
Abstract
Background
Fetal skeletal dysplasia is a diverse group of degenerative diseases of bone and cartilage disorders that can lead to movement disorder and even death. This study aims to evaluate the diagnostic yield of sonographic examination and genetic testing for fetal skeletal dysplasia.
Methods
From September 2015 to April 2021, the study investigated 24 cases with suspected short-limb fetuses, which were obtained from Tongji Hospital affiliated to Tongji Medical College of Huazhong University of Science and Technology. To identify the causative gene, multiple approaches (including karyotype analysis, copy number variations and whole exome sequencing) were performed on these fetuses. And further segregation analysis of the candidate variant was performed in parents by using Sanger sequencing.
Results
① Out of 24 cases, likely pathogenic variants in FGFR3, FBN2, COL1A2, CUL7 and DYNC2H1 were detected in 6 cases; pathogenic variants in FGFR3, IMPAD1 and GORAB were identified in other 6 cases; and variants in WNT1, FBN1, OBSL1, COL1A1, DYNC2H1 and NEK1, known as Variant of Undetermined Significance, were found in 4 cases. There were no variants detected in the rest 8 cases by the whole exome sequencing. ② Of 24 cases, 12 (50%) were found to carry variants (pathogenic or likely pathogenic) in seven genes with 12 variants. Four fetuses (16.7%) had variants of uncertain significance.
Conclusion
Genetic testing combining with ultrasound scanning enhances the accurate diagnosis of fatal skeletal dysplasia in utero, and then provides appropriate genetic counseling.
Similar content being viewed by others

Introduction
Fetal skeletal dysplasia (FSD) is one of the most common fetal malformations, of which the incidence rate is approximately 2.4–4.5 out of 10,000 births [1, 2]. It is defined as a group of bone and cartilage disorders with diverse clinical and genetic heterogeneity that can lead to movement disorder and even death. The pathogenicity is closely associated with mutations of the genes encoding collagen (e. g. type I, II, IX, IX, XI collagen) [3]. Current scientific study reported many pathogenic gene variations in the proband with skeletal dysplasia and variants occurred in causative genes of COL1A1, COL1A2, WNT1, OBSL1, FGFR3, IMPAD1, FBN2, and GORAB [4,5,6]. According to the report of "Nosology and Classification of Genetic skeletal disorders: 2019 Revision", 461 disorders classified within 42 different groups have been described based on radiologic, molecular and biochemical criteria [7]. The clinical phenotype is broad and ranges from a mild to a severe form. Genotype and phenotype relationship is unclear. And then an accurate diagnosis of FSD can be challenging due to its rarity and variety of phenotype, especially for fetuses.Therefore, the determination of FSD is challenging due to the diversity of FSD phenotypes, particularly in prenatal diagnosis.
So far, ultrasound is the most common imaging test to detect fetal skeletal abnormality in early pregnancy [8]. However, overlapping features and phenotypic variability of skeletal dyplasias are limited to differentiate FSD and provide a definitive diagnosis via image findings. In addition, phenotypic characteristics of FSD do not manifest until later in pregnancy, which undoubtedly increase the difficulty in definite diagnosis. Recently, with the advance in next-generation sequencing technology, high-throughput sequencing has been considered as an effective method for genetic diagnosis. E.g. whole exome sequencing (WES), is advantageous in identification of De novo and compound heterozygous variants [9, 10]. Suggested by the American College of Medical Genetics and Genomics (ACMG), next-generation sequencing can be considered to increase the sensitivity of diagnosis when the traditional gene testing, such as chromosomal microarray analysis, failed to yield a definitive result for the diagnosis [11].
By analyzing the imaging in terms of ultrasounds and gene variation of 24 cases with suspected fetal skeletal dysplasia, this study successfully obtained some solid evidence for prenatal diagnosis of FSD. Ultrasound scanning combining with genetic test in the first or second trimester of gestation enhances the accurate diagnosis of fatal skeletal dysplasia in utero, then provides appropriate genetic counseling.
