, De Franceschi G, Relini A, Greggio E, Dalla Serra M, Bubacco L. Polverino de Laureto P: α-Synuclein oligomers induced by docosahexaenoic acid affect membrane integrity. PloS One. 2013;8:e82732.
Broersen K, van den Brink D, Fraser G, Goedert M, Davletov B. Alpha-synuclein adopts an alpha-helical conformation in the presence of polyunsaturated fatty acids to hinder micelle formation. Biochemistry. 2006;45:15610–6.
Clark LN, Kisselev S, Park N, Ross B, Verbitsky M, Rios E, et al. Mutations in the Parkinson's disease genes, Leucine Rich Repeat Kinase 2 (LRRK2) and Glucocerebrosidase (GBA), are not associated with essential tremor. Parkinsonism Relat Disord. 2010;16:132–5.
Bae EJ, Lee HJ, Jang YH, Michael S, Masliah E, Min DS, et al. Phospholipase D1 regulates autophagic flux and clearance of α-synuclein aggregates. Cell Death Different. 2014;21:1132–41.
Wood PL, Tippireddy S, Feriante J, Woltjer RL. Augmented frontal cortex diacylglycerol levels in Parkinson's disease and lewy body disease. PloS One. 2018;13:e0191815.
Belarbi K, Cuvelier E, Bonte MA, Desplanque M, Gressier B, Devos D, et al. Glycosphingolipids and neuroinflammation in Parkinson's disease. Mole Neurodegener. 2020;15:59.
Farooqui T, Farooqui AA. Lipid-mediated oxidative stress and inflammation in the pathogenesis of Parkinson's disease. Parkinson's Dis. 2011;2011:247467.
Alessenko A, Albi E. Exploring Sphingolipid Implications in Neurodegeneration. Front Neurol. 2020;11:437.
Paciotti S, Albi E, Parnetti L, Beccari T. Lysosomal ceramide metabolism disorders: implications in Parkinson’s disease. J Clin Med. 2020;9:594.
Henchcliffe C, Dodel R, Beal MF. Biomarkers of Parkinson's disease and Dementia with Lewy bodies. Prog Neurobiol. 2011;95:601–13.
Fais M, Dore A, Galioto M, Galleri G, Crosio C, Iaccarino C. Parkinson's disease-related genes and lipid alteration. Int J Mole Sci. 2021;22(14):7630.
George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–72.
Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9.
Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci U S A. 2001;98:9110–5.
Fu M, Sun T, Bookout A, Downes M, Yu R, Evans R, et al. A Nuclear Receptor Atlas: 3T3-L1 adipogenesis. Mole Endocrinol (Baltimore, Md). 2005;19:2437–50.
Veum VL, Dankel SN, Gjerde J, Nielsen HJ, Solsvik MH, Haugen C, et al. The nuclear receptors NUR77, NURR1 and NOR1 in obesity and during fat loss. Int J Obes (Lond). 2012;36:1195–202.
Holla V, Wu H, Shi Q, Menter D, DuBois R. Nuclear orphan receptor NR4A2 modulates fatty acid oxidation pathways in colorectal cancer. J Biol Chem. 2011;286:30003–9.
Briand O, Helleboid-Chapman A, Ploton M, Hennuyer N, Carpentier R, Pattou F, et al. The nuclear orphan receptor Nur77 is a lipotoxicity sensor regulating glucose-induced insulin secretion in pancreatic β-cells. Mole Endocrinol (Baltimore, Md). 2012;26:399–413.
de Vera I, Giri P, Munoz-Tello P, Brust R, Fuhrmann J, Matta-Camacho E, et al. Identification of a binding site for unsaturated fatty acids in the orphan nuclear receptor Nurr1. ACS Chem Biol. 2016;11:1795–9.
de Vera IMS, Munoz-Tello P, Zheng J, Dharmarajan V, Marciano DP, Matta-Camacho E, et al. Defining a canonical ligand-binding pocket in the orphan nuclear receptor Nurr1. Structure. 2019;27:66–77.e65.
Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7:489–503.
Volakakis N, Joodmardi E, Perlmann T. NR4A orphan nuclear receptors influence retinoic acid and docosahexaenoic acid signaling via up-regulation of fatty acid binding protein 5. Biochem Biophys Res Commun. 2009;390:1186–91.
Honkaniemi J, Sharp FR. Prolonged expression of zinc finger immediate-early gene mRNAs and decreased protein synthesis following kainic acid induced seizures. Eur J Neurosci. 1999;11:10–7.
Honkaniemi J, Sagar SM, Pyykönen I, Hicks KJ, Sharp FR. Focal brain injury induces multiple immediate early genes encoding zinc finger transcription factors. Brain Res Mole Brain Res. 1995;28:157–63.
Loppi S, Kolosowska N, Kärkkäinen O, Korhonen P, Huuskonen M, Grubman A, et al. HX600, a synthetic agonist for RXR-Nurr1 heterodimer complex, prevents ischemia-induced neuronal damage. Brain Behav Immun. 2018;73:670–81.
Spathis AD, Asvos X, Ziavra D, Karampelas T, Topouzis S, Cournia Z, et al. Nurr1:RXRα heterodimer activation as monotherapy for Parkinson's disease. Proc Natl Acad Sci U S A. 2017;114:3999–4004.
Latourelle JC, Dybdahl M, Destefano AL, Myers RH, Lash TL. Estrogen-related and other disease diagnoses preceding Parkinson's disease. Clin Epidemiol. 2010;2:153–70.
Luchetti S, Bossers K, Frajese GV, Swaab DF. Neurosteroid biosynthetic pathway changes in substantia nigra and caudate nucleus in Parkinson's disease. Brain Pathol (Zurich, Switzerland). 2010;20:945–51.
Schaefer S, Vogt T, Nowak T, Kann PH. Pituitary function and the somatotrophic system in patients with idiopathic Parkinson's disease under chronic dopaminergic therapy. J Neuroendocrinol. 2008;20:104–9.
Mohammadi S, Dolatshahi M, Rahmani F. Shedding light on thyroid hormone disorders and Parkinson disease pathology: mechanisms and risk factors. J Endocrinol Invest. 2021;44:1–13.
Matsuda K, Uruno A, Kogure N, Sugawara K, Shimada H, Nezu M, et al. Angiotensin II receptor blockers differentially affect CYP11B2 expression in human adrenal H295R cells. Mole Cell Endocrinol. 2014;383:60–8.
Shimada H, Kogure N, Noro E, Kudo M, Sugawara K, Sato I, et al. High glucose stimulates expression of aldosterone synthase (CYP11B2) and secretion of aldosterone in human adrenal cells. FEBS Open Bio. 2017;7:1410–21.
Pirih FQ, Tang A, Ozkurt IC, Nervina JM, Tetradis S. Nuclear orphan receptor Nurr1 directly transactivates the osteocalcin gene in osteoblasts. J Biol Chem. 2004;279:53167–74.
Peel MT, Ho Y, Liebhaber SA. The transcription factor NR4A2 plays an essential role in driving prolactin expression in female Pituitary Lactotropes. Endocrinology. 2020;161.
Goetz CG, Pal G. Initial management of Parkinson's disease. BMJ. 2014;349:g6258.
Dong J, Cui Y, Li S, Le W. Current Pharmaceutical Treatments and Alternative Therapies of Parkinson's Disease. Curr Neuropharmacol. 2016;14:339–55.
Liu W, Gao Y, Chang N. Nurr1 overexpression exerts neuroprotective and anti-inflammatory roles via down-regulating CCL2 expression in both in vivo and in vitro Parkinson's disease models. Biochem Biophys Res Commun. 2017;482:1312–9.
Oh S, Chang M, Song J, Rhee Y, Joe E, Lee H, et al. Combined Nurr1 and Foxa2 roles in the therapy of Parkinson's disease. EMBO Mole Med. 2015;7:510–25.
Dong J, Li S, Mo JL, Cai HB, Le WD. Nurr1-Based Therapies for Parkinson's Disease. CNS Neurosci Therapeut. 2016;22:351–9.
Wei X, Gao H, Zou J, Liu X, Chen D, Liao J, et al. Contra-directional Coupling of Nur77 and Nurr1 in Neurodegeneration: A Novel Mechanism for Memantine-Induced Anti-inflammation and Anti-mitochondrial Impairment. Mol Neurobiol. 2016;53:5876–92.
Esteves M, Cristóvão AC, Saraiva T, Rocha SM, Baltazar G, Ferreira L, et al. Retinoic acid-loaded polymeric nanoparticles induce neuroprotection in a mouse model for Parkinson's disease. Front Aging Neurosci. 2015;7:20.
Pan T, Xie W, Jankovic J, Le W. Biological effects of pramipexole on dopaminergic neuron-associated genes: relevance to neuroprotection. Neuroscience Lett. 2005;377:106–9.