Materials and methods
Editorial policies and ethical considerations
Our study was approved by the Research Ethics Committee of Tongji Hospital affiliated to Tongji Medical College of Huazhong University of Science and Technology (IRB reference number: M2019036). Informed consents were obtained from all the participants in the study.
Patients information
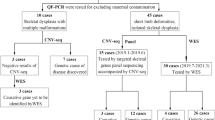
After approval of the Research Ethics Committee of our Hospital, informed consents and samples were obtained from all the participants. Twenty-four cases were included in the study since all fetuses were detected with suspected short-limb from primary ultrasound scanning in the prenatal diagnosis center (one of prenatal diagnosis referral centers in China) of Tongji Hospital affiliated to Tongji Medical College of Huazhong University of Science and Technology between September 2015 and April 2021. Out of 24 cases, the pregnant women in case 9 and 10 had osteogenesis imperfecta, while the clinical phenotypes of the pregnant women and their spouses of the rest cases (22 cases) were all normal. In addition, case 6 had induced labor previously due to the suspected short limbs of the fetus determined only by ultrasound examination.
Ultrasound scanning
The biometric data of the fetal skeleton (including biparietal diameter (BPD), head circumference (HC), measurements of all long bones (including humerus length (HL), femur length (FL)), assessment of skeletal mineralization), and any other abnormalities were collected and analyzed in comparison with normal values [12].
Sample collection
Amniotic fluid and blood samples were retrieved from umbilical cord by amniocentesis and cordocentesis individually under ultrasound at second trimester of gestation. For the cases of which the blood or amniotic fluid samples could not be collected, two pieces of fetal muscle tissue (2 × 2 cm, containing the skin) or umbilical cord (about 3 cm) were sampled after the termination of pregnancy.
Fetal karyotype analysis and genetic analysis
Karyotype analysis was performed on cultured amniotic fluid cells. Fetal genomic DNA was extracted from amniotic fluid samples or the tissues samples of fetuses using a DNA Extraction Kit (TianGen, Beijing, China) according to the manufacturer's instructions and stored at − 20 °C for further analysis. The genomic DNA of the couples in all cases was extracted from whole blood sample using the same protocol/Kit. The WES was performed for all the DNA samples. After fetal DNA was quantified with Nanodrop 2000 (Thermal Fisher Scientific, DE), 3–5 μg DNA was used for sequencing and copy number variations (CNV) test. The sequencing data was used for analyzing likely pathogenic gene variation by the Polyphen 2.0 and SIFT software [13, 14]. Chromosome profiles were finally plotted as copy number (Y-axis) vs. 20- kb count windows (X-axis).
Identified and mapped CNVs were interrogated against publicly available databases, including Decipher, Database of Genomic Variants (DGV), 1000 genomes, and Online Mendelian Inheritance in Man (OMIM), and their pathogenicity assessed according to the guidelines outlined by the American College of Medical Genetics (ACMG) for interpretation of sequence variants. Variants were classifified as either pathogenic, likely pathogenic, variants of uncertain signifificance (VUS), likely benign, or benign [15].
Familial validation
Once the pathogenic gene variations were detectable in the fetal samples, the coding regions of the mutations were identified and further examined for parents by Sanger sequencing [16].
Results
Clinical information and ultrasound findings
Ultrasound imaging revealed normal amniotic fluid volumes and normal appearance of the brain, heart, liver, or kidneys in all cases, whereas the suspected skeletal malformations were detected. The lengths of humerus and femur in 24 cases were found to be either significantly lower than the reference (Mean ± SD) at the same gestation period according to Hadlock’s reference chart. The results of prenatal ultrasound were summarized in Table 1.
Abnormalities by karyotype analysis and copy number variations (CNV)
The chromosome G band karyotype revealed negative in all 24 cases, while CNV was undetected in 22 cases except of case15 and 23. In the case15, we found a 0.2 MB duplication in the chromosome 7 q11.21, considered benign according to the available evidence; In the case 23, a 0.4 MB duplication in the chromosome 18p11.31q11.23 was detected and known as VUS.
Abnormalities detected by whole exome sequencing (WES)
Out of 24 cases, 6 cases were identified with carrying likely pathogenic variants in gene FGFR3, FBN2, COL1A2, CUL7 and DYNC2H1; 6 other cases were considered to carry pathogenic variants and the chromosomes of fetuses were trisomy 13 and trisomy 18, respectively; 4 cases were detected with variants in gene WNT1, FBN1, OBSL1, COL1A1, DYNC2H1 and NEK1, known as VUS; The rest 8 cases showed negative in WES (Table 2), which was further confirmed by Sanger sequencing.
For the fetus of case 4 carried the c.7842 T > A (p. Ala2614Ala) variant in FBN1 gene and the deletion of c.2135-3_2135-2delCA in OBSL1 gene. The Sanger sequencing analysis further revealed that the variant was carried by the father and the deletion was carried by the mother (Fig. 1).

Case 4: A The fetus carried the c.7842 T > A (p. Ala2614Ala) mutation in the FBN1 gene. B The deletion of c.2135-3_2135-2delCA in the OBSL1 gene was detected. The Sanger verification revealed that the mutation was carried by the father while the deletion was carried by the mother
For the fetus of case 6, we detected the variant of c.700G > T (p.E234*) and the deletion of CDS4-5 in IMPAD1 gene. Both Sanger sequencing and qPCR determined that the heterozygous deletion of CDS4-5 and the homozygous variant in gene IMPAD1 were inherited from their parents, resulting to a composite heterozygous variant (Fig. 2).

Case 6: A The fetus carried the c.700G > T (p. E234*) mutation in the IMPAD1 gene and inherited from mother by Sanger Sequencing verification. B The deletion of CDS4-5 in IMPAD1 gene was detected in the fetus and inherited from father by qPCR verification
In the case 9 and 10, the two women were diagnosed with osteogenesis imperfecta before pregnancy. Our analysis detected the variant of c.1118G > C (p. Gly373Ala) in COL1A2 for the fetus in case 9, and variant of c.178C > T (p. Arg60*) in GORAB for the fetus in case 10. The two variants originated from osteogenesis imperfecta and inherited from their mother (Fig. 3, 4).

Case 9: The variant of c.1118G > C (p. Gly373Ala) in COL1A2 gene of the fetus and the mother

Case 10: A The variant of c.178C > T (p. Arg60*) in GORAB gene of the mother. B The heterozygous mutation in GORAB gene of the fetus
Discussion
The prenatal diagnosis of FSD is important at the second trimester of gestation, however, it is still challenging due to diverse clinical and genetic heterogeneity of the disorder. For decades, ultrasound is widely used in the noninvasive detection of FSD. Pajkrt and Chitty [17] found that FL and HL of the fetuses with skeletal dysplasia were 5% shorter than the normal value. Previous reports in China demonstrated that the diagnosis accuracy of continuous sequential follow-up ultrasound was over 80% and 87.2% in the second trimester of gestation [18]. In this study, by ultrasound scanning, we found the HL and FL of the fetuses in the 24 cases was either lower or higher than the normal value except of case 10 (the woman with OI (osteogenesis imperfecta) before pregnancy). The BPD and HC of the fetus were normal in all the 24 cases. In one-year follow-up survey, we found the 8 infants (account for 33.3%) were normal in skeletal development after delivery when the HL and FL of these fetuses was 2SD-3SD lower than the mean value. Differently, when the HL and FL of the fetuses was 3SD less or 3SD more, only two newborns (8.33%) were normal. The results of this ultrasound-only dependent approach are inadequate for diagnosing the disorder and impossible to differentiate the complex types of FSD.