Zhang L, Cen L, Qu S, Wei L, Mo M, Feng J, et al. Enhancing Beta-Catenin Activity via GSK3beta Inhibition Protects PC12 Cells against Rotenone Toxicity through Nurr1 Induction. PloS One. 2016;11:e0152931.
Sim Y, Park G, Eo H, Huh E, Gu PS, Hong SP, et al. Protective effects of a herbal extract combination of Bupleurum falcatum, Paeonia suffruticosa, and Angelica dahurica against MPTP-induced neurotoxicity via regulation of nuclear receptor-related 1 protein. Neuroscience. 2017;340:166–75.
Acknowledgments
We thank the Weidong Le lab team members for their critical discussion regarding these topics.
Funding
This work was supported by the Guangdong Provincial Key R&D Program (2018B030337001) and the Shanghai municipal central government funds for guiding local scientific and technological development (YDZX20213100001002).
Author information
Authors and Affiliations
Contributions
WL, MAN, YT, and SL conceived and designed the manuscript. WL, MAN, YT, SL, and CC performed the literature search, wrote and revised the manuscript. WL provides expertise in the concept of molecular metabolic neurodegeneration in PD. All authors read and approved the final manuscript.
Authors’ information
Professor Weidong Le,
Weidong Le, MD/PhD, is a senior neurologist/neuroscientist. He received a doctoral degree from Shanghai 2nd Medical University in 1988, got postdoctoral training in 1989, and was promoted to Professor of Neurology in 2006 at Baylor College of Medicine. Since then, he has been working as a Director of Neurology Institute in Shanghai Jiao-tong University School of Medicine; Life-time Professor and Director of the Center for Clinical Research on Neurological Diseases, Dalian Medical University; and Professor and Director of Neurology Institute, Sichuan Academy of Medical Science-Sichuan Provincial Hospital. Dr. Le has published over 300 SCI papers in peer-reviewed journals (including Nature, Sciences, JAMA, Lancet Neurology, Brain, Autophagy, and Alzheimer’s & Dementia) with a citation of over 20,000. He is one of the most influenced Neuroscientists/Neurologists in China, as evaluated by Elsevier Publishing Network. He has been awarded over 50 national and international research grants. He serves as the Chief Editor/Associate Editor/Editorial Board Member for Ageing and Neurodegenerative Diseases, Neuroscience Bulletin, Brain Research Bulletin, J. Alzheimer’s Disease, J Neurochemistry, Autophagy, Alzheimer’s & Dementia, Neurodegenerative Disease, and Translational Neurodegeneration.
Professor Song Li,
Prof. Li received his Ph.D. degree from Shenyang Pharmaceutical University, China, and post-doc training at the University of South Florida, USA. Prof. Li has published numerous SCI papers in peer-reviewed journals. He is now engaged in neuropsychopharmacology research, especially the pathogenic mechanisms and innovative therapy of neurodegenerative diseases, including Parkinson’s disease and Alzheimer’s disease.
Murad Al-Nusaif MBBS, MMed,
Resident and researcher in the Liaoning provincial key laboratory for research on the pathogenic mechanisms of neurological diseases, the center for clinical research in neurological diseases and neurological department-first affiliated hospital of Dalian medical university. Dr. Murad has published numerous SCI papers in peer-reviewed journals. He is now engaged in clinical neurological research, especially the pathogenic mechanisms and innovative therapy of Parkinson’s disease, Alzheimer’s disease, Motor neuron disease and Neurovascular disease.
Yuting Yang MBBS, MMed,
Master of neurology medicine Dalian Medical University, Liaoning, Dalian, China. She published many SCI papers in the journal of brain pathology, frontiers in aging neuroscience, and Neuro psycho pharmacotherapy. She is now engaged in neuroscience research, especially the mechanisms and innovative role of Nurr1 in neurodegenerative diseases, including Parkinson’s disease and Alzheimer’s disease.
Cheng Cheng, MS.
Current Ph.D, MS, 2014- Present researcher in the Liaoning provincial key laboratory for research on the pathogenic mechanisms of neurological diseases, - first affiliated hospital of Dalian medical university Cheng focuses on studying the pathophysiological properties of genetic changes associated with Parkinson’s disease (PD). Contributed and published many SCI articles.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Al-Nusaif, M., Yang, Y., Li, S. et al. The role of NURR1 in metabolic abnormalities of Parkinson’s disease. Mol Neurodegeneration 17, 46 (2022). https://doi.org/10.1186/s13024-022-00544-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-022-00544-w