In recent years, NGS (WES and WGS, Whole exome sequencing and Whole gene sequencing) has been applied in the area of disease diagnosis [19]. The variants detection tool needs to optimize since the detection rates of WES could be variable depending on many factors, such as the sample size, the analysis criteria, proband-only or trio WES, and so on [10]. More importantly, the complex of genetic variants was found to be associated with the diverse pathogenicity of FSD.In this study, WES identified well-described variants (pathogenic or likely pathogenic) in 12 out of 24 cases (50%), and VUS in 4 cases(16.67%), rendering a total diagnostic yield of 66.67%. Compared with our findings, three recent cohort studies reported a significantly higher diagnostic rate by WES, which was 80% (12 out of 15), 85% (11 out of 13), and 70% (21 out of 30), respectively [20,21,22].
In case 4, The fetus carried the c.7842 T > A (p. Ala2614Ala) variant in the FBN1 gene and the deletion of c.2135-3_2135-2delCA in OBSL1 gene. The mutation is synonymous mutation and the deletion is in the intron. This variant in FBN1 gene [23] is reported to be associated with Weill Marchesani syndrome (clinical manifestations are short limb deformity, secondary glaucoma, short stature, etc.). The OBSL1 variant [24] is related to 3-M syndrome (clinical manifestations include severe intrauterine growth retardation, short stature, recessive spina bifida, compression deformation of long metaphysis). The inheritance patterns of Weill Marchesani syndrome and 3-M syndrome are both AR (autosomal recessive inheritance). The Sanger analysis [16] indicated that the variant was carried by the father and the deletion was carried by the mother (Fig. 1). The mother of case 4 had natural delivery by vaginal at 39+6 weeks of gestation, and the newborn did not show any abnormality of bone development (up to 18-monthes old) at the end of the following-up survey.
In case 6, we detected the variant of c.700G > T (p.E234*) and the deletion of CDS4-5 in IMPAD1 gene for the fetus. The heterozygous deletion of CDS4-5 and the homozygous variant in IMPAD1 were inherited from their parents, as a compound heterozygote variant (Fig. 2). IMPAD1-related chondrodysplasia is an autosomal recessive disease [25]. The pregnant women had cesarean section at 38 weeks of gestation in this case, and the newborn had typical short limb deformity and died within one month after delivery.
In the study, there were two women (case 9 and 10) diagnosed with osteogenesis imperfecta before pregnancy. We detected the variant of c.1118G > C (p. Gly373Ala) in COL1A2 for the fetus in case 9, and found the variant of c.178C > T (p. Arg60*) in GORAB for the fetus in case 10 (Fig. 3, 4). The two variants related to osteogenesis imperfecta and inherited from their mother, but one genetic type is autosomal dominant (AD) [26] and the other is autosomal recessive (AR) [27]. The fetus of case 9 died before delivery at 35 weeks of gestation, while the newborn of case 10 had no significant abnormality in bone development up to 2-years old in the following-up survey.
In the case 2, 5 and 8, we found the WNT1or COL1A1-related VUS in osteogenesis imperfecta, and DYNC2H1 and NEK1-related asphyxiative hypoplasia of thorax. Notably, the fetuses in these cases all had short lower limbs, which was determined after abortion, while the proteins encoded by the gene variants were predicted to be deleterious using the SIFT and Polyphen analysis. Furthermore, we had negative findings by WES in 8 cases, and the 8 infants were normal in skeletal development after delivery in the telephone follow-up. Additionally, our study has some limitations. For instance, this is a single-center study with a relatively small case number and selected population. Prospective multicenter studies with large sample sizes are needed to obtain more reliable data.
Conclusions
In sum, skeletal dysplasia is mostly hereditary. Our study has obtained 16/24 (66.67%) cases carrying 12 different skeletal dysplasia genotypes, which will broaden the spectrum of FSD in Chinese patients. Genetic diagnosis combining with ultrasound scanning enhances the accurate diagnosis of FSD in utero, then can provide appropriate genetic counseling.
Availability of data and materials
The datasets generated and analyzed during the current study are not publicly available due to patient privacy and confidentiality. Anonymized data can be made available from the corresponding author upon reasonable request.
Abbreviations
- FSD:
-
Fetal skeletal dysplasia
- FGFR3:
-
Fibroblast growth factor receptor3
- FBN1:
-
Fibrillin-1
- FBN2:
-
Fibrillin-2
- COL1A1:
-
Collagen type I alpha 1 chain
- COL1A2:
-
Collagen type I alpha 2 chain
- CUL7:
-
Cullin-7
- DYNC2H1:
-
Dynein cytoplasmic 2 heavy chain 1
- IMPAD1:
-
Inositol monophosphatase domain containing 1
- WNT1:
-
Wnt family member 1
- GORAB:
-
Golgi-associated Rab-binding protein
- OBSL1:
-
Obscurin like cytoskeletal adaptor 1
- NEK1:
-
NIMA (never-in-mitosis A)-related kinase 1
- VUS:
-
Variant of undetermined significance
- WES:
-
Whole exome sequencing
- WGS:
-
Whole gene sequencing
- NIPT:
-
Non-invasive prenatal test
- BPD:
-
Biparietal diameter
- HC:
-
Head circumference
- HL:
-
Humerus length
- FL:
-
Femur length
- NT:
-
Nuchal translucency
- CNV:
-
Copy number variations
- OI:
-
Osteogenesis imperfecta
- NGS:
-
Next-generation sequencing
- AR:
-
Autosomal recessive inheritance
- AD:
-
Autosomal dominant inheritance
References
Liu Y, Wang L, Yang YK, et al. Prenatal diagnosis of fetal skeletal dysplasia using targeted next-generation sequencing: an analysis of 30 cases. Diagn Pathol. 2019;14(1):76–89.
Krakow D, Rimoin DL. The skeletal dysplasias. Genet Med. 2010;12(1):327–41.
Franzone JM, Shah SA, Wallace MJ, Kruse RW. Osteogenesis imperfecta: a pediatric orthopedic perspective. Orthop Clin North Am. 2019;50(2):193–209.
Xu Y, Li L, Wang C, et al. Clinical and molecular characterization and discovery of novel genetic mutations of Chinese Patients with COL2A1-related dysplasia. Int J Biol Sci. 2020;16(5):859–68.
Zheng C, Lin X, Xu X, et al. Suppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias. EBio Med. 2019;40(2):695–709.
Cao YJ, Zhang H, Zhang ZL. Novel mutations in the WNT1, TMEM38B, P4HB, and PLS3 genes in four unrelated chinese families with osteogenesis imperfecta. Endocr Pract. 2019;25(3):230–41.
Mortier GR, Cohn DH, Cormier-Daire V, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179:2393–419.
Yang K, Shen M, Yan Y, et al. Genetic analysis in fetal skeletal dysplasias by trio whole-exome sequencing. Biomed Res Int. 2019;3(10):1155–64.
Fu F, Li R, Li Y, et al. Whole exome sequencing as a diagnostic adjunct to clinical testing in fetuses with structural abnormalities. Ultrasound Obstet Gynecol. 2018;51(4):493–502.
Chandler N, Best S, Hayward J, et al. Rapid prenatal diagnosis using targeted exome sequencing: a cohort study to assess feasibility and potential impact on prenatal counseling and pregnancy management. Genet Med. 2018;20(11):1430–7.
Huang Y, Liu C, Ding H, et al. Exome sequencing in fetuses with short long bones detected by ultrasonography: a retrospective cohort study. Front Genet. 2023;14(2):1032346.
Papageorghiou AT, Fratelli N, Leslie K, et al. Outcome of fetuses with antenatally diagnosed short femur. Ultrasound Obstet Gynecol. 2008;31(5):507–11.
Flanagan SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomark. 2010;14(4):533–7.
Pan Z, Fu Z, Luo C, et al. CDH1 germline mutations in a Chinese cohort with hereditary diffuse gastric cancer. J Cancer Res Clin Oncol. 2022;148(8):2145–51.
Liu W, Shi X, Li Y, et al. The evaluation of genetic diagnosis on high-risk fetal CAKUT. Front Genet. 2022;13(5): 869525.
Crossley BM, Bai J, Glaser A, et al. Guidelines for Sanger sequencing and molecular assay monitoring. J Vet Diagn Invest. 2020;32(6):767–75.
Pajkrt E, Chitty LS. A sonographic approach to the prenatal diagnosis of skeletal dysplasias. Prenat Diagn. 2019;39(9):701–19.
Wang L, Takai Y, Baba K, et al. Can biparietal diameter-to-femur length ratio be a useful sonographic marker for screening thanatophoric dysplasia since the first trimester? A literature review of case reports and a retrospective study based on 10,293 routine fetal biometry measurements. Taiwan J Obstet Gynecol. 2017;56(3):374–8.
Han J, Yang YD, He Y, et al. Rapid prenatal diagnosis of skeletal dysplasia using medical trio exome sequencing: benefit for prenatal counseling and pregnancy management. Prenat Diagn. 2020;40(5):577–84.
Liu J, Huang L, He Z, et al. Clinical value of genetic analysis in prenatal diagnosis of short femur. Mol Genet Genom Med. 2019;7(11): e978.
Peng Y, Yang S, Huang X, et al. Whole exome sequencing analysis in fetal skeletal dysplasia detected by ultrasonography: an analysis of 38 cases. Front Genet. 2021;12(9): 728544.
Tang H, Zhang Q, Xiang J, et al. Whole exome sequencing aids the diagnosis of fetal skeletal dysplasia. Front Genet. 2021;12(3): 599863.
Newell K, Smith W, Ghoshhajra B, et al. Cervical artery dissection expands the cardiovascular phenotype in FBN1-related Weill–Marchesani syndrome. Am J Med Genet A. 2017;173(9):2551–6.
Isik E, Arican D, Atik T, et al. A rare cause of syndromic short stature: 3M syndrome in three families. Am J Med Genet A. 2021;185(2):461–8.
Rosario M, Venselaar H, Knoll U, et al. Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP. Am J Hum Genet. 2011;88(5):608–15.
Augusciak-Duma A, Witecka J, Sieron AL, et al. Mutations in the COL1A1 and COL1A2 genes associated with osteogenesis imperfecta (OI) types I or III. Acta Biochim Pol. 2018;65(1):79–86.
Yang H, Albiol L, Chan WL, et al. Examining tissue composition, whole-bone morphology and mechanical behavior of GorabPrx1 mice tibiae: a mouse model of premature aging. J Biomech. 2017;65(10):145–53.
Acknowledgements
We thank all the subjects who participated in this study.
Funding
This study was supported by The National Key Research and Development Program of China (2018YFC1002904).
Author information
Authors and Affiliations
Contributions
WL, JC and XS performed laboratory investigation and data analysis. WL, YL and FQ over-saw laboratory analyses and provided statistical support. WLand YWwrote and reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Research Ethics Committee of Tongji Hospital affiliated to Tongji Medical College of Huazhong University of Science and Technology (IRB reference number: M2019036). The parents provided a signed informed consent prior to intrauterine diagnosis and sample collection in our country.
Consent for publication
Yes.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, W., Cao, J., Shi, X. et al. Genetic testing and diagnostic strategies of fetal skeletal dysplasia: a preliminary study in Wuhan, China. Orphanet J Rare Dis 18, 336 (2023). https://doi.org/10.1186/s13023-023-02955-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-023-02955-4